

Abstract
A crucial question that must be addressed in the drug development process is whether the proposed therapeutic target will yield the desired effect in the clinical population. Pharmaceutical and biotechnology companies place a large investment on research and development, long before confirmatory data are available from human trials. Basic science has greatly expanded the computable knowledge of disease processes, both through the generation of large omics data sets and a compendium of studies assessing cellular and systemic responses to physiologic and pathophysiologic stimuli. Given inherent uncertainties in drug development, mechanistic systems models can better inform target selection and the decision process for advancing compounds through preclinical and clinical research.
Computational methods have made exciting contributions to pharmaceutical research and development. Computer-aided drug design has been established as a valuable tool for the design of new drugs, with many success stories since the 1980s [1]. Pharmaceutical companies have invested substantially in bioinformatics approaches, and it has been predicted such approaches will have an important role in pharmacogenomics and personalized medicine [2]. Already, the FDA has recognized the importance of informatics approaches to generate novel biomarkers to personalize cancer therapies [3].
Mechanistic modeling approaches can yield insights from data throughout the drug development process. For example, in the context of metabolomics, it is well-established that systems models facilitate insights from high-throughput data [4]. Even when models are not specifically constructed for pairing with high-throughput data, they can be informed from the literature and preclinical studies. Much of the utility of systems modeling for advancing therapeutics lies in the ability to develop hypotheses regarding the characteristics of a disease system. Such approaches to pharmaceutical research parallel systems biology. They are driven by the ability to formulate testable hypotheses, are inherently quantitative because they use a quantitative modeling framework, integrate potentially high dimensional data from multiple sources, and enable global mechanistically based analysis of the physiologic system [5]. Notably, such integrative approaches can assist in translating a result from an in vitro study or animal model to better predict efficacy in a clinical context.
Our purpose is not to provide a comprehensive review of computational methods used in the pharmaceutical industry. For example, we intentionally do not delve into the discussion of data mining approaches or PK/PD modeling. Rather, our focus is large mechanistic models of biological systems [6], especially those with applications in drug development. Such approaches have demonstrated value to industrial research programs [7], and we posit that they will become an integral component of research practice as the pharmaceutical industry transitions to increasing utilization of computational approaches as a component of an evolving research paradigm. Notably, a growing body of literature facilitates discussion of two mechanistic systems modeling methods that can inform drug research and development. One is a biosimulation technique that links clinical disease phenotypes to increasingly granular mathematical representations of pathophysiologic processes. The second constructs functional, computable cellular networks from the molecular building blocks of genes and proteins to elucidate the impact of pathologic or therapeutic alterations on network operating states and hence clinical phenotype. As we will discuss in the case studies, both approaches may directly facilitate the in silico evaluation of systems-level pharmaceutical action, are amenable to intelligent alterations of assumptions to address best-case and worst-case scenarios, identify important preclinical research experiments, provide a method to interpret high-throughput data sets, can guide drug repositioning, and can guide the development of biomarkers. Finally, we discuss how mechanistic systems models can inform the prioritization of research programs to help improve the return on investment for the costly process of drug development.
Clinical phenotype-driven models of disease pathophysiology
Perhaps the most renowned example of a phenotype-driven model of pathophysiology is the minimal model of Bergman and Cobelli, for which clinical results were first published in 1981 [8]. The minimal model is a carefully validated framework [9] that models glucose and insulin dynamics in response to an intravenous glucose tolerance test. Fitting the model to a data set results in parameter estimates that are particularly useful for determining insulin sensitivity and the responsiveness of β cells to glucose on an individual patient basis. While the minimal model reports the disposition index, an indicator of risk for developing type 2 diabetes [10], this simple model cannot be used to investigate the efficacy of many new therapeutics in the absence of clinical data. Although such small-scale models have great utility in extracting important information from data [6,11], it would not be possible to form a priori predictions, for example, of the effects of insulin secretagogues, such as glyburide, on plasma glucose levels. As we will discuss shortly, a different formalism must be developed to achieve the potential for prediction. The components of a phenotype-driven model, required data and represented aspects of the phenotype are described in Fig. 1a.
FIGURE 1. The scope, input and output of two modeling paradigms.
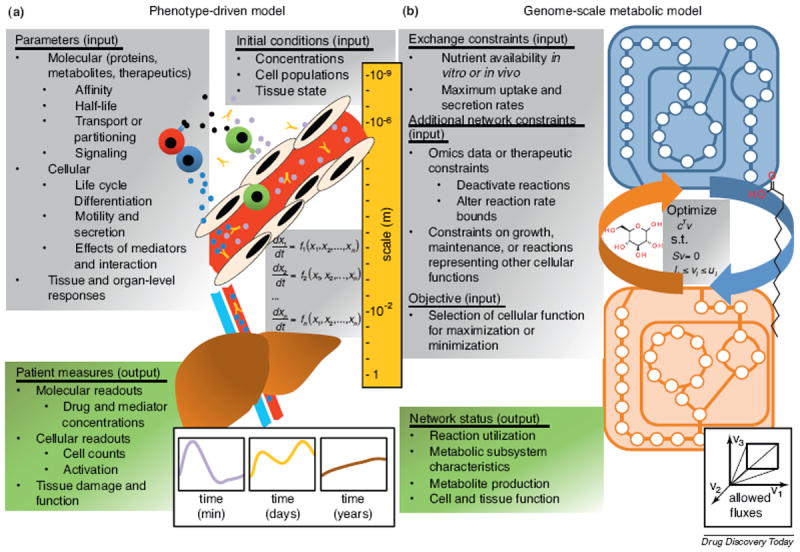
(a) Phenotype-driven models integrate biological processes relevant to disease across length scales, from molecular mediators to tissue responses. Once the model is built and parameters are defined, either from the literature or experimental measures, a simulation solves a large system of ordinary differential equations. The result is the prediction of the dynamics of therapeutic and mediator concentrations, cell populations and composition, and tissue-level function. Simulations must span multiple time scales to capture important events such as the administration of therapeutics as well as clinical disease progression. (b) Genome-scale metabolic models use knowledge of metabolic reactions in a given cell type to construct stoichiometrically defined reaction networks. Reactions are frequently associated with enzymes from known genetic loci, which makes the task of informing the model with high-throughput data, such as proteomics or transcriptomics, more tractable. The network is often assumed to be at steady-state, with constant reaction, metabolite consumption and metabolite production rates. The formulation of cellular goals, or objectives, enables the application of methods from linear programming. Flux balance analysis and related mathematical approaches result in a description of allowable fluxes, the rates at which reactions are used for metabolic conversions, in the network. This description of the network state can be further analyzed to deduce cellular function, including the optimization of goals such as growth, ATP production, or glucose production, as well as the effect of perturbations in the network originating from disease or therapy.
Model building
A process for constructing phenotype-driven models has been developed based on American Diabetes Association guidelines and is depicted in Fig. 2a [12,13]. Ostensibly, constructing such models requires expertise in a therapeutic area and sufficient knowledge of disease pathophysiology. There is some flexibility in deciding which level of mechanistic detail to include when constructing phenotype-driven models. Fortuitously, as computing power has increased, it is possible in practice to construct models with as much detail for which there are data with sufficient confidence and relevance. To be of utility for drug development, the salient features contributing to the disease and accessible to therapeutic targeting, as identified by experts in the therapeutic area, should be integrated into the model. Rather than observing the response of the system to defined perturbations and fitting model parameters accordingly, the goal can be to integrate isolated measures together to predict system responses. Such phenotype-driven, or ‘top-down,’ modeling can therefore become useful for in silico evaluations of the clinical efficacy of a target [14] and for exploring which pathways are most important to mediate a clinical response.
FIGURE 2. Construction of large-scale mechanistic models.
