

Abstract
Rac1, a Rho GTPase, modulates diverse cellular processes and is hyperactive in some cancers. Estrogen receptor-alpha (ER) in concert with intracellular signaling pathways regulates genes associated with cell proliferation, tumor development and breast cancer cell survival. Therefore, we examined the possibility of Rac1 and ER crosstalk in breast cancer cells. We found that Rac1 enhanced ER transcriptional activity in breast cancer cells. Vav3, a Rho guanine nucleotide exchange factor that activates Rac1, was an upstream mediator and P21/Cdc42/Rac1 activating kinase-1 (Pak-1) was a downstream effector of Rac1 enhancement of ER activity. These results suggest that Rac1 may prove to be a therapeutic target. To test this hypothesis, we used a small molecule Rac inhibitor, EHT 1864, and found that EHT 1864 inhibited ER transcriptional activity. Furthermore, EHT 1864 inhibited estrogen-induced cell proliferation in breast cancer cells and decreased tamoxifen resistant breast cancer cell growth. EHT 1864 decreased activity of the promoter of the ER gene resulting in downregulation of ER mRNA and protein levels. Therefore, ER downregulation by EHT 1864 is the likely mechanism of EHT 1864-mediated inhibition of ER activity and estrogen-stimulated breast cancer cell proliferation. Since ER plays a critical role in the pathogenesis of breast cancer and the Rac inhibitor EHT 1864 downregulates ER expression and breast cancer cell proliferation, further investigation of the therapeutic potential of Rac1 targeting in the treatment of breast cancer is warranted.
INTRODUCTION
Rho GTPases are a subgroup of the Ras superfamily and include the well-characterized members, RhoA, Rac1, and Cdc42. Rho proteins cycle between the active state (GTP-bound) and inactive form (GDP-bound) enabling them to act as molecular switches in numerous signaling pathways (Hall. 1998, Van Aelst & D’Souza-Schorey. 1997). Rac1 and other Rho proteins are critical components of key mitogenic pathways providing a link between the cell surface and transcriptional events (Bosco, et al. 2009).
Substantial evidence supports roles for Rho GTPase signaling alterations in cancers (Chan, et al. 2005a, Sahai & Marshall. 2002). Since mutations in Rho proteins are extremely rare, the mechanism for Rho action in cancer likely occurs through the overexpression or hyperactivity of these proteins (Schnelzer, et al. 2000). Increased Rho GTPase activity may be mediated by deregulation of upstream Rho guanine nucleotide exchange factors (GEFs), such as Vav3. Rac1 activation has been implicated in breast cancer cell invasion and metastasis (Burbelo, et al. 2004, Fritz, et al. 2002, Baugher, et al. 2005, Chan, et al. 2005a, Xie & Haslam. 2008). Furthermore, Rho GTPases may also function as mediators of EGFR-stimulated breast cancer cell growth (Yang, et al. 2006).
The effects of Rac1 on estrogen receptor-alpha (ER) transcriptional activity and cell proliferation have not been fully elucidated. One study described Rac1 activation of ER-mediated transcription in ovarian cancer cells, but another study showed that Rac1 inhibits ER transcriptional activity (Lee, et al. 2000, Su, et al. 2001). In addition, introduction of a constitutively active form of Rac1 resulted in resistance to the “pure” ER antagonist ICI 182,780 in an ER positive breast cancer cell line (Cai, et al. 2003). This evidence suggests that Rac1 may play an important role in the pathogenesis of breast cancer. EGFR and Vav3, a growth factor-activated Rho GTPase GEF, both enhance ER transcriptional activity in breast cancer cells (Lange. 2004, Lee, et al. 2001, Lee, et al. 2008). Moreover, Rac1 participates in both MAPK and PI3K pathways, which are involved in crosstalk between EGFR and ER (Ali & Coombes. 2002, Cai, et al. 2003). Therefore, we speculated that inhibition of Rac1 may have an antiproliferative effect in breast cancer cells. EHT 1864 is a Rac small molecule inhibitor that holds Rac in an inert and inactive state and prevents downstream effector binding and activation without disturbing GEF-Rac interactions (Onesto, et al. 2008, Shutes, et al. 2007). EHT 1864 specifically inhibits Rac without interfering with other Rho GTPases such as Rho or Cdc42 (Onesto, et al. 2008, Shutes, et al. 2007).
Currently, patients with ER positive breast cancer are treated with either selective estrogen receptor modulators (SERMs), such as tamoxifen, or aromatase inhibitors, such as anastrozole, which interfere with estrogen synthesis. These therapies are effective because they inhibit the action of ER to promote expression of genes associated with cell proliferation, tumor development, and survival. However, both primary and secondary resistance to these therapies are serious clinical problems and thus it is important to develop new therapeutic strategies that will also target tamoxifen resistant breast cancer (Schiff, et al. 2003). Since the majority of breast cancers express ER, and since Rac1 is a downstream mediator of EGFR, which activates ER, we wanted to determine if there is crosstalk between Rac1 and ER in breast cancer cells.
In this study, we found that Rac1 increased ER transcriptional activity in breast cancer cells. Inhibition of Rac1 by EHT 1864 decreased ER transcriptional activity as well as estrogen-induced breast cancer cell proliferation in ER positive as well as tamoxifen resistant cells. Furthermore, Vav3 was an upstream activator and Pak-1 was a downstream effector of Rac1 enhancement of ER transcriptional activity. We demonstrate that EHT 1864 inhibited ER activity through downregulation of ER mRNA and protein.
MATERIALS AND METHODS
Cell Culture and chemical reagents
The human breast cancer cell lines MDA MB 231, MCF-7, and T47D were kindly provided by Dr. Catherine Welsh (University of Miami, Miami, FL). The MCF-7 tamoxifen-sensitive and MCF-7 tamoxifen-resistant cells were kindly provided by Dr. Rachel Schiff (Baylor College of Medicine, Houston, TX). Cell culture media (RPMI-1640, DMEM/F12 50:50, DMEM, DMEM Glutamax) were obtained from GIBCO-BRL (Gaithersburg, MD USA). Fetal bovine serum (FBS) was obtained from Hyclone (Logan, Utah USA).T47D cells were cultured in RPMI supplemented with 100 IU/ml penicillin, 100ug/ml streptomycin, 2mM L-glutamine (Gibco-BRL, Gaithersburg, MD, USA) and 10% FBS. MCF-7 cells and MDA MB 231 cells were cultured in DMEM/F12 50:50 supplemented with 5% FBS, 100 IU/ml penicillin, 1000ug/ml streptomycin, and 2mM L-glutamine. MCF-7 tamoxifen-sensitive cells were cultured in DMEM Glutamax supplemented with 10% FBS, 100 IU/ml penicillin, 1000ug/ml streptomycin, 2mM L-glutamine, and 15ug/ml Insulin. MCF-7 tamoxifen-resistant cells were cultured in Phenol-Red free DMEM supplemented with 5% charcoal-stripped serum supplemented with 100 IU/ml penicillin, 1000ug/ml streptomycin, 2mM L-glutamine, 15ug/ml Insulin, and 100nM tamoxifen. EHT 1864 and tamoxifen were purchased from Sigma (St. Louis, MO) and estradiol was purchased from Steraloids (Wilton, NW). For estradiol depletion, cells were transferred to medium containing Phenol-Red free DMEM supplemented with 5% charcoal-stripped serum for 24 hours. Assays of ERE luciferease and QPCR of ER target genes were carried out at 24 hours following re-addition of estradiol 1 nM vehicle.
