

Abstract
The nematode Caenorhabditis elegans has emerged as an informative experimental system for analysis of meiosis, in large part because of the advantageous physical organization of meiotic nuclei as a gradient of stages within the germline. Here we provide tools for detailed observational studies of cells within the worm gonad, including techniques for light and electron microscopy.
Keywords: C. elegans, meiosis, nuclear architecture, chromosomes, meiotic prophase, immunofluorescence, fluorescence in situ hybridization, electron microscopy
1. Introduction
Over the last several years, C. elegans has become one of the preeminent organisms for experimental studies of meiosis. This is largely due to the facility of combining powerful molecular genetics with high-resolution cytological analysis in this system. Many key aspects of meiosis, including meiotic entry and exit, sex determination in the germline, chromosome pairing and synapsis, crossover recombination, meiotic checkpoints, chromosome cohesion, and assembly and function of the segregation apparatus, have been investigated in the worm. Available mutants enable the researcher to manipulate most of these processes in versatile ways, facilitating ongoing and future studies. The goal of this chapter is to provide robust techniques for visualizing meiosis in the nematode. We will cover methods for sample preparation and staining to permit subcellular and subnuclear localization of chromosome loci and proteins through fluorescence microscopy, as well as preparation techniques for transmission electron microscopy. These techniques have been developed for C. elegans but can be easily adapted to related species, such as C. briggsae and C. remanei, as well as more distantly related organisms with a similar body plan and composition.
In a wild-type population of C. elegans, most animals are self-fertilizing hermaphrodites, which contain a bilobed gonad. The ovotestes sequentially produces sperm and then oocytes. Males, which arise spontaneously in wild-type populations at frequencies of ~1/500, can be propagated by deliberate matings with hermaphrodites. Oocytes (in hermaphrodites) and spermatocytes (in both males and hermaphrodites) display similar meiotic progression through mid-prophase, then diverge in their chromosome condensation and segregation behavior in that oocytes undergo the classically defined stages of diplonema and diakinesis, while spermatocytes condense their chromosomes and divide twice without any obvious individualization of homologs or bivalents. As in most metazoans, oocytes divide asymmetrically to produce a single haploid female pronucleus that can be incorporated into a new zygote, eliminating three quarters of their genetic material into the polar bodies. By contrast, spermatocytes undergo two symmetrical divisions to generate four potentially functional spermatids.
C. elegans offers the great advantage that all stages of meiosis are present in a convenient arrangement within the gonads of individual adult animals (Fig. 11.1A and B). The gonad contains a monolayer of germ cells arranged around the periphery of a “rachis” comprised largely of extracellular proteins, much as kernels are arranged on an ear of maize. There are no nurse cells, meaning that all of the germline nuclei within the gonad are destined to undergo meiosis. However, a substantial fraction of oocytes will normally exit this pathway at late pachytene and undergo apoptosis, rather than condensing and executing meiotic divisions. Nuclei with defects in chromosome synapsis or recombination are preferentially targeted for apoptosis, but many apparently normal nuclei are also eliminated in wild-type animals.
Fig. 11.1.
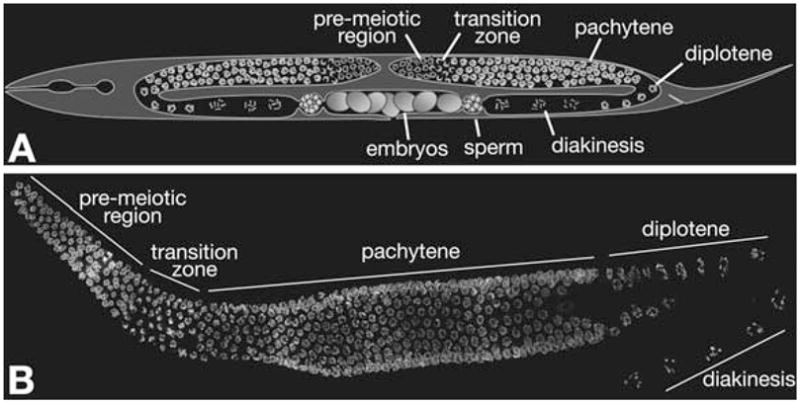
Meiotic progression in C. elegans. (A) Diagram of a C. elegans hermaphrodite. The bi-lobed gonad contains a temporal progression of meiotic nuclei. (B) Whole gonad dissected out of a wild-type worm and stained with DAPI. The stages of meiotic prophase can be clearly identified by the chromatin morphology.
The plasma membranes of meiotic cells are discontinuous, and the gonad is therefore regarded as a syncytium. This organization permits experimental introduction of pharmacological agents, DNA, or fluorescent tracers into the cells by microinjection into the distal region of the gonad. However, membrane-impermeant molecules do not readily enter the nuclei other than during nuclear envelope disassembly during the premeiotic mitoses and meiotic divisions. As nuclei enter and progress through the stages of meiosis, they also advance spatially from the distal to the proximal end of the gonad (defined with respect to the vulva, where embryos emerge, or the spermatheca in males). The gonad therefore contains a gradient of premeiotic and meiotic stages, greatly facilitating analysis of the effects of mutations or other perturbations.
The stages of meiosis can be clearly identified by the morphology of the chromatin within meiotic nuclei, as well as their locations within the gonad (Fig. 11.1B). As nuclei enter meiosis, the chromosomes, which are normally dispersed around the periphery of the nucleus, become somewhat polarized, resulting in a crescent-shaped appearance of nuclei following DNA staining. This region, referred to as the “transition zone” in C. elegans, represents the leptotene and zygotene stages of meiosis, during which homologous chromosomes undergo pairing and synapsis, and recombination initiates through formation of double-strand breaks (DSBs). The cause of the polarization is not fully understood, although it likely involves interactions between the chromosomes and the nuclear envelope. As the chromosomes complete synapsis, they redisperse around the nuclear periphery, and recombination events are completed. This pachytene stage is followed by diplotene, during which the synaptonemal complex begins to disassemble, revealing the physical linkages produced by recombination, or chiasmata. The final stage of prophase is diakinesis, where the chromosomes condense dramatically in preparation for meiotic division. These events are readily observed in oocytes, due to a dramatic expansion of nuclear volume during late prophase, but not in spermatocytes, where both the chromosomes and the nuclei compact prior to segregation.
Oocytes paused at diakinesis are ovulated through the spermatheca, where sperm entry triggers the first meiotic division, followed closely by MII. These division events can be observed in the newly fertilized embryos, which are retained in the uterus of the adult hermaphrodite until after several rounds of mitotic cell division have occurred.
The gonad is optically transparent, as is the entire body of the animal, making the worm amenable to whole-mount immunofluorescence and in situ hybridization techniques for localization of meiotic proteins and DNA sequences. Germline nuclei can also be dissociated prior to staining, but this eliminates the spatial information available within the context of the intact tissue, and we will not detail such techniques here.
In other model systems, fluorescent tagging of proteins is commonly used in cytological observation in fixed or live cells. However, in C. elegans, expression of transgenes in the germline is problematic due to the phenomenon of cosuppression, in which high-copy transgenes not only fail to be expressed but also silence homologous genomic sequences (1). This problem has been circumvented in some cases by introduction of transgenes at low copy number through microparticle bombardment, a.k.a. ballistic or biolistic transformation (2). However, bombardment is an inefficient procedure that has not yet routinely recapitulated the normal expression of genes required during meiosis. For these reasons, we focus here on cytological methods that do not rely on transgene expression.