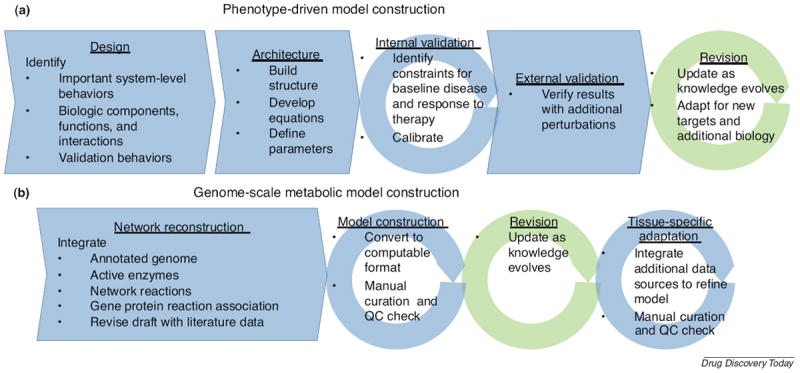
(a) The process of constructing a phenotype-driven model is reviewed here briefly. In the design phase, the model scope, including crucial model behaviors, components and validation behaviors are identified. This phase includes an extensive literature review to ensure important aspects of the disease and validation data are identified. Next, the development of the architecture involves defining the equations that will govern the model behavior and developing parameter estimates. An internal validation step is performed. The calibration may use both manual and automated optimization techniques. Frequently, acceptable perturbations to multiple aspects of the biology, such as therapeutics with distinct mechanisms of action, are used. Often, the goal is to match average population behaviors, and such a carefully calibrated internal validation result serves as a reference virtual patient. During external validation, the responses to additional perturbations (not included in the calibration step) are tested. The model can then be applied for research purposes. However, the model may be recalibrated as additional data become available. An existing model may also be used as a basis for exploring new targets as additional information about relevant pathways becomes available, although such a model enhancement will require a similar, albeit smaller, process of design, architecture development, internal validation and external validation. (b) The process of constructing a genome-scale metabolic network reconstruction is illustrated. First, a general network reconstruction must be built. An annotated draft genome is integrated with databases containing the relevant enzymes and network reactions. After assigning gene protein reaction associations and performing an initial manual curation step, the reconstruction should be converted to a computable format. Once this is done, additional quality control and manual curation steps can be performed, such as verifying that metabolic pathways support fluxes and inspecting dead-end reactions. Once these steps are satisfactorily completed, one has a high-quality metabolic network reconstruction that may be revised as the knowledge base grows. For application to different therapeutic areas, tissue-specific adaptations of the generic model must be constructed.
As illustrated in Fig. 2a, a large system of deterministic ordinary differential equations is developed to track the status of the system with time. The size of these large mechanistic models varies roughly between ten and hundreds of ordinary differential equations [6]. The literature is used to derive parameter estimates directly or define reasonable boundaries for parameter constraints, which must then be optimized. Hundreds to thousands of parameter values must be assigned to run large mechanistic biosimulations. Fortunately, despite potential uncertainty in parameter estimates, detailed investigation of the properties of systems models suggests validation through evaluation of model predictions is a sound strategy that enables reliable biological investigation [15].
An important consideration is the time and cost associated with the development of the phenotype-driven models. Depending on the scope of the model, it can take a skilled team a year or more to develop a comprehensive, functional model consistent with available data. It has been reported that on average, the development of a new drug requires approximately $800 million and 14 years [16-18]. While the investment to develop an informative simulation is small relative to the drug development project, it is a challenge that must be taken into consideration if one is planning to use such a tool to guide research and decision-making. The data in Table 1 may assist in the development effort. If an existing model can be adapted for application, it may help to decrease the development time.
TABLE 1.
Systems models with demonstrated application to drug development
| Therapeutic area | Companya | Platform | Refs | Notes |
|---|---|---|---|---|
| Anemia | Entelos | PhysioLab | [84] | |
| Anemia (hemolytic) | Academic | Multipleb | [23] | Short investigation of glutathione synthetase and glutamate-cysteine ligase in context of full Recon 1 |
| Asthma | Entelos | PhysioLab | [5,82,85-87] | c |
| Cancer | Academic | Multipleb | [41,44,88-90] | d |
| Cardiovascular | Entelos | PhysioLab | [49] | |
| Cardiovascular | Academic | Multipleb | [23] | Short investigation of HMG-CoA inhibition in context of full Recon 1 |
| Central nervous system | Rhenovia Pharma SAS | RHEDDOS | [91] | |
| Central nervous system | Academic | Multipleb | [36] | d |
| Type 1 diabetes | Entelos | PhysioLab | [13] | NOD mouse model |
| Type 2 diabetes | Entelos | PhysioLab | [92,93] | c |
| Type 2 diabetes | Academic, GT Life Sciences, Intrexon, Genomatica | Multipleb | [42] | |
| Drug-induced liver injury | Entelos | PhysioLab | [94] | |
| Leigh’s syndrome | Academic | Multipleb | [95] | Mitochondrial focusd |
| Infectious disease and biodefense | Academic | Multipleb | A. baumannii [96], M. tuberculosis [33,97-100], B. cenocepacia [101], F. tularensis [102], L. major [103,104], V. vulnificus [105], P. aeruginosa [106], S. aureus [107] and S. Typhimurium [108]e | |
| RAAS | Entelos | PhysioLab | [109,110] | |
| Rheumatoid arthritis | Entelos | PhysioLab | [5,22,69,111,112] | |
| Sepsis | Immunetrics | [113-115] |
We only include large mechanistic disease models reported in journal publications or at scientific conferences.
GEMs are often available in Systems Biology Markup Language (SBML) format, which can be used with several software packages, including MATLAB [116]. The COnstraint-Based Reconstruction and Analysis (COBRA) toolbox is one especially useful tool for simulating network states [117].
Of note, smaller, ‘medium scale’ models that capture mechanistic aspects of pathophysiology for use in drug development have been reported at conferences by Rosa & Co. for asthma [118] and diabetes [119-123].
Metabolic network model informed with tissue-specific data.
Although not purely human simulation, we include several examples of probing for metabolic drug targets in lethal pathogens or metabolic network integration with the host to illustrate another important application of metabolic systems modeling.
Therapeutic applications
Mechanistic systems modeling has been employed in several therapeutic areas, as illustrated in Table 1. The phenotype-driven models are very specialized for each therapeutic area, as they are designed to capture relationships between unique clinical outputs and disease mechanisms. The models may contain sufficient detail to simulate the effects of existing therapies in addition to investigational compounds, and they can enable in silico screens of multiple targets. Several examples to introduce how model outputs can be used to inform drug development decisions are given in Table 2. Smaller models with a minimal representation of pathophysiology [8], and shorter model development timelines, can be employed in a complementary manner to the larger systems models discussed here. On a related note, how best to use statistical versus mechanistic approaches to optimally benefit drug discovery and development is a topic of recent discussion at scientific conferences [19,20] and the development and refinement of systems models as organizational core assets to help guide target selection in a therapeutic area has been proposed in the context of biodesign [21]. The question of how to integrate the efforts of different modeling groups is a challenge that must be addressed to fully realize the value offered by each approach.
TABLE 2.
Examples illustrating how model outputs yield insights relevant to drug development decisions
| Model type | Primary model outputs | Drug development insight | Study description and reference |
|---|---|---|---|
| Genome-scale metabolic model | Biomass production (cellular growth rate) | Preclinical target identification | It was computationally predicted that the loss of function in fumarate hydratase that occurs in hereditary leiomyomatosis and renal-cell cancer results in susceptibility to disruption of a heme degradation pathway. This result was then verified in vitro [89]. |
| Phenotype-driven model | Forced Expiratory Volume in One Second (FEV1) and the activity of 501 pathways in multiple virtual patients | Preclinical target verification | Phosphodiesterase-4 (PDE4) was evaluated as target for the treatment of asthma. Biosimulation identified pathway-level effects of PDE4 most predictive of improvements in FEV1, yielding pathway-level outcomes that can be monitored during an in vitro screen to verify advancement to clinical trials [5,82]. |
| Phenotype-driven model | Mortality, organ failure and a panel of 18 analytes | Clinical trial design and marker identification | The model of severe sepsis was trained with data from patients with community-acquired pneumonia that underwent a 30-day hospital stay and treatment. Thirty-day mortality following discharge was predicted with 84% overall accuracy. The model training process identified analytes likely important for the prediction of trial outcomes (mortality). The accuracy of the model with a ‘virtual clinician’ suggests the model can be used to help design trial treatment protocols [115]. |
| Phenotype-driven model | ACR-N score, JSN score and BES in response to multiple therapeutics in a cohort of virtual patients | Clinical trial design and competitive differentiation | Simulations were run for a large set of virtual patients. Virtual patients that responded well to NKG2D inhibition, but not currently approved therapies, were identified, illustrating anti-NKG2D will potentially fulfill an unmet clinical need in rheumatoid arthritis therapy. Additionally, simulations predicted the need to maintain methotrexate therapy for optimal efficacy, an important consideration for trial design. Subsequent in silico studies were proposed to identify how common these patients are in the clinic and also to identify stratifying biomarkers [112]. |
An excellent study illustrating the investigation that can be performed with large phenotype-driven models was published by Rullman et al. for the case of a model of rheumatoid arthritis [22]. Notably, development of the rheumatoid arthritis model began with the goal of simulating the essential aspects of the clinical manifestation of rheumatoid arthritis, inflammation and the degradation of bone and cartilage, and developed a mathematical framework with sufficient resolution to capture the dynamics of cellular behaviors and the cytokine network. First, a model constraint and calibration process was performed. The parameters for a simulated patient were adjusted to yield average clinical responses to therapies for which clinical trial data existed (e.g. methotrexate, etanercept and anakinra). Comparison of the result for the calibrated model for cyclosporin A against trial data then served as a check of the model calibration. The calibration solution that was validated against known therapeutic responses and constrained with acceptable cellular concentrations, mediator concentrations and cellular activities became a ‘reference virtual patient’ that formed the basis for subsequent analyses.