Plasmids
The 3X ERE-luciferase (ERE-Luc) reporter plasmid was provided by Dr. Zafar Nawaz (University of Miami). Constitutively active (Ca) Vav3 (Ca Vav3) was constructed as previously described (Lyons & Burnstein. 2006, Lyons, et al. 2008). Ca Rac1 (CaRacQ61L) and Ca Rac1/PakEDM (CaRacQ61L/43D) were gifts from Dr. Channing Der (University of North Carolina-Chapel Hill). The ER reporter plasmids [ER wild type luciferase (WT ER3500-210LUC) and ER delta enhancer luciferase (delta Enh ER3500-135LUC)] were kindly provided by Dr. Ronald Weigel (deConinck, et al. 1995).
Lentivirus production and generation of stable cell lines
Constitutively activate Rac1 Q61L was cloned into pQCXIN (BD Biosciences, Palo Alto, CA) by PCR amplification of PCGN caRac1 vector and verified by sequencing. For viral production, GP2–293 cells at 60–80% confluency in 100-mm dishes were transfected with 7.5 μg VSV-G and 12.5 μg pQCXIN (encoding empty vector or caRac1 ) using CalPhos kit (Clontech, Palo Alto, CA). Forty-eight hours after transfection, media containing viral particles were collected and filtered through a 0.45-μm cellulose acetate filter and stored at –80 C. For selection, cells were infected overnight with appropriate constructs 24 hours after seeding and cultured in 750 μg/mL G418 48 hours after infection for 8-10 days.
Reporter gene assays and transfections
All transfections were carried out using the cationic lipid reagent Lipofectamine (Invitrogen Life Technologies, Grand island, NY) according to the manufacturer’s instructions. Cells were plated in 35mm dishes 16-20 hours prior to transfection. Immediately prior to transfection, media were replaced with unsupplemented DMEM. Cells were transfected with: 1.5ug reporter [ER wild type luciferase (WT) or ER delta enhancer luciferase (delta Enh)], or 1.5ug ERE-luc, and 125ng Ca Rac1, Ca Vav, or Ca Rac1/PakEDM. Following a 4-5 hour incubation with DNA/lipid complexes, cells were transferred to estrogen depleted media (phenol red free media supplemented with 2% charcoal stripped serum) for 24 hours and then treated with vehicle or 1nM estradiol in the absence or presence of 10 μM EHT 1864 for an additional 24 hours. 48 hours following transfection, cells were harvested, lysed and assessed for luciferase activity using the Promega luciferase assay kit.
RNA Extraction and Real-Time RT-PCR
Total RNA was harvested using the trizol method according to the manufacturer’s protocol (Invitrogen Life Technologies). 500ng total RNA was reverse transcribed using cDNA archive kit (Applied Biosystems). Real-time PCR was performed using ABI Prism 7700. Taqman probes from Applied Biosystems for 18S, Rac1, HPRT, ER, and pS2 were used. 100 ng cDNA was used for qPCR (except 1 ng cDNA was used for 18S the endogenous control). The comparative threshold cycle (Ct) method was used to determine the relative expression level of mRNA. This involved comparing the Ct values of the treated samples with a control sample. The Ct values of both the control and the samples of interest were normalized to 18S (an endogenous housekeeping gene). Relative mRNA levels were determined by the following equation:
Western Blot Analysis
Cells were grown to ~80% confluence in 60mm plates. 24 hours prior to harvest, cells were treated with vehicle or 1 nM estradiol in the presence or absence of 10 μM EHT 1864 and phenol red free media supplemented with 2% charcoal stripped serum. 40 μg total protein from each sample was resolved on 12% sodium dodecyl sulfate-polyacrylamide gels and transferred onto nitrocellulose membranes. Membranes were blocked with 5% nonfat dry milk in Tris-buffered saline [20 mM Tris base (pH 7.5) and 50 mM NaCl and 2.5mM EDTA] containing 0.1% Tween 20 (TBS-T), then probed with anti-ER (HC-20;1:200) or anti-actin (1:500) from Santa Cruz Biotechnology. Primary antibodies were diluted in 5% nonfat dry milk in TBS-T. After washing in TBS-T, membranes were incubated with their appropriate horseradish peroxidase-conjugated secondary antibodies (Santa Cruz) and developed using an enhanced chemiluminescence detection system (Amersham Biosciences, Arlington Heights, IL) according to the instructions of the manufacturer.
Cell Proliferation Assay
Cells were plated at an initial density of 20,000 (MCF7 cells), 10,000 (T47D cells), 10,000 (MDAMB231), 10,000 (Ca Rac1 expressing and control T47D), cells per well in a 24-well dish containing media supplemented with 10% charcoal stripped serum. The following day, cells were treated with vehicle, 1nM estradiol, 10μM EHT 1864, and/or 500nM tamoxifen for various time periods. After the appropriate treatment period, adherent and floating cells were collected and viable cells counted by trypan blue exclusion using a hemocytometer. Experiments were performed in triplicate.
GST protein purification
Rac/Cdc42 binding domain (p21-binding domain, PBD) of Pak1 coupled to GST (PBD-GST) was a gift from Dr. Martin Schwartz (University of Virginia, Charlottesville, VA). PBD-GST protein was purified according to the manufacturer’s protocol (Amersham). BL-21 competent cells were transformed with plasmids encoding GST proteins. Gene expression was induced with100mM isopropyl β-D-thiogalactosidase for 90 min. Cell lysates were added to glutathione columns then eluted with reduced glutathione. Protein concentration was determined and purified proteins were visualized by Coomassie stain.
Rac GTPase activity “pull down” assay
Rac1 activity was assessed by pull-down assay using PBD-GST as described previously (Knight-Krajewski, et al. 2004). Briefly, cells were harvested and lysed in Mg 2+ lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 10 mM MgCl2, 0.5 % NP-40, 10% glycerol, 10 μL/mL protease inhibitor) containing 60 μg/300 μL PBD-GST. Cell lysates containing 1 mg total protein were added to 100 μl glutathione sepharose beads and rotated gently for 30 min at 4°C. Sepharose beads were pelleted by centrifugation and complexes were washed four times with lysis buffer. GTP-bound Rac1/Cdc42 was eluted and separated along with 5% inputs by SDS-PAGE. Proteins were transferred to nitrocellulose membranes and analyzed using anti-Rac1 antibody (Upstate).