The following sections outline methods to perform immunofluorescence and fluorescence in situ hybridization (FISH) to localize proteins and DNA sequences in meiotic nuclei, and preparation methods for electron microscopic analysis of the worm gonad. Where appropriate, we have also suggested variations that may help to optimize these protocols for particular experimental goals. Complementary methods for analysis of recombination in C. elegans are presented elsewhere in this volume (Chapter 7).
2. Materials
2.1. Light Microscopy
2.1.1. Immunofluorescence
Egg Buffer (10×): 250 mM HEPES-NaOH, pH 7.4, 1.18 M NaCl, 480 mM KCl, 20 mM EDTA, 5 mM EGTA. Sterilize by filtration and store at room temperature (see Note 1).
NPG-Glycerol mounting medium: 2 g N-propyl gallate, 50 g optical-grade glycerol. Dissolve by mixing slowly on nutator or rotary mixer overnight, and store at room temperature. Dissolution of NPG may also be expedited by heating the solution gently in a waterbath or microwave oven.
Flat aluminum block, such as those typically supplied with dry heat blocks, chilled on a bed of dry ice.
Egg Buffer + Tween-20 + azide (EBTA) (1 mL): 100 μL 10× Egg Buffer, 10 μL 10% Tween-20 (make a 10% (v/v) solution in H2O, sterile filter and store at room temperature), 40 μL 0.5 M sodium azide (toxic), 850 μL H2O. Make fresh on the day of use.
Fixative solution (for 3.7% formaldehyde final): 100 μL 10× Egg Buffer, 200 μL 37% formaldehyde, 700 μL H2O. Make fresh on the day of use (see Note 2).
Methanol. Stored in Coplin jar at −20°C (see Note 3).
10× PBS (Phosphate-Buffered Saline): Per liter, add 80 g NaCl, 2 g KCl, 14.4 g Na2HPO4, 2.4 g KH2PO4. Adjust pH to 7.4 with HCl.
PBST (1 L): 100 mL 10× PBS, 890 mL H2O, 10 mL 10% Tween-20.
Blocking agents: normal serum from the species used for secondary antibody production or bovine serum albumin (BSA).
Primary antibodies (see Note 4).
Secondary antibodies.
DAPI or Hoechst dyes for fluorescent counterstaining of DNA.
Mounting Medium: 35 μL 2 M Tris base (not pH-adjusted), 450 μL NPG-Glycerol. Make fresh on the day of use. Mix this solution using a P-1000 tip that has been cut to a larger bore with a razor blade, vortex to mix further, then spin for 1–2 min at top speed to remove bubbles. Optional: Using a finely pulled glass capillary, sparge the solution with nitrogen to reduce oxygen content, then spin to remove bubbles.
18 × 18 mm coverslips No. 1.
22 × 22 mm coverslips No. 1½.
No. 11 Scalpel blades. We prefer Feather brand (available from Electron Microscopy Sciences), which are more consistently sharp than other commercial brands.
Polylysine-subbed slides, or commercial charged slides such as Histobond (Marienfeld) or Superfrost Plus (Erie Scientific) (75 × 25 × 1 mm).
Humid Chamber: a flat, waterproof container (e.g., Tupper-ware box) with moist paper towels in the bottom covered with Parafilm.
Nail polish (we prefer to use clear Sally Hansen Hard-as-Nails, but other brands/colors are also acceptable).
2.1.2. Fluorescence In Situ Hybridization (FISH)
Egg Buffer (10×): (see Section 2.1.1)
NPG-Glycerol mounting medium: (see Section 2.1.1)
Hybridization solution: 5 mL formamide, 1.5 mL 20× SSC, 1.0 g dextran sulfate, H2O added to a total vol of 10.0 mL. Prepare in advance (dextran sulfate takes a long time to dissolve). Store at 4°C. Stable for several months. 20× SSC is 3 M NaCl, 0.3 M sodium citrate, pH 7.
Flat aluminum block chilled on a bed of dry ice.
Egg Buffer + Tween-20 + azide (EBTA) (1 mL): (see Section 2.1.1)
1.25× EB (1 mL): 100 μL 10× Egg Buffer, 700 μL H2O. Aliquot 32 μL of 1.25× EB into each of several 0.5-mL Eppendorf tubes, one tube per sample that will be fixed. Make fresh on the day of use.
4% (w/v) Ethylene glycol bis[succinimidylsuccinate] (EGS) in dimethyl formamide (DMF): Weigh by difference into a 1.5-mL screw-cap conical tube approximately 2–4 mg EGS (Pierce), stored at room temperature over Drierite dessicant. This cross-linking agent is not soluble in aqueous medium, so it is dissolved in DMF and then added to buffer just before use. Add DMF (toxic/mutagenic) as required to yield a 4.0% w/v solution – i.e., 25 μL DMF per mg of EGS (use plain old bottled DMF, not special-grade dry DMF in ampules, since it will not dissolve EGS as well). 8 μL of this solution is used per sample. Make fresh on the day of use.
Methanol. Stored in Coplin jar at −20° (see Note 3).
2x SSCT (per liter): 100 mL 20× SSC, 1 mL Tween-20. The pH does not usually require adjusting, but should be ~7.
Egg Buffer + 3.7% formaldehyde (EBF) (50 mL): 5 mL 10× Egg Buffer, 5 mL 37% formaldehyde, 40 mL H2O. Make fresh on the day of use.
Formamide (Fluka 47670; substitute other brands at your own risk). Toxic. Store at 4°C.
DAPI or Hoechst dyes for fluorescent counterstaining of DNA.
Mounting Medium: (see Section 2.1.1).
18 × 18 mm coverslips No. 1.
22 × 22 mm coverslips No. 1½.
No. 11 Scalpel blades (Feather brand).
Histobond slides (75 × 25 × 1 mm).
Humid Chamber: a large Tupperware container with moist paper towels in the bottom covered with Parafilm.
Heatblock set at 95°C with flat surface facing up.
Nail polish (Sally Hansen Hard-as-Nails, clear).
2.1.3. FISH Probe Labeling
Restriction Enzymes (New England Biolabs): AluI, HaeIII, Mbo1, MseI, MspI, RsaI.
Glycogen (molecular biology grade). Glycogen is added to ethanol precipitation reactions to promote precipitation of small DNA fragments and to produce a visible pellet.
3 M sodium acetate (for DNA precipitation).
Ethanol (95–100%).
3.3 μL 1 mM aminoallyl-dUTP (aa-dUTP). This amine-modified nucleotide is available in powder form from Sigma, or from Ambion as a 50 mM stock solution (nucleotides are more stable in solution than as lyophilized solids, so this may be preferable). If you buy the solid form, dissolve 1 mg in 164 μL water to make a 10 mM stock, aliquot 20 μL per tube, and store −80°C. Keep a working aliquot at −20°C and dilute 1:10 to make a 1 mM working stock when needed.
6.6 μL 1 mM unlabeled dTTP (freshly diluted from concentrated stock solution).