The reference virtual patient was then used to evaluate efficacy of new targets in the context of the representation of the pathophysiological system, to perform in silico screens of multiple targets, and was amenable to alterations of assumptions to evaluate alternate scenarios of efficacy. For example, Rullman et al. analyzed the effect of knocking out either IL-12 or IL-15 in rheumatoid arthritis, an assumption equivalent to adding a blocking monoclonal antibody, assuming sufficient dosing and transport to the synovial tissue. The simulation results for the IL-15 knockout were in qualitative agreement with clinical data from a phase 2 trial. Although confirmatory data were not yet available for IL-12, it was predicted that inhibition of IL-12 would be less efficacious than the current first-line therapy, methotrexate. In all, the effect of individually inhibiting 31 targets was simulated, with varying assumptions of the roles for each target in the pathway. To clarify, the literature data around a particular target can be quantitatively variable. The approach allowed Rullman et al. to postulate how variability, whether due to data uncertainties or true biologic heterogeneity, affected predicted efficacy in best- and worst-case scenarios. Thus, once construction of a mechanistic systems model is complete, it is relatively straightforward to interrogate and to form recommendations for drug targets, or combinations of drug targets, that are clinically efficacious in the context of the pathophysiological system. The leading targets can be used to guide subsequent experiments to verify the predictions of effects on downstream processes at the preclinical stage and cross-validate model assumptions.
Biological network simulations
Simulations of biological networks offer an alternative method to investigate disease pathophysiology and to evaluate the effects of pharmaceuticals in the context of a mechanistic biological system. Rather than initiating development with the disease phenotype and reconstructing the relevant physiology, a model of the entire network is developed. The activity of the network in disease and health is simulated, and modulation of the disease network by therapy can be investigated. There are several cellular networks relevant to drug discovery, including interaction networks, regulatory networks, signaling networks and metabolic networks. Integrating all components of cellular function will be a challenge and a thorough review of each is beyond the scope of this review. We choose to focus here on metabolic networks. Arguably, metabolic networks are the most complete, and a full computable model of the human metabolic network has been developed for study [23]. Metabolic networks therefore represent a relatively mature knowledge base available in a computable format and are ready to facilitate rigorous, holistic study. Notably, the metabolome is smaller and downstream of both the genome and proteome [24]. We might therefore expect metabolism to carry information more closely related to the operating state of a cell and to be perturbed in disease states. The components, scope and output of genome-scale metabolic models (GEMs) are described in Fig. 1b.
Model building
The process for constructing a GEM is depicted in Fig. 2b and has been reviewed extensively [4,25-27]. Notably, GEMs were initially developed for bacteria for reasons that arguably include the early availability of genomic data and relative ease of experimentally verifying model growth phenotype predictions [26]. One important distinction for the human, a multicellular organism, is the metabolic adaptation necessary to support specialized tissue functions. For example, the active metabolic pathways in an adipocyte are different from the liver.
The construction and mathematical basis of GEMs is different from the phenotype-driven models discussed previously. GEM construction can be described as a bottom-up, informatics-driven process because it begins with large lists of cellular parts: the annotated genome, metabolic reactions and enzyme components [27]. However, as reviewed previously and demonstrated again when constructing a human GEM [23,27], manual curation and validation are crucial to obtain a high-quality reconstruction. Mathematically, the metabolic conversion reaction network is defined by a large stoichiometric matrix of metabolic transformations in the cell. To date, investigations have primarily focused on steady-state network properties. Reaction flux constraints are used to guide the analysis, and detailed knowledge of the kinetic rates of individual reactions is therefore unnecessary. This constraint-based approach instead characterizes the possible operating states of the network at steady-state. Flux balance analysis and approaches to characterize metabolic network properties have been reviewed in detail elsewhere [28]. Although some progress has been made toward kinetic mass action stoichiometric simulations [29], steady-state-based approaches have already yielded insights as will be discussed shortly.
Similar to phenotype-driven modeling, it is generally preferable to adapt an existing model to expedite research. Fortunately, this requirement has been taken into account when constructing the human GEM, H. sapiens Recon 1, which broadly incorporates metabolic reactions from all tissues. Similar to other biochemically, genetically and genomically structured (BiGG) genome-scale reconstructions, Recon 1 includes gene-protein mappings [30]. Gene-protein mappings make it possible to adapt the pathways included in the simulation to obtain tissue-specific representations of metabolism based on available RNA expression microarray data. Therefore, in addition to predicting metabolic fluxes, the GEM provides a ‘context for content,’ enabling the researcher to better interpret large data sets in the context of the metabolic system [4,26,31].
Omics data integration has proven to be a crucial step in developing tissue-specific models of metabolism. For example, Recon 1 was informed with microarray and proteomic data to develop reaction flux predictions for the heart, kidney, brain, liver, lung, pancreas, prostate, spleen, skeletal muscle and thymus [32]. Additional reports have used omics data sets to develop computable models of the macrophage [33,34], kidney [35], brain [36], liver [37] and erythrocyte [38]. A third model of liver metabolism has also been constructed by integrating Recon 1 with an independent network reconstruction of human metabolism [39] and extensive manual testing of the resulting reconstruction [40]. The extensively validated model of liver metabolism has also facilitated the development of new algorithms for tissue-specific model development by serving as a reference [41]. Additionally, models of the adipocyte, hepatocyte and myocyte have been metabolically integrated together [42], an important advance for modeling complex diseases. Manual curation is a labor-intensive but crucial component of the process, because fully automated methods may miss crucial organ functions. For example, purely automated methods yielded a kidney that lacked the ability to excrete urea [35]. Notably, unless a high purity of cell-type specific RNA has been obtained, the result of customization may be a network that represents a compendium of local tissue functions. Table 3 describes additional studies and resources for integrating omics data sets into analysis with GEMs.
TABLE 3.
Methods for informing GEMs with omics data sets
| Algorithm name and reference | Brief description |
|---|---|
| None specified [124] | Transcript absent decisions from microarray data are used to eliminate network reactions |
| iMATa (integrated Metabolic Analysis Tool) [125] | Predicts a set of reaction fluxes based on microarray and proteomic data |
| GIMMEb (Gene Inactivity Moderated by Metabolism and Expression) [126] | Uses microarray data, an expression threshold, and required metabolic functions to propose allowed network reactions and penalties |
| E-Flux [127] | Alters allowed reaction flux ranges based on gene expression data |
| MBA (Model Building Algorithm) [37] | Proposes a parsimonious set of reactions that is consistent with multiple high-throughput data sources |
| IOMAc (Integrative Omics-Metabolic Analysis) [128] | Predicts reaction fluxes that are consistent with proteomics and metabolomics measurements |
| tFBA [129] | Maximizes consistency of reaction fluxes with changes in gene expression |
| MADEd (Metabolic Adjustment by Differential Expression) [130] | Uses statistical changes in metabolic gene expression to enable network reactions |
| INIT (Integrative Network Inference for Tissues) [41] | Uses the Human Protein Atlas as a primary data source to create tissue-specific models and also allows for the accumulation of network metabolites |
| GIMMEp (Gene Inactivity Moderated by Metabolism and Expression by Proteome) [34] | Expands on the original GIMME method by integrating proteomic data to identify additional required metabolic functions |
iMAT is available online (www.cs.technion.ac.il/~tomersh/methods.html). Versions of the iMAT algorithm are also available in the COBRA toolbox and TIGER.