Analysis of ER degradation
T47D cells were estrogen-depleted in 10% charcoal stripped serum for 24 hours followed by treatment with vehicle or 10 μM EHT 1864 for 1 hour prior to cycloheximide addition. Cells were lysed in ice-cold lysis buffer containing: 50 mM HEPES, pH 7.5; 150 mM NaCl; 1 mM EDTA, pH 8.0; 2.5 mM EGTA, pH 8.0; 10% glycerol; 10 mM β-glycerophosphate; 1 mM NaF; 0.1% Tween-20; 1 mM PMSF; 0.1 mM Na2VO4; 0.5 mM DTT; and 0.02 mg/ml each of aprotinin, leupepsin, and pepstatin. Western blots used 20–100 μg protein per lane. The ER-alpha t1/2 was determined by cycloheximide chase, with addition of 100 μg cycloheximide considered t = 0. Cells were lysed at the times indicated in the figure legend and ER was blotted as previously described (Chen, et al. 2009, Chen, et al. 2010, Chu, et al. 2007, Sun, et al. 2007).
Chromatin Immunoprecipitation (ChIP)
T47D cells were plated 7-8 × 106 cells/150mm plate in phenol red-free DMEM with 5% charcoal stripped serum and grown for 3 days before treatment with vehicle or 10 μM EHT 1864 for 24 hours. After 24 hours, cells were treated with vehicle or 1 nM estradiol for 1 hour. Chromatin was then crosslinked with 1% formaldehyde for 10 minutes at room temperature. The crosslinked chromatin was then sonicated, diluted, and immunoprecipitated with ER (HC-20) antibody (Santa Cruz Biotechnology) or normal rabbit IgG control at 4°C overnight. Protein A agarose beads with salmon sperm DNA were added and then washed with a low salt buffer, followed by a high salt buffer, then LiCl buffer, and finally TE buffer. The protein DNA complexes were eluted and the crosslinks reversed. The DNA fragments were purified with the Qiagen PCR purification kit. The fragments were then analyzed by real time PCR. Real time PCR was performed using an icycler iQ PCR detection system (Bio-Rad) with iQ SyberGreen supermix (Bio-Rad). Primers for pS2 and GREB1 promoters were described previously (Dhananjayan, et al. 2006, Sun, et al. 2007).
RESULTS
Rac1 enhances ER transcriptional activity
To determine if there is crosstalk between Rac1 and ER, we created derivative cell lines of T47D and MCF7 cells that express either constitutively active (Ca) Rac1 or vector (as a control) and performed ERE reporter gene assays. As expected, Rac1 “pull down” activity assays showed elevated active Rac1 levels in the Ca Rac1 expressing cells compared to control (Fig 1A). PC3, a prostate cancer cell line that has relatively high Rac1 activity, was used as a positive control (Knight-Krajewski, et al. 2004). The Ca Rac1-expressing cells had active Rac1 levels comparable to PC3 cells, suggesting that Rac1 activity in the derivative breast cancer cells was in a range relevant to that observed in cancer cells (Fig 1A). Reporter gene assays revealed that ER transcriptional activity was increased in cells expressing Ca Rac1 compared to control cells in both T47D and MCF7 cells (Fig 1B-C). ER transcriptional activity in estrogen-depleted Ca Rac1 expressing cells for 24 hours was increased in both the absence and presence of estradiol, suggesting that Rac1 enhances both ligand-dependent as well as ligand-independent receptor activity. Ca Rac1 enhancement of the ligand-independent activation of ERE-luciferase activity may be due to the persistence of very low levels of estrogens in the stripped serum and/or due to true ligand-independent activation of ER. To confirm and extend the results of the reporter gene assays, we examined ER regulation of a native ER target gene (pS2) and found that pS2 mRNA levels were increased in cells expressing Ca Rac1 compared to control in both the presence and absence of estradiol (Fig 1D).
Figure 1. Rac1 enhances ER transcriptional activity in breast cancer cells.


(A) MCF7 and T47D cells were infected with control vector or a constitutively active (Ca) Rac1 retroviral vector and then selected with G418. Rac1 activity (Rac1-GTP) was determined in these cells by pull down assays. PC3 cells, which have relatively high Rac1 activity, were used as a positive control. MCF7 (B) and T47D (C) cells expressing either control vector or Ca Rac1 were transfected with a 3X ERE-luciferase reporter plasmid then estrogen deprived for 24 hours and then treated with vehicle or 1nM estradiol for an additional 24 hours. 48 hours following transfection, luciferase activity was measured. (D) T47D cells expressing control vector or Ca Rac1 were treated with vehicle or 1 nM estradiol for 24 hours. RNA was extracted, reverse transcribed to cDNA, and real time PCR was performed to determine pS2 mRNA and 18S mRNA (control) levels. (E) RNA was extracted from T47D and MCF7 cells expressing control vector or Ca Rac1, reverse transcribed to cDNA, and real time PCR was performed to determine HPRT mRNA and 18S mRNA (control) levels. Data (B-D) represent the mean of three experiments performed in triplicate ± SEM. P-values were determined by student t-test. *P-value < 0.05, **P-value < 0.01.
Since Ca Rac1 enhanced ER activity in the presence and absence of estradiol we wanted to verify that these effects were not due to global activation of transcription. HPRT encodes an enzyme involved in the purine synthesis pathway and is not regulated by ER (de Kok, et al. 2005). Ca Rac1 did not alter HPRT mRNA levels compared to control in either MCF7 or T47D cells consistent with a selective effect of Rac1 on ER-mediated transcription (Fig 1E).
The Rac inhibitor, EHT 1864, decreases ER transcriptional activity and ER recruitment to target gene promoters
Since Rac1 enhanced ER transcriptional activity in breast cancer cells, we examined the effect of Rac1 inhibition on ER-mediated responses. EHT 1864 is a small molecule Rac inhibitor that blocks Rac GTPase activity and prevents Rac activation of downstream effectors (Onesto, et al. 2008, Shutes, et al. 2007). Reporter gene assays in MCF7 and T47D cells showed that even in the absence of Rac1 overexpression, EHT 1864 inhibited estradiol-stimulated ER transcriptional activity (Fig 2A-B). EHT 1864 also modestly inhibited ER activity in the estradiol-deprived cells. Both effects were more notable in T47D cells compared to MCF7 cells. Similarly, EHT 1864 treatment decreased pS2 but not HPRT mRNA levels in T47D and MCF7 cells, confirming our finding that EHT 1864 selectively impaired ER transcriptional activity (Fig 2C-E).
Figure 2. The Rac inhibitor, EHT 1864, decreases ER transcriptional activity and ER recruitment to target gene promoters in breast cancer cells.