Recombinant terminal deoxynucleotidyl transferase (TdT). Roche supplies this enzyme at high concentration (400 U/μL) with a 5× TdT reaction buffer (contains Tris pH 7.2, potassium cacodylate, and BSA) and 25 mM CoCl2 solution.
1 M bicarbonate/carbonate buffer: dissolve 0.1 g sodium carbonate and 0.75 g sodium bicarbonate in 10 mL of water. Sterilize this solution using a syringe filter. This will have a pH of ~9. This can be stored frozen in aliquots, but if stored at room temperature the pH will become more acidic with time due to reaction with CO2 in the air.
Fluorescent NHS-esters (reactive dyes for probe labeling) (see Note 5).
1 M glycine-HCl pH 8.
2.2. Electron Microscopy
2.2.1. High-Pressure Freezing (HPF)
High-pressure freezer (see Note 6).
Specimen carriers (see Note 7).
20% BSA in M9 buffer. M9 buffer (per liter) contains 3 g KH2PO4, 6 g Na2HPO4, 5 g NaCl, 1 mL 1 M MgSO4 (see Note 8).
Paper points (Ted Pella).
1-hexadecene.
60 mm Petri dish lined with filter paper.
0.5–10 μL Pipetman with tips.
Fine forceps.
Worm plates with thick E. coli lawn.
4-liter Dewar filled with liquid nitrogen.
2.2.2. Freeze Substitution
2.2.3. Embedding
2.2.3.1. Epoxy Resins
An epoxy resin of your choice such as Epon, or Epon-Araldite. We do not recommend Spurr’s resin unless mixed 1:1 with Epon.
Microscope slides. We prefer those with frosted or white ends so they can easily be labeled with a pencil. Do not use ink, as it can be dissolved if resin contacts the label.
Parafilm.
Teflon release agent (Miller-Stephenson).
Dry acetone, preferably in 100 mL unopened bottles (Electron Microscopy Sciences).
A piece of cardboard about 5 × 7 in. with two 6-in. wooden sticks (Electron Microscopy Sciences) taped about 2 in. apart oriented parallel to the long direction of the cardboard.
2.2.3.2. Methacrylate Resins
LR White resin (hard grade) (see Note 11).
Flat-bottomed embedding capsules (Ted Pella).
Aclar plastic (Ted Pella, Inc., Redding, CA; cat. no. 10501-10).
Office paper punch.
Fine forceps or needles.
Parafilm.
Vacuum embedding oven set at 60°C.
2.2.4. Remounting Worms for Sectioning
Epoxy 907 Adhesive System (Miller-Stephenson).
No. 11 scalpel blades.
Light microscope with DIC or phase optics.
Blank epoxy blocks for remounting. Both BEEM capsule and flat-embed mold geometries are recommended.
Jeweler’s saw with fine blade.
Table-top vise.
Double-edged razor blades.
3. Methods
3.1. Light Microscopy
3.1.1. Immunofluorescence
Before you start: make EBT and fixative solutions and place flat aluminum block on dry ice so it starts cooling.
Pick 15–25 worms into a 30-μL drop of EBT on an 18 × 18 mm coverslip on top of a glass slide (see Note 12). Use a scalpel blade to nick each worm immediately behind the pharynx and/or just before the tail to release the gonad arms.
Pipette 30 μL of fixative solution into the drop of dissected worms. Pipette up and down a few times to mix and extrude more gonads. Let worms fix for 4–5 min total (see Note 2).
Pipette off all but 15 μL, being careful to leave the tissue behind. Pick up the coverslip by touching the drop with the center of a HistoBond slide. Invert slide and wick away excess liquid from the edges of the coverslip using a torn piece of absorbent paper. The more liquid you remove, the better the worms will stick to the slide.
Freeze-crack the samples. Freeze the sample by placing it on the aluminum block in dry ice for >5 min. Carefully flick off the cover slip by catching the edge with a fresh razor blade.
Place the slide immediately in methanol (prechilled to −20°C) for 1 min, then move to a Coplin jar of PBST at room temperature (see Note 3).
Repeat Steps 2 through 6 until all samples are dissected.
Wash slides 3× (10 min/wash) by moving slides to fresh Coplin jars of PBST.
Block slides in a 1:10 dilution of normal serum proteins (typically supplied at 6% w/vol following reconstitution) in PBST or 0.5% BSA in PBST. Pipette 50–100 μL of block onto the slide near the worms and cover with a Parafilm coverslip (just a piece of Parafilm cut to size). Place slides in a humid chamber for >30 min at room temp.
Primary Antibody. Remove the Parafilm coverslip by placing each slide in a Coplin jar of PBST and letting it float off. Apply 50–100 μL of primary antibody (see Note 4) in PBST (or PBST plus block), cover with a Parafilm coverslip, and incubate in a humid chamber for 2 h at room temperature or overnight at 4°C.
Wash slides 3× (10 min/wash) by moving slides to fresh Coplin jars of PBST.
Secondary Antibody. Remove the Parafilm coverslip as in Step 10. Apply 50–100 μL of secondary antibody in PBST (or PBST plus block), cover with a Parafilm coverslip, and incubate in a humid chamber for 2 h at room temperature or overnight at 4°C.
Wash and DAPI stain samples (see Note 13). Remove the Parafilm coverslip as in Step 10. Wash slides 10 min in a fresh Coplin jar of PBST, then move to Coplin jar of PBST plus 0.5 μg/mL DAPI (add 5 μL of 5 mg/mL stock to 50 mL PBST in a Coplin jar) and incubate for >10 min. Finally, wash slides >30 min in a fresh Coplin jar of PBST.
Mount slides. Pipette 10–12 μL of mounting medium onto a 22 × 22 mm coverslip. Remove the slide from buffer, remove as much buffer as possible without desiccating the tissue sample by wiping with a tissue. Be careful not to wipe off your worms. Invert the slide and touch it to the mounting medium. Invert the slide again, and carefully wick off any excess mounting medium. Seal with nail polish.
Observe slides (see Note 14).
3.1.2. Fluorescence In Situ Hybridization (FISH)
Before you begin dissections: prepare EBTA, 1.25× EB, and 4% (w/vol) EGS in DMF. Also, place aluminum block on dry ice, flat side up, for freezing slides.
Pick 15–25 worms into a 30-μL drop of EBTA on a 18 × 18 mm coverslip on top of a glass slide (see Note 12). Use a scalpel blade to nick each worm immediately behind the pharynx and/or just before the tail to release the gonad arms.
To a 32-μL aliquot of 1.25× EB, add 8.0 μL 4% EGS in DMF. Transfer 30 μL of this solution to the drop with the dissected worms. Pipette up and down a few times to mix and extrude more gonads.
Using a P-20 Pipetman, remove all but ~15 μL, being careful to leave the tissue behind. Pick up the coverslip by touching the drop with the center of a HistoBond slide. Invert slide and wick away excess liquid from the edges of the coverslip using a torn piece of absorbent paper. The more liquid you remove, the better the worms will stick to the slide.
Incubate in a humid chamber for 30 min at room temperature.
Freeze-crack the samples. Freeze the sample by placing it on the aluminum block in dry ice for >1 min. Carefully flick off the cover slip by catching the edge with a fresh razor blade.