GIMME is available in the COBRA toolbox [117] (http://opencobra.sourceforge.net/openCOBRA) as well as in TIGER.
IOMA is available online (http://www.cs.technion.ac.il/~tomersh/methods.html).
MADE is available online (http://bme.virginia.edu/csbl/downloads.php) as well as in TIGER (Toolbox for Integrating Genome-scale metabolic models, Expression data and transcriptional Regulatory networks) [131] (https://bitbucket.org/csbl/tiger/downloads).
Therapeutic applications
Before exploring the application of computable metabolic systems models, it is important to gain an appreciation for the translationally relevant questions that they can address. As these models are constructed in a bottom-up fashion, they may not directly simulate clinical endpoints. Using the example of type 2 diabetes, a clinical measure of primary interest would be the peak plasma glucose following an oral glucose tolerance test. Although metabolic network models are not constructed with such endpoints in mind, they can be used, for example, to formulate detailed hypotheses regarding the effects of alterations in hepatic glucose metabolism.
Therapeutic areas that have been explored with tissue-specific adaptations of GEMs are included in Table 1. A breadth of in silico techniques that can be applied to interpret cancer etiology and develop therapies have been reviewed elsewhere [43], and we discuss here a recent study by Folger et al. to highlight the power and utility of GEM-guided analyses [44]. Cancer is a promising therapeutic area for the application of GEM-approaches. Metabolic alterations are known to occur in cancer, suggesting that metabolic pathways can be targeted that would be specifically deleterious to cancer growth. Folger et al. adapted Recon 1 using the methodologies mentioned previously [37] to create two metabolic representations of cancer. First, a generic model of cancer metabolism was constructed using a core set of reactions deduced from shared expression profiles among the NCI-60 collection of cell lines. Second, a model of nonsmall cell lung cancer was constructed. Notably, the study by Folger et al. demonstrates the utility of the human GEM to interpret high-throughput data in the context of target discovery. The phenotypic results of shRNA gene silencing experiments are available for the NCI-60 lines and served as model validation. In the generic model, all 199 genes that supported optimization of a cancer growth objective were found to be experimentally essential. As expected, the nonsmall cell lung cancer model outperformed the generic model when compared with essentiality data specific to the cell line. Having thus validated the model, Folger et al. developed predictions for cancer targets using a cytostatic score, a ratio that takes into account the cytostatic effect on cancer cells while minimally perturbing crucial ATP flux for the entire human network. They predicted 52 single gene targets that have a high cytostatic score. Notably, 13 of the 52 cytostatic single gene targets are targeted by approved noncancer drugs, suggesting these drugs might be repositioned for cancer. An intriguing possibility for targeting cancer is to exploit synthetic lethality, targeting pairs of genes that when knocked out only in combination result in the arrest of metabolic function. Many more synthetic lethal gene pairs with a high cytostatic score were discovered. Of the 133 synthetic lethal combinations with a high cytostatic score, 99 were not predicted to reduce the growth of healthy cells and eight were predicted to be potentially damaging to liver function. Additionally, synthetic lethal combinations with one downregulated partner in cancer may be viable therapeutic targets. Of 342 total synthetic lethal combinations, 72 pairs were identified that fit this criterion. Folger et al. also highlight the iterative nature of model construction, noting that revised tissue models may alter the synthetic lethal predictions. Future revisions to Recon 1 may further improve predictions.
Triangulation of translational systems biology models: could the phase 3 failure of torcetrapib have been prevented?
A unique example illustrating how different systems simulation methodologies can be integrated into decision support processes in the pharmaceutical industry can be found among the cholesterol-modifying drugs that have been developed to reduce the risk of cardiovascular events. Pfizer’s Lipitor® (atorvastatin) is a tremendously successful drug that blocks cholesterol synthesis in the liver through competitive inhibition of HMG-CoA reductase [45]. In some patient populations, Lipitor has been demonstrated to reduce cardiovascular events such as heart attacks and strokes [46]. Annual sales were $12.8 billion in 2008 alone [Doing Things Differently, Pfizer Annual Review 2008]. Furthermore, it has been observed that a reduction in the levels of cholesterol-ester transfer protein (CETP) results in an increase in HDL-C and a reduction in LDL-C [47], a cholesterol profile normally associated with a reduced risk of cardiovascular events. Pfizer developed a CETP inhibitor, torcetrapib, to be taken in combination with the highly successful atorvastatin. However, the clinical program was terminated after a large phase 3 trial enrolling 15,000 patients (Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events, ILLUMINATE) found an increase in blood pressure and mortality in the group treated with combination atorvastatin and torcetrapib [48].
The late-stage failure of torcetrapib highlights the risks taken by the pharmaceutical industry when developing novel therapeutics. Importantly, the outcome has gained the attention of several translational systems biology studies, affording a unique opportunity to directly compare the predictions offered by each. Each provides unique hypotheses relevant to the safety and efficacy of torcetrapib.
A study by Powell et al. [49] employing phenotype-driven simulation methods predicted that the atorvastatin/torcetrapib combination would not improve atherosclerotic endpoints, represented by atheroma volume, as measured clinically by intravital ultrasound (IVUS). Notably, as the model employed by Powell et al. only accounts for the action on the target and not the specific structure of the inhibitor, the predicted lack of an improvement in atheroma volume is a potential deficiency of other CETP inhibitors, and not specific to torcetrapib. Although the CETP inhibitor altered the plasma HDL cholesterol profile, the net catabolism of cholesterol did not increase. A follow-up study by Wahba et al. [50] in a virtual patient cohort included two additional CETP inhibitors, dalcetrapib (JTT-705) and anacetrapib (MK-0859), with background statin therapy and found little improvement in the hazard ratio for coronary heart disease risk. However, the results suggested a subpopulation of virtual patients with high triglyceride and low HDL levels would benefit from CETP inhibition. Wahba et al.’s study was timely because it was presented as dalcetrapib and anacetrapib were undergoing phase 3 clinical trials (http://clinicaltrials.gov/, trials NCT01323153, NCT01059682 and NCT01252953) [51,52]. Subsequently, Roche terminated phase 3 trials for dalcetrapib due to a lack of efficacy (http://www.roche.com/media/media_releases/med-cor-2012-05-07.htm).
Although structural biology is not a focus of the current review, a study by Xie et al. formulated a salient hypothesis regarding the hypertensive side effect of torcetrapib [53], as observed in the ILLUMINATE trial. Xie et al. employed a novel method whereby the binding site structure for torcetrapib on CETP was extracted, the binding pocket was used to search for local structural homology in additional proteins with structural information using a novel alignment method, and high-ranking alignments were used for docking studies with ligand. CETP inhibitors were predicted to interact with several members of the nuclear receptor family involved in the regulation of the renin angiotensin aldosterone system (RAAS), inflammation and proliferation: LXRα/β, PPARα/γ/δ, GCR, RXR and VDR. Torcetrapib was predicted to bind the most strongly to the broadest array of positive regulators of RAAS and weakly to the negative regulator, suggesting torcetrapib would have the most pronounced effect on blood pressure. The result is consistent with the phase 2 data available from the two other CETP inhibitors, which have not been reported to exhibit a hypertensive effect [54-59].
A simulation of renal metabolic function integrating structural biology methods was performed to evaluate the effect of torce-trapib on the renal regulation of blood pressure [35]. A model of renal metabolism was constructed from Recon 1, and the structures of the enzymes mediating the metabolic reactions were screened for similarity to the binding site for torcetrapib to CETP. A novel optimization objective was developed to ensure the kidney processed metabolites involved in the regulation of blood pressure and had an adequate supply of renal ATP. Perhaps most interesting, the inhibition of prostaglandin I2 synthase by torcetrapib was predicted to strongly disrupt PGI2 production, a potent vasodilator capable of reducing blood pressure, and peroxisomal acylcoenzyme A oxidase 1, which was predicted to affect renal absorption of citrate and amino acids. Notably, in order of decreasing affinity, anacetrapib, torcetrapib and dalcetrapib were predicted to more strongly bind PTGIS than the endogenous ligand, prostaglandin H2. Chang et al. also extended their approach to investigate pharmacogenomics, and made observations regarding genetic alterations that would yield renal metabolism more susceptible to the off-target effects of the CETP inhibitors. The similar predicted effect of anacetrapib and torcetrapib on blood pressure, given the enhanced binding relative to the endogenous ligands of PTGIS and ACOX1, does not appear to be entirely consistent with the lack of an observed effect of anacetrapib on blood pressure in clinical trials so far. However, these postulated mechanisms may still have a role in the clinical phenotype given differences in PK and ADME have not been included when applying and interpreting the stoichiometric model. There also may exist concurrent and dominant effects due to nuclear receptor binding, as discussed previously.