MCF7 (A) and T47D (B) cells were transfected with a 3X ERE-luciferase reporter plasmid. Cells were estrogen deprived for 10 hours then treated with either vehicle or 1 nM estradiol in the presence or absence of 10 μM EHT 1864. Luciferase activity was determined 48 hours later. MCF7 (C) and T47D (D) cells were treated with vehicle or 1nM estradiol in the presence or absence of 10 μM EHT 1864 for 24 hours. RNA was extracted, reverse-transcribed to cDNA, and real time PCR was performed to determine pS2 mRNA and 18S mRNA (control) levels. (E) T47D cells were treated with vehicle or 10 μM EHT 1864 for 24 hours. HPRT mRNA and 18S mRNA (control) levels were examined. (F and G) T47D cells deprived of estrogen were treated with vehicle or 10 μM EHT 1864 for 24 hours. After 24 hours, cells were treated with vehicle, 1 nM estradiol, +/− 500nM tamoxifen for 1 hour and then subjected to chromatin immunoprecipitation as described in methods. ER recruitment to the pS2 promoter (F) or the GREB1 promoter (G) was determined by real time PCR. Data represent the mean of three experiments performed in triplicate ± SEM. P-values were determined by student t-test. *P-value < 0.05, **P-value < 0.01.
To determine if EHT 1864 also disrupts ER recruitment to target gene promoters, we performed chromatin immunoprecipitation. As expected, estradiol treatment increased ER recruitment to the promoters of the ER target genes pS2 and GREB1, while tamoxifen inhibited receptor recruitment (Fig 2F-G). Consistent with EHT 1864 inhibition of ER transcriptional activity, there was significantly less ER recruited to the pS2 and GREB1 promoters following EHT 1864 treatment (Fig 2F-G).
EHT 1864 inhibits estrogen-induced breast cancer cell proliferation
Since EHT 1864 inhibited ER activity, we wanted to determine if Rac1 inhibition would also decrease breast cancer cell proliferation. EHT 1864 significantly inhibited the rate of T47D and MCF7 cell proliferation in the presence or absence of estradiol (Fig 3A-B). While EHT 1864 also inhibited MDA MB 231 cells, an ER negative breast cancer cell line, the extent of inhibition was substantially less than that observed in the ER positive, estradiol-stimulated MCF7 and T47D cells (Fig 3C). In the presence of estradiol, there was approximately 90% decrease in the T47D cell number, 80% in MCF7 cells, and about 50% growth inhibition in MDA MB 231 following EHT 1864 treatment. Therefore, EHT 1864’s anti-proliferative effects appear to be at least partially mediated by inhibiting ER activity.
Figure 3. EHT 1864 inhibits estrogen-induced breast cancer cell proliferation.

MCF7 (A), T47D (B) and MDA MB 231 (C) cells were treated with either vehicle, 1 nM estradiol and/or 10 μM EHT 1864. Total cells that excluded trypan blue were counted on the indicated days. (D) T47D cells were treated with vehicle, 500 nM tamoxifen, 10 μM EHT 1864 or tamoxifen plus EHT 1864 in the presence and absence of 1 nM estradiol. Cells were counted as described above 5 days after treatment. P-values were determined by comparing each treatment group to control using student t-test. * P < 0.05.
Tamoxifen, a potent ER blocking drug, is of important therapeutic value in breast cancer. Since EHT 1864 inhibits estrogen-induced breast cancer cell proliferation, we tested whether the combination of tamoxifen and EHT 1864 would cause a greater inhibition of cell proliferation. However, there was no additive effect on cell proliferation from the combination of EHT 1864 and tamoxifen treatment in T47D cells (Fig 3D) or MCF7 cells (data not shown). There was also no additive inhibition of cell proliferation at lower (suboptimal) concentrations of each drug in either T47D or MCF7 cells (data not shown). Together, these results reveal that EHT 1864 treatment not only diminished ER-dependent gene transcription but also breast cancer cell proliferation.
Vav3, a Rho GTPase GEF, is an upstream activator of Rac1 enhancement of ER transcriptional activity
In addition to its well-recognized role as an activator of Rho GTPases, Vav3 has been shown to increase ER transcriptional activity (Lee, et al. 2008). Therefore, the role of Vav3 in Rac1 enhancement of ER transcriptional activity was examined. As expected, constitutively active (Ca) Vav3 increased ER transcriptional activity in breast cancer cells (Fig 4A). EHT 1864 prevented Ca Vav3-mediated stimulation of ER activity in the absence and presence of estradiol, thus Rac1 action appears to be required for Vav3-mediated stimulation of ER activity (Fig 4A).
Figure 4. Vav3 is an upstream activator and Pak-1 is a downstream effector of Rac1 enhancement of ER transcriptional activity.

(A) T47D cells were transfected with a 3X ERE-luciferase reporter plasmid and empty vector or Ca Vav3. Cells were treated with either vehicle or 1 nM estradiol in the presence or absence of 10 μM EHT 1864. (B) T47D cells were transfected with 3X ERE-luciferase and empty vector, Ca Rac1 or Ca Rac1 that is deficient in binding Pak-1 (Ca Rac1/PakEDM). Cells were then estrogen-deprived for 24 hours and then treated with either vehicle or 1 nM estradiol. Luciferase activity was determined 48 hours after transfection. Data represent the mean of three experiments performed in triplicate ± SEM. P-values were determined by student t-test. *P-value < 0.05, **P-value < 0.01.
Pak-1 is a downstream mediator of Rac1-dependent ER transcriptional activation
Pak-1, a well-characterized downstream effector of Rac1, has been shown to mediate growth factor effects on motility and invasiveness in breast cancer cells (Adam, et al. 1998, Manser, et al. 1994, Vadlamudi, et al. 2000). Therefore, we investigated the role of Pak-1 on Rac1 and ER crosstalk. To determine if Pak-1 is a downstream effector, a Ca Rac1 effector domain mutant (EDM) that is selectively unable to bind and activate Pak-1 (Ca Rac1/PakEDM) was transfected into T47D cells (Westwick, et al. 1997). In contrast to Ca Rac1, the Ca Rac1/PakEDM construct did not enhance ER transcriptional activity indicating that Pak-1 may be a key mediator of Rac1 stimulation of ER activity via direct Rac1-Pak-1 interaction (Fig 4B).
EHT 1864 downregulates ER mRNA and protein levels
We next addressed the molecular mechanism whereby EHT 1864 inhibits ER activity. One possible mechanism is through downregulation of ER levels. As expected, estradiol treatment resulted in decreased ER protein levels. Following EHT 1864 treatment in both the presence and absence of estradiol, there was a significant decrease in ER protein levels in both MCF7 and T47D cells (Fig 5A). RT-qPCR analysis revealed that EHT 1864 treatment reduced ER mRNA levels in both the presence and absence of estradiol in MCF7 and T47D cells without any evidence of global transcriptional inhibition since HPRT levels were unchanged (Fig 5B-C). Estradiol treatment did not change ER mRNA levels (Fig 5B-C).
Figure 5. EHT 1864 downregulates ER mRNA and protein levels.