Place the slide immediately in −20° methanol for 1 min, then move to a Coplin jar of 2x SSCT at room temperature (see Note 3).
Repeat Steps 2 through 7 until all samples are dissected.
Remove each slide from 2x SSCT and drain. Wearing gloves, place in EBF in Coplin jar. Wait 5 min.
Carefully dip slides into a beaker filled with 2xSSCT to remove fixative. Transfer slides to Coplin jar filled with 2xSSCT and wash for 5 min. Wash with one change of 2xSSCT for 5 min.
Prepare 100 mL of 50% formamide in 2xSSCT and divide equally between two Coplin jars. Label jars No. 1 and No. 2.
Transfer slides to jar No. 1. Wait 5–10 min and move slides to jar No. 2. Place jar No. 2 at 37°C overnight.
Remove jar with slides from 37°C while preparing probe solution.
Make up probe solution [per slide: 20–500 ng of each probe (see Section 3.1.3, Step 9)] diluted into a total vol of 15 μL hybridization solution.
For each sample: pipette 15 μL probe solution onto a 22 × 22 mm coverslip. Wearing GLOVES: Drain slides and wick away as much liquid as possible without damaging or drying out sample. Touch the worms to the drop of probe solution on the coverslip, allow the liquid to spread out, and invert the slides. Seal coverslips with nail polish.
Denature slides (two at a time) on a 95°C heat block for 3 min.
Place in a humid chamber at 37°C overnight (see Note 15).
Carefully remove coverslip by using a sharp razor blade to cut off nail polish.
Wash slides in 50% formamide in 2xSSCT at 37°C for at least an hour, first in jar No. 1 then in jar No. 2 (see Note 15).
Wash away formamide and DAPI stain samples (see Note 13). Wash slides 10 min in a fresh Coplin jar of 2x SSCT, then move to Coplin jar of 2x SSCT plus 0.5 μg/mL DAPI (add 5 μL of 5 mg/mL DAPI stock to 50 mL 2x SSCT in a Coplin jar and mix). Incubate for >10 min. Finally, wash slides >30 min in a fresh Coplin jar of 2x SSCT.
Mount slides. Pipette 10–12 μL of Mounting Medium onto a 22 × 22 mm coverslip. Remove the slide from buffer, remove as much buffer as possible without desiccating the tissue sample by wiping with a tissue. Be careful not to wipe off your worms. Invert the slide and touch it to the mounting medium. Invert the slide again, and carefully wick off any excess mounting medium. Seal with nail polish.
Make a sacrifice to the FISH gods, skip around the room, or perform other rituals as desired.
Observe slides (see Note 14).
3.1.3. FISH Probe Labeling
Generating DNA for probe synthesis: probes can be generated from most DNA sources (PCR products, plasmids, cosmids, oligonucleotides, etc) (see Note 16). For probes consisting of DNA fragments longer than ~100 bp, the DNA must be digested into small fragments. This is most reliably and economically achieved using multiple restriction endonucleases. To digest 30 μg DNA (scale up or down as appropriate, do not exceed ~0.1 μg/mL DNA in digest): Dilute 30 μg probe DNA to a vol of 250 μL with H2O, add 30 μL NEB Buffer 2 (10×), 3 μL NEB 10 mg/mL BSA (100×), and 30–60 units each of the following six enzymes: AluI, HaeIII, Mbo1, MseI, MspI, and RsaI (note: you may choose to substitute Tsp509 I for this enzyme mixture to digest the C. elegans 5S rDNA repeat). Add water to a final vol of 300 μL. Mix and place at 37°C for ≥2 h (overnight digestion is fine).
Check digest by loading 0.25–5 μg DNA onto a 2% gel and compare to low MW markers. The average fragment size should be ≤150 bp.
Ethanol precipitate digested DNA: Add 1 μL 20 mg/mL glycogen, 30 μL 3 M sodium acetate (don’t use ammonium acetate, since ammonium ions will compete for dye in the conjugation reaction), and 750 μL 95–100% ethanol. Chill at −20°C or colder for 10 min (or longer). Spin down 15 min at maximum speed, wash pellet carefully with cold 70% ethanol, and air-dry pellet or remove residual ethanol in a centrifugal evaporator. Resuspend DNA in 30 μL H2O.
If DNA concentration was not accurately known prior to digestion, you should now measure OD260 of a 1:200 dilution to calculate actual DNA concentration.
Tailing 10 μg of DNA with aa-dUTP (can be scaled up or down as desired): Dilute 10 μg fragmented DNA or oligonucleotide with H2O to a total vol of 50 μL. If DNA is double-stranded: Cap tube and place tube in a 95°C water bath for 2 min, then chill immediately on ice; spin briefly to collect condensed water from sides of tube (single-stranded DNA is a preferred substrate for TdT). Add at room temperature: 20 μL Roche 5× TdT reaction buffer (contains Tris-HCl pH 7.2, potassium cacodylate, and BSA), 20 μL Roche 25 mM CoCl2 solution (or other CoCl2 stock to 5 mM final), 3.3 μL 1 mM aa-dUTP, 6.6 μL 1 mM unlabeled dTTP, 2 μL (800 units) recombinant Terminal deoxynucleotidyl Transferase (TdT). Incubate for 1 h at 37°C.
Ethanol precipitate digested DNA: Add EDTA to 5 mM final to chelate the cobalt. Add 1 μL 20 mg/mL glycogen, 10 μL 3 M sodium acetate (don’t use ammonium acetate), and 500 μL 95–100% ethanol. Chill at −20°C or colder for 10 min (or longer). Spin down 15 min at maximum speed, wash pellet carefully with cold 70% ethanol, and air-dry pellet (or remove residual ethanol in a centrifugal evaporator) (see Note 17).
Conjugation with dye: resuspend 10 μg of aa-labeled DNA in 10 μL H2O. To one aliquot of dry dye (see Note 5), add 5 μL of DNA (5 μg) in water and 3 μL of 1 M bicarbonate/carbonate buffer. If using Alexa dyes, also add 1.5 μL high-quality DMSO to increase the solubility of the dye. Mix thoroughly by tapping, spin the tube briefly, and incubate the reaction shielded from light for 1 h at room temperature (longer is fine but probably doesn’t improve incorporation). Add 1 μL 1 M glycine-HCl pH 8 to quench the remaining reactive dye. Incubate a further 15 min at room temperature.
Ethanol precipitate digested DNA: Dilute labeling reaction to 100 μL with H2O, add 1 μL 20 mg/mL glycogen, 100 μL 4 M ammonium acetate, and 500 μL 95–100% ethanol. Chill at −20°C or colder for 10 min (or longer). Spin down 15 min at maximum speed to pellet labeled DNA, wash pellet carefully with cold 75% ethanol (labeled DNA may be slightly soluble in 70% ethanol due to hydrophobic properties of the dyes), and air-dry pellet (or remove residual ethanol in a centrifugal evaporator) (see Note 18).
Resuspend the labeled probe DNA in hybridization buffer (see Note 19). It is convenient to dissolve probes at a concentration such that a standard volume (typically 0.5 μL) is used for each sample to be hybridized. Complex, single-copy probes (such as cosmid mixtures or BACs) should be stored at concentrations as high as 1 μg/μL, while high-copy probes (targeting rDNA or other repetitive elements) can be made to 40 ng/μL, since less is needed per hybridization. Store probes in nondefrosting −20°C or −80°C freezer.