Had the safety predictions for CETP inhibitors been made in advance of clinical studies, they might have brought attention to potential issues. The binding studies raise an unaddressed possibility for long-term effects of CETP inhibitors on blood pressure, inflammation and cancer. These results raise the concern that systems biology approaches may hinder the development of clinically safe and beneficial pharmaceuticals by falsely predicting side effects or yielding overly pessimistic predictions for efficacy. Therefore, just as reliable in vitro systems are used to verify predictions for compound effects on pathways are accurate, methods to verify whether predicted side effects could manifest clinically should be used. There are existing tools that may help, such as physiologically based pharmacokinetic (PBPK) methods to help predict the drug concentration in tissues [60]. Analysis of the interaction network of the off-target proteins may also yield insights into the potential for side effects [61-63]. Analyses may suggest additional, precautionary endpoints to monitor in phase 1 trials, hopefully avoiding failures in later stages.
Biomarkers
In the Critical Paths Initiative, the FDA has emphasized the importance of biomarkers in the drug development process and has established programs to foster the development of new biomarkers [64]. Biomarkers present the opportunity to identify patients at the greatest risk for rapid disease progression, stratify patients to select for those most likely to respond to an investigational therapy, and potentially may serve as surrogate endpoints for clinical trials. However, one must be very cautious when applying surrogate endpoints. The previous example of CETP inhibitors illustrates the danger. Although HDL-C is known to negatively correlate with the risk for atherosclerosis and cardiovascular events, thus far there is much debate regarding whether HDL-C directly modulated through CETP inhibition in the human will result in an improvement in plaque-related endpoints [65]. Also, as emphasized by Powell et al. [49], recent studies targeting cardiovascular risk factors did not result in an improvement in cardiovascular outcomes, despite the desired improvements in the targeted risk factors themselves [66-68].
These caveats aside, the mechanistic systems modeling approaches discussed here have served as important contributors to biomarker identification. For example, a novel biomarker predictive of bone erosion in rheumatoid arthritis was discovered using the model of rheumatoid arthritis discussed previously [69]. The marker, CXCL13, is a cytokine primarily involved as a chemotactic factor for B cells. Mechanistically, one initially would not suspect processes primarily mediated through macrophage-derived osteoclastic as well as osteoblastic functions would be predicted by CXCL13. The result, which was confirmed clinically, illustrates the ability of mechanistic modeling of pathophysiology of large systems to yield unexpected and practical results.
Using Recon 1, Shlomi et al. describe a computational method to develop predictions for markers of genetic metabolic diseases [70]. By cross-referencing the reactions in the model with the Online Mendelian Inheritance in Man (OMIM) database, 304 metabolic disorders were tested for biomarkers. By quantifying differences in the transport reaction flux into the cell, 176 diseases were predicted to yield at least one distinguishing analyte. A manual inspection of metabolic disorders in the OMIM database with a high-quality set of 17 disorders of amino acid metabolism suggested a precision of 0.76 and recall of 0.56. The result is good considering the number of assumptions that were made. Fluxes, not concentrations, of metabolites are used as proxies for the change in metabolite in body fluids. The entire human GEM was used without developing tissue-specific models or accounting for the exchange of metabolites across tissues through the serum. The lack of regulatory constraints could also lead to false predictions. Despite the simplifications, the method resulted in a roughly tenfold enhancement in biomarker detection performance, as compared with randomly generated biomarker sets. The approach therefore already has provided suitable biomarker suggestions.
Discussion
Moving beyond hypothesis testing, when there is sufficient confidence in the simulation predictions, mechanistic systems simulations can be used as much more than a tool to guide preclinical research. It has been reported that failing an unsuccessful compound early, in phase 1 rather than phase 3, may substantially reduce the costs of drug development. A roughly 10% improvement in the success rate of phase 3 trials, from better lead selection or failing bad compounds early, is predicted to result in an average cost reduction of greater than $200 million per approved therapeutic [71]. Reliable bad news should be greeted as actionable information, but other options must be available for an organization to respond in an optimal fashion to predictions of failure. Indeed, as pharmaceutical companies are highly incentivized to pursue high risk targets to minimize competition [72], data used to improve estimates of a probability of success measure have tangible benefits. The best scenario is one where the systems modeling results are informing decisions across therapeutic areas in an organization. The concept is illustrated in Fig. 3. In the unfortunate scenario that all of the current lead candidates in a therapeutic area lack potential, resources can be more fully redirected to the development of new leads, other therapeutic areas where there is a better potential for the portfolio, or to acquisitions.
FIGURE 3.
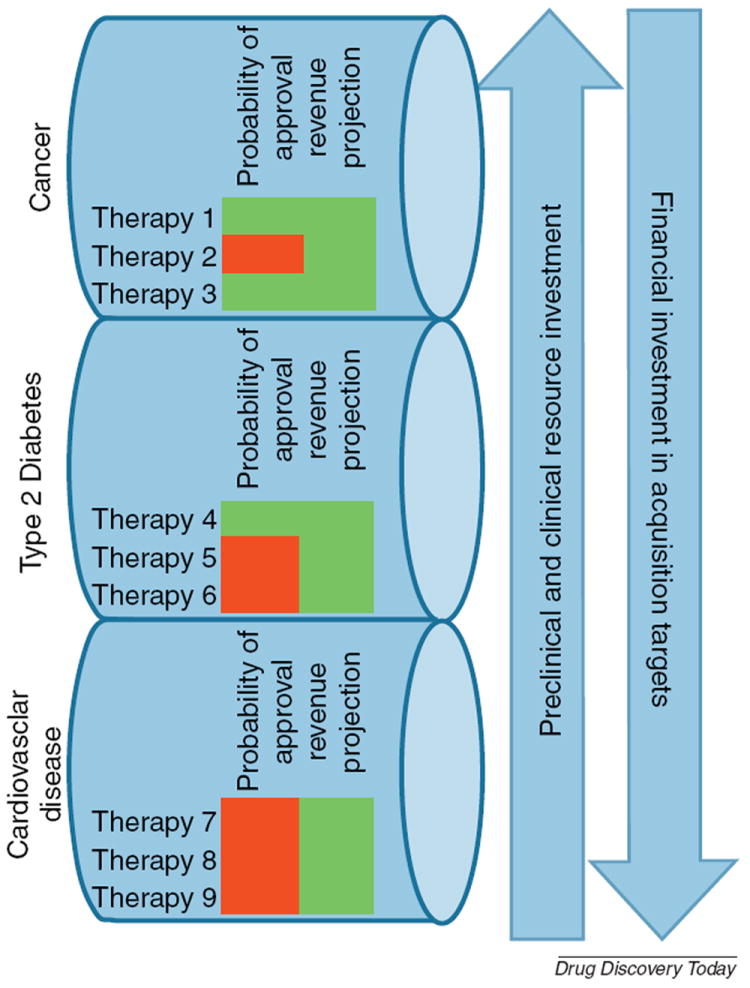
Portfolio and organization-level pipeline management decisions can be improved using systems modeling approaches. It is important to note that predictions derived from modeling yield increased or decreased confidence in the clinical success of a therapy, and therefore may be incorporated directly into corporate strategy. A hypothetical scenario is shown where a company is developing nine leads, all of which have been vetted for high potential returns, across three therapeutic areas. Modeling has improved confidence in the likelihood of success of two cancer targets, and dedicated research and development resources can be allocated as needed to maximize productivity through lower risk, high reward projects. In this scenario, the cardiovascular leads under development have not demonstrated great potential for translating through the pipeline through phase 3 successfully, and the company may want to explore acquisition options.
The modeling approaches reviewed here each offer unique advantages and compromises. For example, clinical phenotype-driven models enable the inclusion of complex interaction networks of varying scales. Intercellular and interorgan feedback mechanisms can be simulated. It is also straightforward to deduce the clinical phenotype from a simulation result because clinical outputs are used to guide model creation. However, it can be challenging to inform such models with the omics data sets that have been increasingly adopted by industry.