(A) MCF7 and T47D cells were deprived of estrogen for 24 hours and then treated with vehicle or 1nM estradiol in the presence or absence of 10 μM EHT 1864 for 24 hours. Cells were lysed and ER protein levels were determined by western blot. This is a representative blot from 3 experiments. MCF7 (B) and T47D (C) cells were treated as in panel (A). RNA was extracted, reverse transcribed to cDNA, and real time PCR was performed to determine ER mRNA and 18S mRNA (control) levels. Data represents the mean of three experiments performed in triplicate ± SEM. P-values were determined by student t-test.*P-value < 0.05.
To determine if decreased ER protein levels were due to transcriptional downregulation of the ER gene or due to decreased ER protein stability, we first tested the drug’s effect on the promoter for the ER gene. The construct for the promoter of the ER gene contains the requisite regions for ER expression in breast cancer cells but does not contain ER binding sites (deConinck, et al. 1995). When estrogen-deprived cells were treated with EHT 1864 in the presence or absence of estradiol for 24 hours, EHT 1864 inhibited activity of the wild type ER gene promoter but did not affect activity of a reporter plasmid that lacked the enhancer region of the ER gene promoter (Fig 6A).
Figure 6. EHT 1864 inhibits activity of the promoter of the ER gene but does not disrupt ER stability.

(A) T47D cells were deprived of estrogen for 24 hours and then transfected with a WT ER gene promoter-luciferase or the ER gene promoter lacking the enhancer (delta enhancer) reporter plasmid. Cells were treated with either vehicle or 1 nM estradiol in the presence or absence of 10 μM EHT 1864. Luciferase activity was determined 48 hours after transfection and normalized for protein. Results are reported as values relative to ER-luciferase WT set at 100%. Data represent the mean of three experiments performed in triplicate ± SEM. (B) T47D cells were deprived of estradiol for 24 hours and then treated with vehicle or 10 μM EHT 1864 for 24 hours. ER protein levels were determined by western blot and actin was used as a loading control. (C) T47D cells were deprived of estradiol for 24 hours and then pretreated with vehicle or 10 μM EHT 1864 for 1 hour followed by addition of cycloheximide. Cells were harvested and protein lysates obtained at the indicated times after cycloheximide treatment. ER protein levels were determined by western blot and actin was used as a loading control. This is a representative experiment of two experiments. P-values were determined by student t-test. *P-value < 0.05.
We next examined whether EHT 1864-mediated ER loss might be due to de-stabilization of the ER protein. ER protein stability was assayed in estrogen-deprived cells following 24 hours of EHT 1864 treatment. It is noteworthy that when ER-positive MCF-7 breast cancer cells are estrogen deprived, ER protein is highly stable, with an ER t1/2 in excess of 10 hours, as previously demonstrated (Chu, et al. 2007). Our cycloheximide chase showed that while EHT 1864 treatment dramatically reduced ER steady state levels, there was no significant effect of EHT 1864 on ER protein degradation rate (Fig 6B-C). Thus, EHT 1864-mediated down regulation of ER appears to be primarily through decreased transcription of the ER gene.
EHT 1864 inhibits tamoxifen resistant breast cancer cell proliferation
Breast cancer cells, following extended treatment with tamoxifen, can overcome the antagonistic effects of tamoxifen on ER resulting in drug resistance. Therefore, it is important to identify novel drug targets that may be effective in treatment of tamoxifen resistant breast cancer. Rac1 inhibition may be a useful strategy to inhibit growth of tamoxifen resistant cells. To test this, we determined whether EHT 1864 inhibits the growth of a tamoxifen resistant cell line, MCF7 TAMR. EHT 1864 inhibited the proliferation of these cells, whereas, as expected, tamoxifen did not (Fig 7A-B). Furthermore, T47D cells expressing Ca Rac1 were more resistant to tamoxifen treatment compared to control cells (Fig 7C). These results suggest that inhibition of Rac1 may be a potential therapeutic strategy for ER positive, tamoxifen-resistant breast cancer.
Figure 7. EHT 1864 inhibits tamoxifen resistant breast cancer cell proliferation and active Rac1 confers partial tamoxifen resistance.

MCF7 tamoxifen sensitive (TAMS) (A) and MCF7 tamoxifen resistant (TAMR) (B) cells were treated with either vehicle, 100 nM tamoxifen, 10 μM EHT 1864, and 100 nM tamoxifen plus 10 μM EHT 1864. Cells were counted following 5 day treatment. P-values were determined by student t-test. **P-value <0.01. (C) T47D cells stably expressing either vector or Ca Rac1 were treated with either vehicle or 100 nM tamoxifen. Cells were counted after 6 days treatment. This is a representative experiment from 3 experiments performed in triplicate. Data are plotted as % of vehicle-treated cells. P-value determined by comparing Ca Rac1 expressing cells to control using student t-test. *P-value < 0.05.
DISCUSSION
Rac1 is an important mediator of signaling pathways including those that promote cell proliferation, migration, and cancer cell invasiveness (Baugher, et al. 2005, Chan, et al. 2005b, Fritz, et al. 2002). We identified a novel crosstalk between Rac1 and ER signaling in breast cancer cells in which Vav3 served as an upstream activator and Pak-1, a downstream effector of Rac1 enhancement of ER activity. Further, inhibition of this cross-talk with a Rac inhibitor, EHT 1864, decreased ER transcriptional activity through a mechanism involving transcriptional repression of the ER promoter. In line with the possibility that Rac1 may be an effective therapeutic target in breast cancer, EHT 1864 significantly downregulated ER mRNA and protein levels leading to decreased ER transcriptional activity as well as estradiol-stimulated breast cancer cell proliferation. The effect of EHT 1864 on our novel model of crosstalk between Rac1 and ER is illustrated in Fig 8.
Figure 8. Effects of Rac1 inhibition on ER levels.
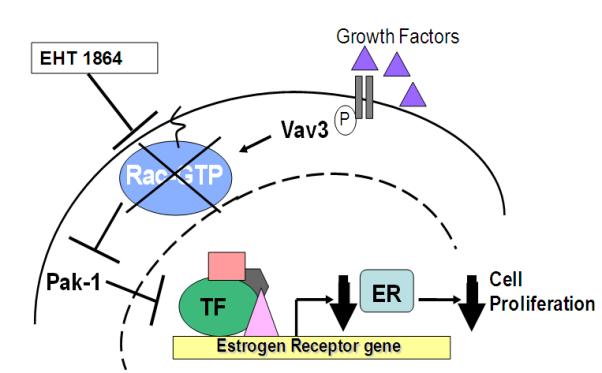
Vav3 activates Rac1 by promoting GTP binding. However, in the presence of EHT 1864, Rac1 fails to bind and activate downstream effectors resulting in ER mRNA downregulation. The decreased ER levels results in lowered ER transcriptional activity and ultimately inhibits breast cancer cell proliferation.
Rac1 stimulates migration and invasion in breast cancer cells due to its well-established effects on cytoskeleton remodeling. One study showed that Rac1 participates in estrogen-stimulated MCF7 cell growth and that these proliferative effects were dependent on the composition of the extracellular matrix (Xie & Haslam. 2008). Estrogen-induced MCF7 cell proliferation was significantly increased when cells were cultured on collagen I compared to laminin and this was attributed to collagen I activation of Rac1 and JNK signaling (Xie & Haslam. 2008). These effects on cell proliferation were attributed to Rac1 regulation of cyclin D1 mRNA levels independent of ER (Xie & Haslam. 2008).