3.2. Electron Microscopy
3.2.1. High-Pressure Freezing
Start up the high-pressure freezer according to instructions.
Place a piece of filter paper in a small Petri dish and saturate the filter paper with 1-hexadecene. Place specimen planchettes on the filter paper, coating both sides if you are using a BAL-TEC or Wohlwend system, just the cup side if you are using the Leica system. Wick or wipe off the excess 1-hexadecene prior to loading the worms and/or filler solution. 1-Hexadecene acts as a release agent so that your sample will come out of the metal planchette easily.
Select a type of specimen planchette that is appropriate for the stage of worm you are freezing. In general, the depth of the planchette should be about the same as the thickness of the worm. For gravid adults, use a 100 μm deep well.
If using 20% BSA as a filler, add just enough to fill the cavity of the specimen planchette. Try to make sure that the BSA doesn’t touch the rim of the planchette because that will allow the worms to crawl out of the center of the cup.
Pick 10–15 worms of the appropriate stage and/or phenotype and transfer them to the BSA in the cup.
If necessary, use paper point wicks or a micropipette to adjust the level in the cup so that it is just barely overfilled. Under no circumstances should you underfill the cup and trap air because the air will interfere with the heat transfer during freezing.
Depending on the type of freezer you are using, either (1) place a flat-bottomed top planchette on the cup with worms (BAL-TEC and Wohlwend machines), or (2) secure the planchette in a freezing pod (Leica).
Freeze.
Remove planchette from specimen holder tip under liquid nitrogen and transfer to storage vial or into frozen fixative vial for freeze substitution (see Note 20).
3.2.2. Freeze Substitution
-
1a
Fill the automatic freeze substitution (AFS) device with liquid nitrogen, set the program (see Note 10), and cool to − 90°C.
-
1b
If you don’t have an AFS, pre-cool a dry block heater (with 13-mm holes) on dry ice in a box until a dummy vial with acetone cools to −78°C.
-
2
Transfer the cryovials with samples and fixative to the AFS at −90°C, or the dry ice box at −78°C.
-
3
When the program ends, or the dry ice device warms to room temperature, proceed to the embedding process.
3.2.3. Embedding
3.2.3.1. Epoxy Resins
Inspect the tubes to see if the worms have separated from the specimen planchettes or not. If using E. coli as a filler, there is a good chance that the worms may be loose in the bottom of the tube. To ensure that the worms aren’t lost during subsequent solution changes, transfer to a 1.5 mL Eppendorf tube so the samples can be concentrated by either gravity or centrifugation prior to solution transfers.
Rinse the samples a minimum of three times (5 min each) in pure acetone.
Infiltrate in a graded series of resins. We usually start with 25% resin in acetone for 1 h. At this step, it is convenient to remove the worms from the carriers. This can be done by gently nudging the disk of BSA/worms in the cup with fine forceps or a needle. In most cases, the BSA will be fixed around the worms and you will have a solid, disk-shaped object to handle. If you choose to freeze worms surrounded by E. coli, then the worms will often separate from the bacteria at this step and you should transfer them to an Eppendorf tube for further processing so they can be spun down between solution changes.
Continue the infiltration by incubating in 50% resin-acetone mix for 2 h, 75% for 4 h, then a brief rinse in pure resin followed by incubation overnight in pure resin. All steps should be done on a rocker or rotator to facilitate fluid exchange.
Carry out one more exchange of pure resin before the final embedding steps.
For each worm sample to be prepared, prepare two Teflon-coated slides. This is done by dipping slides in a solution of Miller-Stephenson MS-143 V TFE release agent, letting them dry until they are an opaque white, then wiping them clean with paper or cloth.
Label two slides for each sample and on one of the set place a double thickness of Parafilm (about 5 × 10 mm in size) at each end of the clear part of the slide.
Put about 300 μL of resin in a strip between the two pieces of Parafilm.
Using the needle tool, or a sharpened toothpick, transfer the worms to the strip of resin. If the worms were frozen in BSA, they may all be stuck together in a disk of fixed BSA. Using a pair of No. 11 scalpel tips it is possible, with practice, to cut out individual worms from the disk. Worms frozen in E. coli will separate easily, if they did not spontaneously separate during earlier stages of resin infiltration. In either case, spread out individual worms so they are well-separated but not at the edge of the slide. Let them settle to the surface of the slide.
Carefully place the other slide of this set on top of the slide with resin and worms so that only the clear portions of the slide overlap. This is best done by resting one end of the top slide on the piece of Parafilm nearest the labeled end and lowering it slowly so the resin spreads out to the edges of the slides. If the worms were resting on the bottom slide, they should not move much toward the edges.
Place the slide astride the two sticks on the cardboard. When all slides are done, put into an embedding oven to cure. Propping the slides on the sticks makes it easy to remove the slides in case excess resin spills onto the cardboard.
3.2.3.2. Methacrylate Resins
Check the tubes to see if worms have separated from the planchettes and/or filler (see Section 3.4.3.1, Step 1).
Rinse as above for epoxy resins, but after the final acetone rinse, rinse for 5 min in a 1:1 mixture of pure ethanol and pure acetone, then two rinses for 5 min each in pure ethanol (see Note 21).
Infiltrate for 1 hr or more in a 1:1 solution of pure ethanol and LR White Hard Grade.
Rinse twice in pure LR White for 5 min, then pure LR White overnight on a rotator.
Prepare flat-bottomed resin capsules by first punching out disks of Aclar with the hole punch and inserting one Aclar disk in the bottom of each capsule. You can use one capsule per worm, or group three to four worms in one capsule.
Label each capsule with the identifying information.
Transfer the worms to a watch glass or glass spot plate or some small dish that you can see through. Regular plastic Petri dishes may react with LR White and are not recommended. Because the worms are colorless, you will need some dish that you can use with transmitted illumination.
Separate the worms as best you can from each other using needles or No. 11 scalpel blades.
Fill the capsule about half full with LR White.
Transfer one to four worms to each capsule, separating them from each other as best you can and making sure they are on the Aclar disk.
Fill the capsule and put a small square of Parafilm over the top before putting on the top. This will help seal the capsule from oxygen during polymerization.
Polymerize in a 60°C oven for 2 days, preferably a vacuum oven or one that can be filled with a gas such as argon or nitrogen.
3.2.4. Remounting Worms for Sectioning
3.2.4.1. Epoxy Resin
Remove slides with worms from the embedding oven and let cool to room temperature.
Remove excess resin from the edges of the slides with a razor blade or scalpel.
Place a single-edged razor blade between the slides and gently push so that one of the slides separates from the polymerized resin. Remove that slide completely. The remaining slide will have a thin layer of polymerized resin containing the worms.
Make a small mark near one of the worms with a marker pen or scalpel tip.
Find that mark and the nearby worm using bright-field illumination on a compound light microscope. Using the fine focus knob, determine whether the worms are on the top side of the resin, or on the bottom (i.e., nearest the slide). Mark the slide accordingly, e.g., as “top” or “bottom.” This will be extremely helpful when remounting for sectioning.