Computable network models enhance the ability to functionally interpret networks. For example, GEMs have elucidated fundamental theories of how cells may respond to alterations in dietary conditions, including the feasibility of gluconeogenesis [73] and epigenetic pathologies [74]. Metabolic models integrating the interaction of multiple, distinct cell types have recently been reported [33,36,42], and several therapeutic areas that will benefit from further efforts in this direction. Metabolic reconstructions may have a role in translating therapeutics into humans in early research: it is noteworthy that a model of murine metabolism has recently been published by refining Recon 1 in the context of murine-specific data [75]. There has also been recent work investigating therapeutic modulation of metabolic network state to a healthy phenotype [76-78]. Ultimately this work may further contribute to the development of drug targets in additional therapeutic areas, as opposed, for example, to therapeutic areas such as cancer or infectious disease, where the primary goal is to selectively disrupt metabolism. It has also been noted that our understanding of metabolism in simple bacteria such as Bacillus subtilus is incomplete [26,79], and it is very possible entirely new metabolic pathways in the human will be discovered as GEMs help to uncover knowledge gaps. Two knowledge areas that could use refinement in human GEMs are intracellular transport and lipid metabolism. Intercompartmental transport reactions may have important effects on the network [80], but have not been well described in the primary literature. Notably, a further discussion of applying Recon 1 to study pathophysiology has been published [81].
Systems modeling approaches may have an important role in the personalization of medicine. It has been established that mechanistic systems models can be used to identify biomarkers of responding subpopulations [82]. Although we have not discussed population-level measures in detail, systems modeling approaches already have demonstrated promise informing therapeutic decisions with statistical calibration of population clinical measures, reflecting underlying mechanistic heterogeneity [49,82]. Admittedly, many challenges remain for the acceptance of personalized medicine approaches [83]. It very possible insights and biomarkers to stratify patients will be discovered by merging omics data sets with the systems modeling techniques discussed here. For example, the NCI-60 lines used by Folger et al. represent a first approach at mining for drug targets in cancer using customized metabolic models [44]. Future efforts may explore how alterations in expression patterns, for example in different breast cancers, impact which targets will yield the best clinical results.
Concluding remarks
Systems modeling techniques have made contributions and exhibit additional promise across therapeutic areas of interest to pharmaceutical companies, including asthma, cancer, cardiovascular disease, diabetes and rheumatoid arthritis. Notably, systems models directly facilitate in silico target evaluation, help to address best-case and worst-case scenarios, help to identify and prioritize pre-clinical research, provide a method to interpret high-throughput data sets, can guide drug repositioning, and can guide the development of biomarkers. The variety of mechanistic systems modeling approaches available can be complementary in their application, as illustrated by the studies with torcetrapib. Systems models are integrative in their incorporation of mechanistic knowledge; likewise, they should be applied in an integrative manner across a pharmaceutical research pipeline. Then, they may help to alleviate the productivity crisis experienced by the pharmaceutical industry.
Systems modeling requires an appropriate business and research environment to be an effective strategy for an organization. In an internally competitive research environment, predicted therapeutic outcomes will probably bias the acceptance of the predictions and the ability of an organization to respond optimally. However, in a cooperative research culture, broadly applying system models across compounds in a therapeutic area will help to select those that exhibit the most promise of advancing the overall strength of the research program. Predictions giving an indication of clinical efficacy can help to inform how resources for preclinical and clinical research and development are prioritized.
Acknowledgments
Brian Schmidt would like to acknowledge discussion with former and present members of the Systems Biology Research Group at the University of California San Diego, including Roger Chang, Dr Nathan Lewis, Dr Monica Mo, Joshua Lerman, Dr Hojung Nam, Aarash Bordbar, Dr Pep Charusanti and Dr Daniel Hyduke, as well as Dr Jason Chan of Entelos. The authors wish to acknowledge funding from the National Institutes of Health (R01 GM088244 to J.P.) and Cystic Fibrosis Research Foundation (grant 1060 to J.P.).
Biographies

Dr Brian J. Schmidt completed his B.S. in Chemical Engineering at the University of Pittsburgh and his Ph.D. in Biomedical Engineering at the University of Virginia. He is currently a Postdoctoral Scholar in Dr Bernhard Ø. Palsson’s Systems Biology Research Group at the University of California, San Diego. His research interests include the integration of omics data sets with genome-scale models of metabolism as well as the application of systems modeling and analysis methods to develop novel insights into infection and disease. Previously, at Entelos, he developed and applied biosimulation platforms to formulate predictions of disease progression and pharmaceutical efficacy in a variety of therapeutic areas, especially rheumatoid arthritis.

Dr Jason A. Papin is an Associate Professor of Biomedical Engineering at the University of Virginia. He completed his B.S., M.S. and Ph.D. in Bioengineering at the University of California, San Diego. He has extensive experience with the development and application of systems biology methods. His research interests include the development of new network reconstructions as well as the incorporation of high-throughput data with integrated signaling, metabolic and regulatory network reconstructions. He employs these tools to study fundamental problems in infectious disease, cancer and bioenergy.

Dr Cynthia J. Musante is a Senior Principal Research Scientist at Pfizer with over 12 years of experience applying modeling and simulation approaches to help predict the efficacy of lead compounds in pharmaceutical research and development and design clinical trials. Her current research focuses on the treatment of cardiovascular, metabolic and endocrine diseases. She completed her B.A. in Mathematics at Westfield State College and her Ph.D. in Applied Mathematics at North Carolina State University.
References
- 1.Kubinyi H. Success stories of computer-aided design. In: Ekins S, editor. Computer Applications in Pharmaceutical Research and Development. John Wiley & Sons, Inc; 2006. [Google Scholar]
- 2.Fernald GH, et al. Bioinformatics challenges for personalized medicine. Bioinformatics. 2011;27:1741–1748. doi: 10.1093/bioinformatics/btr295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khleif SN, et al. AACR-FDA-NCI Cancer Biomarkers Collaborative consensus report: advancing the use of biomarkers in cancer drug development. Clin Cancer Res. 2010;16:3299–3318. doi: 10.1158/1078-0432.CCR-10-0880. [DOI] [PubMed] [Google Scholar]
- 4.Oberhardt MA, et al. Applications of genome-scale metabolic reconstructions. Mol Syst Biol. 2009;5:320. doi: 10.1038/msb.2009.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michelson S, et al. Target identification and validation using human simulation models. In: León D, Markel S, editors. In Silico Technologies in Drug Target Identification and Validation. Vol. 6. CRC Press/Taylor & Francis Group; 2006. [Google Scholar]
- 6.Musante CJ, et al. Small- and large-scale biosimulation applied to drug discovery and development. Drug Discov Today. 2002;7(20 Suppl):S192–S196. doi: 10.1016/s1359-6446(02)02442-x. [DOI] [PubMed] [Google Scholar]
- 7.Aslam S, et al. Biosimulation: advancements in the pathway of drug discovery and development. IJPSR. 2010;3:99–105. [Google Scholar]
- 8.Bergman RN, et al. Physiologic evaluation of factors controlling glucose tolerance in man: measurement of insulin sensitivity and beta-cell glucose sensitivity from the response to intravenous glucose. J Clin Invest. 1981;68:1456–1467. doi: 10.1172/JCI110398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergman RN, et al. Quantitative estimation of insulin sensitivity. Am J Physiol. 1979;236:E667–E677. doi: 10.1152/ajpendo.1979.236.6.E667. [DOI] [PubMed] [Google Scholar]