We showed that EHT 1864 inhibited estrogen-stimulated ER positive breast cancer cells to a greater extent than an ER negative cell line, which expresses comparable Rac1 levels (data not shown), suggesting that inhibition of Rac1 and its resultant suppression of ER levels contributed to EHT 1864 anti-proliferative effects. The mechanisms of EHT 1864-mediated growth inhibition of breast cancer cells require further work. There was no additive inhibition of estrogen-stimulated cell proliferation using a combination of EHT 1864 and tamoxifen perhaps because EHT 1864 downregulation of ER decreased the target for tamoxifen (ER) thereby compromising the effectiveness of this drug. Furthermore, tamoxifen and EHT 1864 may not additively decrease cell proliferation because they both inhibit ER. Since Rac1 is a well known mediator of cell migration and invasion, Rac1 inhibition is likely to have a broader effect than tamoxifen beyond reducing ER levels. EHT 1864 inhibits membrane lamellipodia formation in fibroblasts and prevents Ras and Rac1-dependent transformation (Onesto, et al. 2008, Shutes, et al. 2007). Rac1 may integrate ER signaling and other mitogenic pathways with cytoskeleton remodeling to increase breast cancer aggressiveness. Thus, by targeting multiple critical pathways involved in the pathogenesis and progression of breast cancer, Rac1 inhibition may be an effective therapeutic option.
Our findings also suggest that Rac1 may mediate crosstalk between growth factors and ER signaling in breast cancer since Vav3, a growth factor-activated GEF, stimulated Rac1 enhancement of ER activity. While ligand-independent activation of ER by growth factors can occur through phosphorylation of ER by downstream kinases such as MAPK, glycogen synthase kinase-3, protein kinase A and PI3K/AKT (Rayala, et al. 2006b), we identified an alternative mechanism by which Vav3 and Rac1 may activate ER. Vav3 is a Rho GTPase GEF that is activated by a variety of growth factor receptors including EGFR and stimulates Rac1 activity. Vav3 is upregulated in breast cancer compared to benign mammary tissue (Lee, et al. 2008). One study showed that Vav3 enhances ER transcriptional activity; however, the authors proposed that Vav3 mediates its effects through PI3K signaling (Lee, et al. 2008). Here we showed that inhibition of Rac1 blocked Vav3 enhancement of ER transcriptional activity, implicating Rac1 as a key mediator of Vav3 and possibly growth factor-induced ER activity. Further studies are necessary to test how Rac1 may mediate growth factor enhancement of ER activity. PI3K signaling may also independently play a role in mediating Vav3 effects on ER; however, this is complicated by the fact that Rac1 regulates PI3K as well as MAPK signaling (Bosco, et al. 2009). Therefore, Vav3 activation of Rac1 may stimulate different signaling pathways, including PI3K, to ultimately increase ER levels and ER transcriptional activity.
One signaling protein that appears to be required for Rac1 and ER crosstalk is Pak-1, a well-characterized downstream effector of Rac1. We found that Rac1 activation of Pak-1 was necessary for Rac1-mediated increase in ER transcriptional activity. Therefore, Pak-1 may be responsible for regulating ER mRNA expression downstream of Rac1. Pak-1 regulates gene expression in part through phosphorylation of factors such as FOXO1, a forkhead box transcription factor (Guo & Sonenshein. 2004, Mazumdar & Kumar. 2003). However, Pak-1 has not been shown to affect FOXO3A, which stimulates ER expression (Guo & Sonenshein. 2004, Masiello, et al. 2002). Therefore, it is unclear whether forkhead box transcription factors play a role in Rac1 regulation of ER gene expression.
Pak-1 interacts with and phosphorylates ER at serine 305 leading to phosphorylation at serine 118, which is required for full ER transcriptional activity (Balasenthil, et al. 2004, Wang, et al. 2002). While phosphorylation of ER by Pak-1 may contribute to Rac1 regulation of ER, we have identified a possible novel mechanism by which Rac1, via Pak-1, affects ER levels. Importantly, this mechanism can be therapeutically targeted.
Pak-1 is implicated in breast cancer progression to tamoxifen resistance by stimulating ER transactivation (Rayala, et al. 2006b). We found that EHT 1864 inhibition of Rac1 (and likely the downstream effector, Pak-1) decreased the proliferation of tamoxifen resistant breast cancer cells. We also showed that over expression of Ca Rac1 in breast cancer cells lead to decreased tamoxifen-mediated growth inhibition consistent with a role for Rac1 in tamoxifen resistance.
Inhibition of Rac1 is recognized as a therapeutic target in cancer due to its effects on cell migration and invasion. Furthermore, we found that inhibiting Rac1 also had anti-proliferative effects, which were in part mediated by decreasing ER levels in ER positive breast cancer cells that are either tamoxifen sensitive or resistant. Thus, inhibition of Rac1 may represent a new therapeutic opportunity in the treatment of ER positive breast cancer even in the setting of tamoxifen resistance.
ACKNOWLEDGEMENTS
We thank Drs. Catherine Welsh, Rachel Schiff, Channing Der, Martin Schwartz, Ronald Weigel, and Zafar Nawaz for generously providing reagents. Dr. Omar Flores, Ms. Jessica McCarrick, and Dr. Shuyun Rao provided expert assistance. We are grateful to Dr. Dorraya El-Ashry for helpful advice and comments on the manuscript.
FUNDING: This work was supported in part by funding from a University of Miami Braman Family Breast Cancer Research Institute developmental grant and NIH RO1 CA132200 (to KLB). AER was supported by grant F30 ES014989-01 from the National Institute of Environmental Health Sciences. MIG was supported by training grant T32 HL007188 from the National Institute of Heart, Lung, and Blood.
Abbreviations
- ER-alpha
estrogen receptor
- GEF
guanine nucleotide exchange factor
- EGFR
epidermal growth factor receptor
- Pak-1
P21/Cdc42/Rac1 activating kinase-1
- JNK
Jun N-terminal kinase
- ECM
extracellular matrix
- SERM
selective estrogen receptor modulator
Footnotes
Publisher's Disclaimer: Disclaimer. This is not the definitive version of record of this article. This manuscript has been accepted for publication in Endocrine-Related Cancer, but the version presented here has not yet been copy edited, formatted or proofed. Consequently, the Society for Endocrinology accepts no responsibility for any errors or omissions it may contain. The definitive version is now freely available at http://dx.doi.org/10.1677/ERC-10-0049.R1.
DECLARATION OF INTEREST: Dr. Laurent Desire is an employee of ExonHit Therapeutics.