Scan the slide on the microscope to check the worms and to identify those that have a favorable orientation for your purposes – for example, if you plan to access a particular region of the gonad.
Cut out selected worms from the slide using a No. 11 scalpel blade, and remount on blank epoxy stubs for sectioning. Because you know which side of the thin layer of epoxy the worm is on (“top” or “bottom”), be sure to remount it so that the worm is away from the remount glue and at the surface of the remounted chip. This will enable you to cut sections of worm as soon as you begin sectioning. If the remount glue is not going to be sectioned, then any two-part epoxy, or even superglue may work. However, if remounting requires that some of the glue be sectioned (e.g., to cut cross-sections through the worm) then we recommend using Miller-Stephenson 907 two-part epoxy because it sections well and is stable under the electron beam.
Because worms processed at low temperature tend to retain more cytoplasm, we recommend cutting fairly thin sections, perhaps 50–60 nm. Thicker sections may yield images that are difficult to interpret; however, they may be satisfactory for certain cytoplasmic features.
To find particular features such as the transition zone of the gonad, it may be necessary to screen serial EM sections, or to screen blocks with semi-thick sections in the light microscope, until the right region is located and thin sections can be taken.
3.2.4.2. Methacrylate Resin
Remove the polymerized resin from the flat-bottomed capsule. The capsule plastic is very hard and is best removed by placing the capsule in a vise and sawing along the sides with a jewelers saw. Using razor blades is not advised, since it is both more difficult and there is a risk of injury.
Remove the resin around the edges of the Aclar disk and peel off the Aclar.
Worms can be observed in a light microscope by propping up the block on a slide and focusing on the worm at the surface. An inverted microscope is handy because you can simply place the flat top surface of the block with the worm directly on the slide.
If a polymerized block contains only a single worm and you want longitudinal sections, simply trim and section. If you want cross sections, or if there is more than one worm on the Aclar disk, you will need to cut off the tip (1–2 mm) of the block with the jeweler’s saw, divide the disk with a razor blade so that there is one worm per section, and (if you want cross sections) remount on the side of a flat-embedding mold blank block with remount glue.
Footnotes
Egg buffer was developed by Lois Edgar for isolation and manipulation of blastomeres from living C. elegans embryos. We adapted this buffer for gonad dissection on the assumption that it recapitulates the internal environment of the gonad fairly well, and have found that it yields good chromosome and nuclear morphology. Other buffers we have tested, including “Buffer A” or sperm salts, have produced unsatisfactory morphological preservation based on comparisons between fixed and living tissue. Azide is included in the dissection buffer to rapidly paralyze worms, making them easier to dissect; levamisole or other agents may be substituted. Nonionic detergent (Tween-20 or Triton X-100) is included in the dissection buffer to reduce surface tension and minimize sticking of the tissue to the scalpel blade, as well as facilitating membrane permeabilization. Detergents are also included throughout staining protocols to reduce surface tension of the staining solutions, which enhances retention of the tissue on slides.
C. elegans germline tissue is unusually difficult to fix consistently so as to preserve fine structure while permitting antibody diffusion and epitope accessibility. We do not know why this is the case, but plausible explanations include a high internal protein concentration and/or the composition of the muscular gonad sheath, which may be easily crosslinked by aldehyde fixatives. Many sensitive antibodies will not show specific staining following even brief fixation in 3.7% formaldehyde. We therefore routinely fix at a final concentration of 1% formaldehyde. We have not observed any reproducible differences in fixation using commercial formalin (37% aqueous formaldehyde stabilized with methanol) versus formaldehyde freshly prepared from paraformaldehyde. In some cases, it may be useful to eliminate formaldehyde fixation entirely; following dissection and freeze-cracking, the tissue can be simply fixed by immersion in cold ethanol, methanol, or dimethylformamide (DMF). Sample morphology is less stable and more variable when the formaldehyde fixation step is eliminated, but specific antibodies (e.g., anti-H3meK9 (Upstate, Lake Placid NY)) and cellular structures (e.g. microtubules) are particularly sensitive to formaldehyde fixation. An alternative or adjunct to reducing fixative concentration is to permeabilize the gonad after fixation by treatment with collagenase (we prefer Type 3 from Worthington Biochemical Corporation). However, since collagenase treatment adds several additional variables to the procedure, we do not do this routinely.
Methanol can be substituted with 95% or absolute ethanol or with dimethylformamide (toxic/mutagenic). We have not seen dramatic differences among these three solvents, and we prefer to use methanol or ethanol because of the toxicity of DMF.
Primary antibodies can be preadsorbed against whole worms to remove some background staining. This procedure follows the immunofluorescence protocol closely except that the fixation steps are performed in a microcentrifuge tube. This can be done using mutant worms lacking the protein of interest, but preadsorbtion against wild-type animals is frequently an effective way to suppress background staining without reducing the signal.
Wash worms off of at least one 60-mm plate of gravid adult worms using EBT. Transfer to a 1.5-mL microfuge tube.
Spin at maximum speed in a microcentrifuge for 30 s. Discard supernatant. Add 1 mL fresh EBT and mix.
Spin down and discard supernatant. Wash once with 50% EBT/50% fixative solution for 5 min.
Spin down worms and discard supernatant. Freeze tube on dry ice.
Add 500 μL methanol prechilled to −20°C, mix, and leave on ice for 1 min.
Spin down and remove supernatant. Add 500 μL 1× PBST. Leave on nutator for 30 min at room temperature.
Spin down and remove supernatant. Add 500 μL 1× PBST containing blocking agent (0.5% w/vol serum proteins or BSA). Leave on nutator for 30 min at room temperature.
Spin down and remove supernatant. Add 500 μL 1× PBST containing blocking agent, then 50 μL primary antibody. Leave on nutator overnight at 4°C.
Spin at maximum speed in microfuge. Transfer supernatant to a fresh tube and discard worms. Since the antibody is already diluted 1:10 in this preadsorbtion step, adjust subsequent dilutions accordingly.
In choosing dyes, a major consideration should be the wavelength of the excitation lasers and/or filters on your microscope system. Fluorescent NHS-esters are sold by Invitrogen (the Alexa dyes) and GE Biosciences (Cy3 and Cy5). They are sold in convenient multipacks marketed for labeling of FISH or microarray probes. It’s cheaper to buy in bulk quantities (e.g. 5 mg of dye, which is sufficient for about 100 5-μg DNA labelings), but some loss of activity is expected during aliquoting. Follow manufacturers’ recommendations for aliquoting and storage. Alternatively, aliquots can be prepared by dissolving dyes in dry DMSO, aliquoting appropriate quantities to individual tubes, and evaporating the DMSO under vacuum in a centrifugal evaporator or lyophilizer (this takes a while, since DMSO is not very volatile). Store lyophilized dyes protected from light and desiccated, preferably in a vacuum desiccator, at 4°C; they must be scrupulously free of water and solvents to minimize degradation.