- 10.Bergman RN, et al. The evolution of beta-cell dysfunction and insulin resistance in type 2 diabetes. Eur J Clin Invest. 2002;32(Suppl. 3):35–45. doi: 10.1046/j.1365-2362.32.s3.5.x. [DOI] [PubMed] [Google Scholar]
- 11.D’Alessio DA, et al. Enteral enhancement of glucose disposition by both insulin-dependent and insulin-independent processes. A physiological role of glucagon-like peptide I. Diabetes. 1995;44:1433–1437. doi: 10.2337/diab.44.12.1433. [DOI] [PubMed] [Google Scholar]
- 12.ADA. Guidelines for computer modeling of diabetes and its complications. Diabetes Care. 2004;27:2262–2265. doi: 10.2337/diacare.27.9.2262. [DOI] [PubMed] [Google Scholar]
- 13.Shoda L, et al. The Type 1 Diabetes PhysioLab Platform: a validated physiologically based mathematical model of pathogenesis in the non-obese diabetic mouse. Clin Exp Immunol. 2010;161:250–267. doi: 10.1111/j.1365-2249.2010.04166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michelson S, et al. In silico prediction of clinical efficacy. Curr Opin Biotechnol. 2006;17:666–670. doi: 10.1016/j.copbio.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 15.Gutenkunst RN, et al. Universally sloppy parameter sensitivities in systems biology models. PLoS Comput Biol. 2007;3:1871–1878. doi: 10.1371/journal.pcbi.0030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DiMasi JA, et al. The price of innovation: new estimates of drug development costs. J Health Econ. 2003;22:151–185. doi: 10.1016/S0167-6296(02)00126-1. [DOI] [PubMed] [Google Scholar]
- 17.Adams CP, Brantner VV. Estimating the cost of new drug development: is it really 802 million dollars? Health Aff (Millwood) 2006;25:420–428. doi: 10.1377/hlthaff.25.2.420. [DOI] [PubMed] [Google Scholar]
- 18.Dimasi JA. New drug development in the United States from 1963 to 1999. Clin Pharmacol Ther. 2001;69:286–296. doi: 10.1067/mcp.2001.115132. [DOI] [PubMed] [Google Scholar]
- 19.Friedrich C, et al. Comparison of NLME and mechanistic physiological modeling methods using examples in drug discovery and development. American Conference on Pharmacometrics; San Diego, USA. April 3–10.2011. [Google Scholar]
- 20.Friedrich C, et al. Population Approach Group in Europe. Athens, Greece: 2011. Jun 7–10, Comparison of statistical and physiological modeling methods using examples in drug discovery and development. [Google Scholar]
- 21.Gallen CC. Strategic challenges in neurotherapeutic pharmaceutical development. NeuroRx. 2004;1:165–180. doi: 10.1602/neurorx.1.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rullmann JA, et al. Systems biology for battling rheumatoid arthritis: application of the Entelos PhysioLab platform. IEEE Proc Syst Biol. 2005;152:256–262. doi: 10.1049/ip-syb:20050053. [DOI] [PubMed] [Google Scholar]
- 23.Duarte NC, et al. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci U S A. 2007;104:1777–1782. doi: 10.1073/pnas.0610772104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kell DB. Systems biology, metabolic modelling and metabolomics in drug discovery and development. Drug Discov Today. 2006;11:1085–1092. doi: 10.1016/j.drudis.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Reed JL, et al. Towards multidimensional genome annotation. Nat Rev Genet. 2006;7:130–141. doi: 10.1038/nrg1769. [DOI] [PubMed] [Google Scholar]
- 26.Durot M, et al. Genome-scale models of bacterial metabolism: reconstruction and applications. FEMS Microbiol Rev. 2009;33:164–190. doi: 10.1111/j.1574-6976.2008.00146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feist AM, et al. Reconstruction of biochemical networks in microorganisms. Nat Rev Microbiol. 2009;7:129–143. doi: 10.1038/nrmicro1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raman K, Chandra N. Flux balance analysis of biological systems: applications and challenges. Brief Bioinform. 2009;10:435–449. doi: 10.1093/bib/bbp011. [DOI] [PubMed] [Google Scholar]
- 29.Jamshidi N, Palsson BO. Mass action stoichiometric simulation models: incorporating kinetics and regulation into stoichiometric models. Biophys J. 2010;98:175–185. doi: 10.1016/j.bpj.2009.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schellenberger J, et al. BiGG: a Biochemical Genetic and Genomic knowledgebase of large scale metabolic reconstructions. BMC Bioinformatics. 2010;11:213. doi: 10.1186/1471-2105-11-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mo ML, et al. A genome-scale, constraint-based approach to systems biology of human metabolism. Mol Biosyst. 2007;3:598–603. doi: 10.1039/b705597h. [DOI] [PubMed] [Google Scholar]
- 32.Shlomi T, et al. Network-based prediction of human tissue-specific metabolism. Nat Biotechnol. 2008;26:1003–1010. doi: 10.1038/nbt.1487. [DOI] [PubMed] [Google Scholar]
- 33.Bordbar A, et al. Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions. Mol Syst Biol. 2010;6:422. doi: 10.1038/msb.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bordbar A, et al. Model-driven multi-omic data analysis elucidates metabolic immunomodulators of macrophage activation. Mol Syst Biol. 2012;8:558. doi: 10.1038/msb.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang RL, et al. Drug off-target effects predicted using structural analysis in the context of a metabolic network model. PLoS Comput Biol. 2010;6:E1000938. doi: 10.1371/journal.pcbi.1000938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis NE, et al. Large-scale in silico modeling of metabolic interactions between cell types in the human brain. Nat Biotechnol. 2010;28:1279–1285. doi: 10.1038/nbt.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jerby L, et al. Computational reconstruction of tissue-specific metabolic models: application to human liver metabolism. Mol Syst Biol. 2010;6:401. doi: 10.1038/msb.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bordbar A, et al. iAB-RBC-283: a proteomically derived knowledge-base of erythrocyte metabolism that can be used to simulate its physiological and pathophysiological states. BMC Syst Biol. 2011;5:110. doi: 10.1186/1752-0509-5-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma H, et al. The Edinburgh human metabolic network reconstruction and its functional analysis. Mol Syst Biol. 2007;3:135. doi: 10.1038/msb4100177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gille C, et al. HepatoNet1: a comprehensive metabolic reconstruction of the human hepatocyte for the analysis of liver physiology. Mol Syst Biol. 2010;6:411. doi: 10.1038/msb.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Agren R, et al. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput Biol. 2012;8:E1002518. doi: 10.1371/journal.pcbi.1002518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bordbar A, et al. A multi-tissue type genome-scale metabolic network for analysis of whole-body systems physiology. BMC Syst Biol. 2011;5:180. doi: 10.1186/1752-0509-5-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edelman LB, et al. In silico models of cancer. Wiley Interdiscip Rev Syst Biol Med. 2010;2:438–459. doi: 10.1002/wsbm.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Folger O, et al. Predicting selective drug targets in cancer through metabolic networks. Mol Syst Biol. 2011;7:517. doi: 10.1038/msb.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 2001;292:1160–1164. doi: 10.1126/science.1059344. [DOI] [PubMed] [Google Scholar]
- 46.LaRosa JC, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med. 2005;352:1425–1435. doi: 10.1056/NEJMoa050461. [DOI] [PubMed] [Google Scholar]
- 47.Tall AR, et al. The failure of torcetrapib: was it the molecule or the mechanism? Arterioscler Thromb Vasc Biol. 2007;27:257–260. doi: 10.1161/01.ATV.0000256728.60226.77. [DOI] [PubMed] [Google Scholar]
- 48.Barter PJ, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 49.Powell LM, et al. Application of predictive biosimulation to the study of atherosclerosis: development of the Cardiovascular PhysioLab platform and evaluation of CETP inhibitor therapy. FOSBE 2007. 2007:295–302. [Google Scholar]
- 50.Wahba K, et al. Clinical trial simulations of dyslipidemic patients in a mechanistic model of cardiovascular disease predict little impact on CHD events by CETP inhibitors. AHA Scientific Sessions 2011 [Google Scholar]