REFERENCES
- Adam L, Vadlamudi R, Kondapaka SB, Chernoff J, Mendelsohn J, Kumar R. Heregulin regulates cytoskeletal reorganization and cell migration through the p21-activated kinase-1 via phosphatidylinositol-3 kinase. The Journal of biological chemistry. 1998;273:28238–28246. doi: 10.1074/jbc.273.43.28238. [DOI] [PubMed] [Google Scholar]
- Ali S, Coombes RC. Endocrine-responsive breast cancer and strategies for combating resistance. Nature reviews.Cancer. 2002;2:101–112. doi: 10.1038/nrc721. [DOI] [PubMed] [Google Scholar]
- Balasenthil S, Barnes CJ, Rayala SK, Kumar R. Estrogen receptor activation at serine 305 is sufficient to upregulate cyclin D1 in breast cancer cells. FEBS letters. 2004;567:243–247. doi: 10.1016/j.febslet.2004.04.071. [DOI] [PubMed] [Google Scholar]
- Baugher PJ, Krishnamoorthy L, Price JE, Dharmawardhane SF. Rac1 and Rac3 isoform activation is involved in the invasive and metastatic phenotype of human breast cancer cells. Breast cancer research : BCR. 2005;7:R965–74. doi: 10.1186/bcr1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco EE, Mulloy JC, Zheng Y. Rac1 GTPase: A “rac” of all trades. Cellular and molecular life sciences : CMLS. 2009;66:370–374. doi: 10.1007/s00018-008-8552-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbelo P, Wellstein A, Pestell RG. Altered rho GTPase signaling pathways in breast cancer cells. Breast cancer research and treatment. 2004;84:43–48. doi: 10.1023/B:BREA.0000018422.02237.f9. [DOI] [PubMed] [Google Scholar]
- Cai D, Iyer A, Felekkis KN, Near RI, Luo Z, Chernoff J, Albanese C, Pestell RG, Lerner A. AND-34/BCAR3, a GDP exchange factor whose overexpression confers antiestrogen resistance, activates rac, PAK1, and the cyclin D1 promoter. Cancer research. 2003;63:6802–6808. [PubMed] [Google Scholar]
- Chan AY, Coniglio SJ, Chuang YY, Michaelson D, Knaus UG, Philips MR, Symons M. Roles of the Rac1 and Rac3 GTPases in human tumor cell invasion. Oncogene. 2005b;24:7821–7829. doi: 10.1038/sj.onc.1208909. [DOI] [PubMed] [Google Scholar]
- Chen Y, Guggisberg N, Jorda M, Gonzalez-Angulo A, Hennessy B, Mills GB, Tan CK, Slingerland JM. Combined src and aromatase inhibition impairs human breast cancer growth in vivo and bypass pathways are activated in AZD0530-resistant tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:3396–3405. doi: 10.1158/1078-0432.CCR-08-3127. [DOI] [PubMed] [Google Scholar]
- Chen Y, Alvarez EA, Azzam D, Wander SA, Guggisberg N, Jorda M, Ju Z, Hennessy BT, Slingerland JM. Combined src and ER blockade impairs human breast cancer proliferation in vitro and in vivo. Breast cancer research and treatment. 2010 doi: 10.1007/s10549-010-1024-7. [DOI] [PubMed] [Google Scholar]
- Chu I, Arnaout A, Loiseau S, Sun J, Seth A, McMahon C, Chun K, Hennessy B, Mills GB, Nawaz Z, et al. Src promotes estrogen-dependent estrogen receptor alpha proteolysis in human breast cancer. The Journal of clinical investigation. 2007;117:2205–2215. doi: 10.1172/JCI21739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Come SE, Buzdar AU, Ingle JN, Arteaga CL, Brown M, Dowsett M, Hilsenbeck SG, Kumar R, Johnston SR, Lee AV, et al. Proceedings of the fifth international conference on recent advances and future directions in endocrine therapy for breast cancer: Conference summary statement. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12:997s–1000s. doi: 10.1158/1078-0432.CCR-05-2268. [DOI] [PubMed] [Google Scholar]
- de Kok JB, Roelofs RW, Giesendorf BA, Pennings JL, Waas ET, Feuth T, Swinkels DW, Span PN. Normalization of gene expression measurements in tumor tissues: Comparison of 13 endogenous control genes. Laboratory investigation; a journal of technical methods and pathology. 2005;85:154–159. doi: 10.1038/labinvest.3700208. [DOI] [PubMed] [Google Scholar]
- deConinck EC, McPherson LA, Weigel RJ. Transcriptional regulation of estrogen receptor in breast carcinomas. Molecular and cellular biology. 1995;15:2191–2196. doi: 10.1128/mcb.15.4.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhananjayan SC, Ramamoorthy S, Khan OY, Ismail A, Sun J, Slingerland J, O’Malley BW, Nawaz Z. WW domain binding protein-2, an E6-associated protein interacting protein, acts as a coactivator of estrogen and progesterone receptors. Molecular endocrinology (Baltimore, Md.) 2006;20:2343–2354. doi: 10.1210/me.2005-0533. [DOI] [PubMed] [Google Scholar]
- Fritz G, Brachetti C, Bahlmann F, Schmidt M, Kaina B. Rho GTPases in human breast tumours: Expression and mutation analyses and correlation with clinical parameters. British journal of cancer. 2002;87:635–644. doi: 10.1038/sj.bjc.6600510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Sonenshein GE. Forkhead box transcription factor FOXO3a regulates estrogen receptor alpha expression and is repressed by the her-2/neu/phosphatidylinositol 3-kinase/Akt signaling pathway. Molecular and cellular biology. 2004;24:8681–8690. doi: 10.1128/MCB.24.19.8681-8690.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science (New York, N.Y.) 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Holloway JN, Murthy S, El-Ashry D. A cytoplasmic substrate of mitogen-activated protein kinase is responsible for estrogen receptor-alpha down-regulation in breast cancer cells: The role of nuclear factor-kappaB. Molecular endocrinology (Baltimore, Md.) 2004;18:1396–1410. doi: 10.1210/me.2004-0048. [DOI] [PubMed] [Google Scholar]
- Knight-Krajewski S, Welsh CF, Liu Y, Lyons LS, Faysal JM, Yang ES, Burnstein KL. Deregulation of the rho GTPase, Rac1, suppresses cyclin-dependent kinase inhibitor p21(CIP1) levels in androgen-independent human prostate cancer cells. Oncogene. 2004;23:5513–5522. doi: 10.1038/sj.onc.1207708. [DOI] [PubMed] [Google Scholar]
- Lange CA. Making sense of cross-talk between steroid hormone receptors and intracellular signaling pathways: Who will have the last word? Molecular endocrinology (Baltimore, Md.) 2004;18:269–278. doi: 10.1210/me.2003-0331. [DOI] [PubMed] [Google Scholar]
- Lee AV, Cui X, Oesterreich S. Cross-talk among estrogen receptor, epidermal growth factor, and insulin-like growth factor signaling in breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2001;7:4429s–4435s. discussion 4411s-4412s. [PubMed] [Google Scholar]