Although it is possible to prepare EM samples of worms by methods other than high-pressure freezing, the quality of preservation is so much better by HPF that the method should be used if at all possible. For nonHPF methods, see the article in Methods in Cell Biology by Shai Shaham on the online Wormbook at: http://www.wormbook.org/toc_wormmethods.html. There are three types of high-pressure freezers currently available: the Bal-Tec HPM 010 (Liechtenstein; www.bal-tec.com), the Wohlwend Compact HPM 01 (Sennwald, Switzerland; www.technotradeinc.com), and the Leica EM PACT systems (Vienna, Austria; www.leica-micro-systems.com). Each of these machines has proven itself capable of excellent freezing of C. elegans worms. To see if there is a machine near you, contact the vendors through their respective websites. The necessary details of how to use these machines to prepare worms for EM is beyond the scope of this article. To fill in the gaps of the overview presented here, see the following articles regarding the Bal-Tec HPM 010, with a section specifically for worms (3), the Leica EM PACT system (4), and more detailed instructions specifically for worms (5–7). Bal-Tec, Ag was recently sold to Leica Microsystems, Vienna. The Bal-Tec HPM 010 is no longer available, though the leica HPM 100 may serve as a replacement.
There are numerous options for specimen carriers for each type of high-pressure freezer. The general rule is to use the shallowest holder that will contain your material without compressing or crushing it. For adult worms we usually use 100 μm deep membrane carriers for the Leica HPF machines, and 100 μm deep aluminum carriers for the BAL-TEC and Wohlwend HPF machines. For smaller worm stages, there are custom carriers available for the Bal-Tec/Wohlwend machines, or, one can use EM slot grids of known thickness to form variable-depth spacers (4).
BSA is a good filler for HPF work, but it has the drawback that it will form a solid meshwork around the worms when they are fixed during freeze substitution. One can also use E. coli from worm plates as a filler and it has the advantage that it will separate from individual worms after fixation, making it easy to screen them by light microscopy. The disadvantage of E. coli is that it is not as effective a cryoprotectant as BSA. It is best used with very shallow wells such as those formed when using slot grids as variable spacers.
Leica Microsystems makes an automated freeze-substitution device, as does Boeckeler Instruments. A description of how to use dry ice for freeze substitution can be found in Ref. (8) and http://www.wormbook.org/toc_wormmethods.html.
There are many different freeze-substitution mixtures in the literature, but the most popular use a combination of acetone and osmium tetroxide for morphological studies, and acetone and glutaraldehyde for immunological work. We use a 1% solution of osmium tetroxide in pure acetone plus 0.1% uranyl acetate and 5% water. The water helps to visualize membranes (9). For immunoEM, we use 0.2% glutaraldehyde plus 0.1% uranyl acetate in acetone. Detailed instructions for how to make up these fixatives can be found in Ref. (3).
We prefer to use LR White Hard Grade because it is convenient to work at room temperature where one can see the samples under a stereomicroscope if necessary. Other methacrylate resins include LR Gold and the family of Lowicryl low-temperature resins. The Lowicryls can give quite excellent preservation of ultrastructure and antigenicity, but they are quite volatile and can cause contact dermatitis if adequate protection is not used during handling. For these reasons we prefer to use the LR White resins which were originally designed as a less hazardous alternative to the epoxy resin chemistry.
Age-matched adult worms are important for many types of quantitative or temporal analysis or because the representation of different stages of meiosis within the gonad changes as a function of the age of the animal: pachytene nuclei tend to accumulate over time relative to premeiotic or leptotene/zygotene-stage nuclei. We generally analyze young adults, which can be synchronized to within a few hours of development by picking late L4 larvae 12–24 h prior to dissection. Older adults (2–3 days post-L4) are useful for scoring nuclei at diakinesis, since this stage persists longer as sperm are depleted.
Sometimes it can be advantageous to combine immunofluorescence with FISH. The order in which these steps should be performed will depend on the sensitivity of the antibody to fixation conditions and the demands of the experiment. For best morphological preservation, FISH is performed prior to immunofluorescence, moving from the final wash steps of the FISH protocol (see Section 3.1.2, Step 20) to the blocking step of the immunofluorescence protocol (see Section 3.1.1, Step 9). Because the FISH procedure is more damaging to the tissue, stabilization of the tissue with EGS offers advantages for sample preservation, but is not compatible with all antibodies (see also Note 2). For this reason, we often fix the samples optimally for immunofluorescence, carry out immunofluorescent detection, and then follow with FISH. In this case, after the final washes of the immunofluorescence protocol (see Section 3.1.1, Step 13), the samples are post-fixed with a higher concentration of formaldehyde, and the FISH protocol is then executed (see Section 3.1.2, Step 9). In either case, samples can be couterstained with DAPI or other dyes during the final washes before mounting.
Because of the convenient organization of the gonad, temporal analysis of meiotic nuclei is a common technique used in meiosis in C. elegans. These pseudo-time course experiments are typically executed by dividing each gonad into a number of sections, moving from the proximal (early) to the distal (late) portion of the gonad. Nuclei within each section are scored separately. If the animals are age-matched and the sections are specified in a systematic way, data from corresponding sections in multiple animals can be pooled. We generally divide the gonad into five sections of equal physical length, starting in the premeiotic/mitotic region through to the end of pachytene, because the zones correlate fairly well with the mitotic region in zone 1, the transition zone in zone 2, and early, mid and late pachytene in zones 3–5, respectively (10, 11).
We have adapted our FISH protocol to incorporate microwave irradiation during the prehybridization and hybridization procedure. Microwaves are thought to accelerate diffusion and chemical reactions in a partially heat-independent fashion, although this remains controversial. The rationale for applying this to FISH is that the body of a worm is fairly impermeable to both fixatives and to probes, and microwaves may enable the large probe molecules to gain access to target DNA in the limited time window before chromosomes re-anneal. Empirically, this version of the protocol has given better results to date than any other with respect to both structural preservation of worm nuclei and reproducible, strong FISH signals with low fluorescent background. The procedure is designed around the capabilities of the Ted Pella BioWave microwave oven, which is equipped with variable wattage settings, restrictive-temperature control, and a “ColdSpot” circulating water bath with a flat glass surface that sits on the floor of the microwave oven and is controlled by an external heater/chiller/pump. According to the manufacturer, this circulating water eliminates standing waves and thus “hotspots” within the volume of the oven, resulting in even irradiation throughout. It also allows the temperature of a slide placed directly on the surface of the glass to be controlled fairly precisely and semi-independently of irradiation. The following changes are made to the FISH protocol to utilize the BioWave:
The overnight hybridization step (see Section 3.1.2, Step 17) is replaced by approximately 1.5 h in the BioWave. Prior to denaturation (see Section 3.1.2, Step 16), place the slides on the surface of the ColdSpot, preequilibrated to 30°C. Irradiate slides (10 min ON – 5 min OFF – 10 min ON) at setting No.6 with the temperature probe inserted into the ColdSpot port and a restrictive temperature of 37°C. Denature slides as in Section 3.1.2, Step 16. Return slides to surface of ColdSpot in BioWave. Irradiate slides (10 min ON – 5 min OFF – 10 min ON) at setting No.6 with restrictive temperature at 37°C. Repeat this cycle again for a total of ~1 h of hybridization. Continue with coverslip removal and washing steps (see Section 3.1.2, Step 18).
The final hour of washes in 50% formamide in 2xSSCT (see Section 3.1.2, Step 19) is replaced by incubation with microwave irradiation as follows: Place slides in jar No. 1. Insert temperature probe to the same depth as samples and irradiate with restrictive temperature of 37°C (2 min ON – 2 min OFF – 2 min ON) at setting No. 2. Transfer to jar No. 2 containing fresh 50% formamide/2xSSCT and repeat irradiation. Continue washing and DAPI staining steps (see Section 3.1.2, Step 20).
FISH probes can be generated from repetitive or single-copy regions. For nonrepetitive probes, a probe targeting approximately 5 kb of genomic sequence is (empirically) about the minimal length required to detect a signal above background fluorescence using protocols outlined here. However, probes generated to repetitive regions work more robustly. Two repetitive regions on the X chromosome make very good FISH probes. The sequence 5′-TTTCGCTTAGAGCGATTCCTTACCCTTAAATGGGCGCCGG-3′ is highly enriched near the center of the X chromosome, on cosmid C07D8, and 5′-GACTCCATCCACCAGCACTGCTTCGAGTACGACAGAAAGCACTTC-3′ is highly concentrated in a short (~6-kb) region near the right end of the X chromosome (12). Probes to short, repetitive sequences, including these can be generated as synthetic oligonucleotides and end-labeled with aa-dUTP without digestion (see Section 3.1.3, Step 5). A probe to the C. elegans 5S rDNA repeat is a robust tool to label this region on the right arm of chromosome V. Such a probe can be synthesized by amplification of the 1-kb repeated sequence using the following primers: 5′-TACTTGGATCGGAGAC GGCC-3′ and 5′-CTAACTGGACTCAACGTTGC-3′. This product can be digested to appropriately small fragment sizes using a single enzyme, Tsp509 I.
This same tailing protocol (omitting the dye conjugation step) may be used to incorporate other nucleotide analogs, including digoxigenin-dUTP or fluorescent dNTPs (biotinylated probes tend to be unsatisfactory in C. elegans due to high background staining). Digoxigenin-labeled FISH probes work more robustly than those labeled directly with fluorophores because of the secondary amplification of the signal by an anti-dig antibody. However, use of hapten-labeled probes adds extra steps to the FISH procedure (anti-dig immunofluorescence after the final washes in 50% formamide in 2xSSCT) and multiple digoxigenin probes cannot be used together. When using fluorescent dNTPs, we mix the modified nucleotide with a two-fold excess of unlabeled nucleotide, for two reasons: (1) The enzyme will be unhappy if it only has the modified substrate to incorporate, and (2) incorporation of fluorophores at too high a density will cause quenching of the fluorescence.
If the precipitated DNA is not obviously colored (particularly for Cy3- and Cy5-labeled probes), the labeling has probably not gone well. The terminal transferase reaction is more finicky than the dye conjugation step, so when poor dye incorporation is observed, DNA should probably be re-purified and/or relabeled with aa-dUTP.
Storing probes in hybridization buffer is convenient because an arbitrary amount can be mixed with additional hybridization solution to titrate the optimal amount of probe. In addition, hybridization solution does not freeze at −20°C due to the presence of 10% dextran sulfate. This may minimize probe degradation over long-term storage by eliminating freeze–thaw cycles. Probes may also be reconstituted in water or buffer, in which case the hybridization stock solution should be made more concentrated to accommodate dilution by probe.
Frozen material can be stored in liquid nitrogen for indefinite periods of time. We prefer to transfer the samples to vials with frozen fixative and use these for storage. This eliminates one transfer step when one is ready to begin freeze substitution. For longer-term storage, we use empty (no fixative) cryovials with small holes in the top so that they will stay filled with liquid nitrogen.
In our opinion, this is an optional step, as samples infiltrated with acetone:LR White mixtures work perfectly well for immunolabeling. However, the makers of LR White recommend ethanol, so it can also be done this way. In our experience, the use of ethanol for freeze substitution gives less satisfactory results than acetone.
References
- 1.Dernburg AF, Zalevsky J, Colaiacovo MP, Villeneuve AM. Transgene-mediated cosuppression in the C. elegans germ line. Genes Dev. 2000;14:1578–1583. [PMC free article] [PubMed] [Google Scholar]
- 2.Wilm T, Demel P, Koop HU, Schnabel H, Schnabel R. Ballistic transformation of Caenorhabditis elegans. Gene. 1999;229:31–35. doi: 10.1016/s0378-1119(99)00043-8. [DOI] [PubMed] [Google Scholar]
- 3.McDonald K. High-pressure freezing for preservation of high resolution fine structure and antigenicity for immunolabeling. Methods Mol Biol. 1999;117:77–97. doi: 10.1385/1-59259-201-5:77. [DOI] [PubMed] [Google Scholar]
- 4.McDonald KL, Morphew M, Verkade P, Muller-Reichert T. Recent advances in high-pressure freezing: equipment- and specimen-loading methods. Methods Mol Biol. 2007;369:143–173. doi: 10.1007/978-1-59745-294-6_8. [DOI] [PubMed] [Google Scholar]
- 5.McDonald K. Cryopreparation methods for electron microscopy of selected model systems. Methods Cell Biol. 2007;79:23–56. doi: 10.1016/S0091-679X(06)79002-1. [DOI] [PubMed] [Google Scholar]
- 6.Muller-Reichert T, Hohenberg H, O’Toole ET, McDonald K. Cryoimmobilization and three-dimensional visualization of C. elegans ultrastructure. J Microsc. 2003;212:71–80. doi: 10.1046/j.1365-2818.2003.01250.x. [DOI] [PubMed] [Google Scholar]
- 7.Muller-Reichert T, Srayko M, Hyman A, O’Toole ET, McDonald K. Correlative light and electron microscopy of early Caenorhabditis elegans embryos in mitosis. Methods Cell Biol. 2007;79:101–119. doi: 10.1016/S0091-679X(06)79004-5. [DOI] [PubMed] [Google Scholar]
- 8.McDonald KL. Electron microscopy and EM immunocytochemistry. Methods Cell Biol. 1994;44:411–444. doi: 10.1016/s0091-679x(08)60926-7. [DOI] [PubMed] [Google Scholar]
- 9.Walther P, Ziegler A. Freeze substitution of high-pressure frozen samples: the visibility of biological membranes is improved when the substitution medium contains water. J Microsc. 2002;208:3–10. doi: 10.1046/j.1365-2818.2002.01064.x. [DOI] [PubMed] [Google Scholar]
- 10.MacQueen AJ, Colaiacovo MP, McDonald K, Villeneuve AM. Synapsis-dependent and -independent mechanisms stabilize homolog pairing during meiotic prophase in C. elegans. Genes Dev. 2002;16:2428–2442. doi: 10.1101/gad.1011602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MacQueen AJ, Villeneuve AM. Nuclear reorganization and homologous chromosome pairing during meiotic prophase require C. elegans chk-2. Genes Dev. 2001;15:1674–1687. doi: 10.1101/gad.902601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lieb JD, de Solorzano CO, Rodriguez EG, Jones A, Angelo M, Lockett S, Meyer BJ. The Caenorhabditis elegans dosage compensation machinery is recruited to X chromosome DNA attached to an autosome. Genetics. 2000;156:1603–1621. doi: 10.1093/genetics/156.4.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]