- Lee H, Jiang F, Wang Q, Nicosia SV, Yang J, Su B, Bai W. MEKK1 activation of human estrogen receptor alpha and stimulation of the agonistic activity of 4-hydroxytamoxifen in endometrial and ovarian cancer cells. Molecular endocrinology (Baltimore, Md.) 2000;14:1882–1896. doi: 10.1210/mend.14.11.0554. [DOI] [PubMed] [Google Scholar]
- Lee K, Liu Y, Mo JQ, Zhang J, Dong Z, Lu S. Vav3 oncogene activates estrogen receptor and its overexpression may be involved in human breast cancer. BMC cancer. 2008;8:158. doi: 10.1186/1471-2407-8-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons LS, Burnstein KL. Vav3, a rho GTPase guanine nucleotide exchange factor, increases during progression to androgen independence in prostate cancer cells and potentiates androgen receptor transcriptional activity. Molecular endocrinology (Baltimore, Md.) 2006;20:1061–1072. doi: 10.1210/me.2005-0346. [DOI] [PubMed] [Google Scholar]
- Lyons LS, Rao S, Balkan W, Faysal J, Maiorino CA, Burnstein KL. Ligand-independent activation of androgen receptors by rho GTPase signaling in prostate cancer. Molecular endocrinology (Baltimore, Md.) 2008;22:597–608. doi: 10.1210/me.2007-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manser E, Leung T, Salihuddin H, Zhao ZS, Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–46. doi: 10.1038/367040a0. [DOI] [PubMed] [Google Scholar]
- Masiello D, Cheng S, Bubley GJ, Lu ML, Balk SP. Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. The Journal of biological chemistry. 2002;277:26321–26326. doi: 10.1074/jbc.M203310200. [DOI] [PubMed] [Google Scholar]
- Mazumdar A, Kumar R. Estrogen regulation of Pak1 and FKHR pathways in breast cancer cells. FEBS letters. 2003;535:6–10. doi: 10.1016/s0014-5793(02)03846-2. [DOI] [PubMed] [Google Scholar]
- Onesto C, Shutes A, Picard V, Schweighoffer F, Der CJ. Characterization of EHT 1864, a novel small molecule inhibitor of rac family small GTPases. Methods in enzymology. 2008;439:111–129. doi: 10.1016/S0076-6879(07)00409-0. [DOI] [PubMed] [Google Scholar]
- Rayala SK, Molli PR, Kumar R. Nuclear p21-activated kinase 1 in breast cancer packs off tamoxifen sensitivity. Cancer research. 2006a;66:5985–5988. doi: 10.1158/0008-5472.CAN-06-0978. [DOI] [PubMed] [Google Scholar]
- Rayala SK, Talukder AH, Balasenthil S, Tharakan R, Barnes CJ, Wang RA, Aldaz CM, Khan S, Kumar R. P21-activated kinase 1 regulation of estrogen receptor-alpha activation involves serine 305 activation linked with serine 118 phosphorylation. Cancer research. 2006b;66:1694–1701. doi: 10.1158/0008-5472.CAN-05-2922. [DOI] [PubMed] [Google Scholar]
- Sahai E, Marshall CJ. RHO-GTPases and cancer. Nature reviews.Cancer. 2002;2:133–142. doi: 10.1038/nrc725. [DOI] [PubMed] [Google Scholar]
- Schiff R, Chamness GC, Brown PH. Advances in breast cancer treatment and prevention: Preclinical studies on aromatase inhibitors and new selective estrogen receptor modulators (SERMs) Breast cancer research : BCR. 2003;5:228–231. doi: 10.1186/bcr626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff R, Massarweh SA, Shou J, Bharwani L, Arpino G, Rimawi M, Osborne CK. Advanced concepts in estrogen receptor biology and breast cancer endocrine resistance: Implicated role of growth factor signaling and estrogen receptor coregulators. Cancer chemotherapy and pharmacology. 2005;56(Suppl 1):10–20. doi: 10.1007/s00280-005-0108-2. [DOI] [PubMed] [Google Scholar]
- Schnelzer A, Prechtel D, Knaus U, Dehne K, Gerhard M, Graeff H, Harbeck N, Schmitt M, Lengyel E. Rac1 in human breast cancer: Overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene. 2000;19:3013–3020. doi: 10.1038/sj.onc.1203621. [DOI] [PubMed] [Google Scholar]
- Shutes A, Onesto C, Picard V, Leblond B, Schweighoffer F, Der CJ. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of rac family small GTPases. The Journal of biological chemistry. 2007;282:35666–35678. doi: 10.1074/jbc.M703571200. [DOI] [PubMed] [Google Scholar]
- Su LF, Knoblauch R, Garabedian MJ. Rho GTPases as modulators of the estrogen receptor transcriptional response. The Journal of biological chemistry. 2001;276:3231–3237. doi: 10.1074/jbc.M005547200. [DOI] [PubMed] [Google Scholar]
- Sun J, Nawaz Z, Slingerland JM. Long-range activation of GREB1 by estrogen receptor via three distal consensus estrogen-responsive elements in breast cancer cells. Molecular endocrinology (Baltimore, Md.) 2007;21:2651–2662. doi: 10.1210/me.2007-0082. [DOI] [PubMed] [Google Scholar]
- Vadlamudi RK, Adam L, Wang RA, Mandal M, Nguyen D, Sahin A, Chernoff J, Hung MC, Kumar R. Regulatable expression of p21-activated kinase-1 promotes anchorage-independent growth and abnormal organization of mitotic spindles in human epithelial breast cancer cells. The Journal of biological chemistry. 2000;275:36238–36244. doi: 10.1074/jbc.M002138200. [DOI] [PubMed] [Google Scholar]
- Van Aelst L, D’Souza-Schorey C. Rho GTPases and signaling networks. Genes & development. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- Wang RA, Mazumdar A, Vadlamudi RK, Kumar R. P21-activated kinase-1 phosphorylates and transactivates estrogen receptor-alpha and promotes hyperplasia in mammary epithelium. The EMBO journal. 2002;21:5437–5447. doi: 10.1093/emboj/cdf543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwick JK, Lambert QT, Clark GJ, Symons M, Van Aelst L, Pestell RG, Der CJ. Rac regulation of transformation, gene expression, and actin organization by multiple, PAK-independent pathways. Molecular and cellular biology. 1997;17:1324–1335. doi: 10.1128/mcb.17.3.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie JW, Haslam SZ. Extracellular matrix, Rac1 signaling, and estrogen-induced proliferation in MCF-7 breast cancer cells. Breast cancer research and treatment. 2008;110:257–268. doi: 10.1007/s10549-007-9719-0. [DOI] [PubMed] [Google Scholar]
- Yang C, Liu Y, Lemmon MA, Kazanietz MG. Essential role for rac in heregulin beta1 mitogenic signaling: A mechanism that involves epidermal growth factor receptor and is independent of ErbB4. Molecular and cellular biology. 2006;26:831–842. doi: 10.1128/MCB.26.3.831-842.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]