- 51.Schwartz GG, et al. Rationale and design of the dal-OUTCOMES trial: efficacy and safety of dalcetrapib in patients with recent acute coronary syndrome. Am Heart J. 2009;158:896–901.e3. doi: 10.1016/j.ahj.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 52.Cannon CP, et al. Design of the DEFINE trial: determining the EFficacy and tolerability of CETP INhibition with AnacEtrapib. Am Heart J. 2009;158:513–519.e3. doi: 10.1016/j.ahj.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 53.Xie L, et al. Drug discovery using chemical systems biology: identification of the protein–ligand binding network to explain the side effects of CETP inhibitors. PLoS Comput Biol. 2009;5:E1000387. doi: 10.1371/journal.pcbi.1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stein EA, et al. Safety and tolerability of dalcetrapib (RO4607381/JTT-705): results from a 48-week trial. Eur Heart J. 2010;31:480–488. doi: 10.1093/eurheartj/ehp601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stein EA, et al. Safety and tolerability of dalcetrapib. Am J Cardiol. 2009;104:82–91. doi: 10.1016/j.amjcard.2009.02.061. [DOI] [PubMed] [Google Scholar]
- 56.Robinson JG. Dalcetrapib: a review of Phase II data. Expert Opin Investig Drugs. 2010;19:795–805. doi: 10.1517/13543784.2010.488219. [DOI] [PubMed] [Google Scholar]
- 57.Krishna R, et al. Effect of the cholesteryl ester transfer protein inhibitor, anacetrapib, on lipoproteins in patients with dyslipidaemia and on 24-h ambulatory blood pressure in healthy individuals: two double-blind, randomised placebo-controlled phase I studies. Lancet. 2007;370:1907–1914. doi: 10.1016/S0140-6736(07)61813-3. [DOI] [PubMed] [Google Scholar]
- 58.Bloomfield D, et al. Efficacy and safety of the cholesteryl ester transfer protein inhibitor anacetrapib as monotherapy and coadministered with atorvastatin in dyslipidemic patients. Am Heart J. 2009;157:352–360.e2. doi: 10.1016/j.ahj.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 59.Cannon CP, et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363:2406–2415. doi: 10.1056/NEJMoa1009744. [DOI] [PubMed] [Google Scholar]
- 60.Poulin P, Theil FP. Prediction of pharmacokinetics prior to in vivo studies. II. Generic physiologically based pharmacokinetic models of drug disposition. J Pharm Sci. 2002;91:1358–1370. doi: 10.1002/jps.10128. [DOI] [PubMed] [Google Scholar]
- 61.Hwang WC, et al. Identification of information flow-modulating drug targets: a novel bridging paradigm for drug discovery. Clin Pharmacol Ther. 2008;84:563–572. doi: 10.1038/clpt.2008.129. [DOI] [PubMed] [Google Scholar]
- 62.Berger SI, Iyengar R. Network analyses in systems pharmacology. Bioinformatics. 2009;25:2466–2472. doi: 10.1093/bioinformatics/btp465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berger SI, Iyengar R. Role of systems pharmacology in understanding drug adverse events. Wiley Interdiscip Rev Syst Biol Med. 2011;3:129–135. doi: 10.1002/wsbm.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mahajan R, Gupta K. Food and drug administration’s critical path initiative and innovations in drug development paradigm: challenges, progress, and controversies. J Pharm Bioallied Sci. 2010;2:307–313. doi: 10.4103/0975-7406.72130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.von Eckardstein A. Mulling over the odds of CETP inhibition. Eur Heart J. 2010;31:390–393. doi: 10.1093/eurheartj/ehp394. [DOI] [PubMed] [Google Scholar]
- 66.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 67.Gulseth MP. Ximelagatran: an orally active direct thrombin inhibitor. Am J Health Syst Pharm. 2005;62:1451–1467. doi: 10.2146/ajhp040534. [DOI] [PubMed] [Google Scholar]
- 68.Vickers MR, et al. Main morbidities recorded in the women’s international study of long duration oestrogen after menopause (WISDOM): a randomised controlled trial of hormone replacement therapy in postmenopausal women. BMJ. 2007;335:239. doi: 10.1136/bmj.39266.425069.AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meeuwisse CM, et al. Identification of CXCL13 as a marker for rheumatoid arthritis outcome using an in silico model of the rheumatic joint. Arthritis Rheum. 2011;63:1265–1273. doi: 10.1002/art.30273. [DOI] [PubMed] [Google Scholar]
- 70.Shlomi T, et al. Predicting metabolic biomarkers of human inborn errors of metabolism. Mol Syst Biol. 2009;5:263. doi: 10.1038/msb.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.DiMasi JA. The value of improving the productivity of the drug development process: faster times and better decisions. Pharmacoeconomics. 2002;20(Suppl. 3):1–10. doi: 10.2165/00019053-200220003-00001. [DOI] [PubMed] [Google Scholar]
- 72.Pammolli F, et al. The productivity crisis in pharmaceutical R&D. Nat Rev Drug Discov. 2011;10:428–438. doi: 10.1038/nrd3405. [DOI] [PubMed] [Google Scholar]
- 73.Kaleta C, et al. In silico evidence for gluconeogenesis from fatty acids in humans. PLoS Comput Biol. 2011;7:E1002116. doi: 10.1371/journal.pcbi.1002116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sigurdsson MI, et al. Genome-scale network analysis of imprinted human metabolic genes. Epigenetics. 2009;4:43–46. doi: 10.4161/epi.4.1.7603. [DOI] [PubMed] [Google Scholar]
- 75.Sigurdsson MI, et al. A detailed genome-wide reconstruction of mouse metabolism based on human Recon 1. BMC Syst Biol. 2010;4:140. doi: 10.1186/1752-0509-4-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Motter AE. Improved network performance via antagonism: from synthetic rescues to multi-drug combinations. Bioessays. 2010;32:236–245. doi: 10.1002/bies.200900128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li Z, et al. Two-stage flux balance analysis of metabolic networks for drug target identification. BMC Syst Biol. 2011;5(Suppl. 1):S11. doi: 10.1186/1752-0509-5-S1-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li Z, et al. Drug target identification based on flux balance analysis of metabolic networks. International Conference on Computational Systems Biology. 2010:331–338. [Google Scholar]
- 79.Oh YK, et al. Genome-scale reconstruction of metabolic network in Bacillus subtilis based on high-throughput phenotyping and gene essentiality data. J Biol Chem. 2007;282:28791–28799. doi: 10.1074/jbc.M703759200. [DOI] [PubMed] [Google Scholar]
- 80.Mintz-Oron S, et al. Network-based prediction of metabolic enzymes’ subcellular localization. Bioinformatics. 2009;25:i247–i252. doi: 10.1093/bioinformatics/btp209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bordbar A, Palsson BO. Using the reconstructed genome-scale human metabolic network to study physiology and pathology. J Intern Med. 2012;271:131–141. doi: 10.1111/j.1365-2796.2011.02494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bangs A. Predictive biosimulation and virtual patients in pharmaceutical R and D. Stud Health Technol Inform. 2005;111:37–42. [PubMed] [Google Scholar]
- 83.Gonzalez-Angulo AM, et al. Future of personalized medicine in oncology: a systems biology approach. J Clin Oncol. 2010;28:2777–2783. doi: 10.1200/JCO.2009.27.0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moore K, et al. Use of a large biological model with single-dose clinical data to predict hemoglobin (Hb) response in patients with anemia. American Conference on Pharmacometrics; San Diego, USA. April 3–6.2011. [Google Scholar]
- 85.Stokes C. Biological systems modeling: powerful discipline for biomedical e-R&D. AIChE J. 2000;46:430–433. [Google Scholar]
- 86.Stokes CL, et al. A computer model of chronic asthma with application to clinical studies: example of treatment of exercise-induced asthma. J Allergy Clin Immunol. 2001;107:A933. [Google Scholar]
- 87.Lewis AK, et al. The role of beta2-adrenergic receptor polymorphisms in clinical outcomes following chronic beta2-agonist use. Am J Respir Crit Care Med. 2001;163:A143. [Google Scholar]
- 88.Li L, et al. Predicting enzyme targets for cancer drugs by profiling human metabolic reactions in NCI-60 cell lines. BMC Bioinformatics. 2010;11:501. doi: 10.1186/1471-2105-11-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Frezza C, et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature. 2011;477:225–230. doi: 10.1038/nature10363. [DOI] [PubMed] [Google Scholar]
- 90.Shlomi T, et al. Genome-scale metabolic modeling elucidates the role of proliferative adaptation in causing the Warburg effect. PLoS Comput Biol. 2011;7:E1002018. doi: 10.1371/journal.pcbi.1002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ambert N, et al. Computational studies of NMDA receptors: differential effects of neuronal activity on efficacy of competitive and non-competitive antagonists. Open Access Bioinformatics. 2010;2:113–125. doi: 10.2147/OAB.S7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Waters SB, et al. Treatment with sitagliptin or metformin does not increase body weight despite predicted reductions in urinary glucose excretion. J Diabetes Sci Technol. 2009;3:68–82. doi: 10.1177/193229680900300108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kansal AR, Trimmer J. Application of predictive biosimulation within pharmaceutical clinical development: examples of significance for translational medicine and clinical trial design. IEEE Proc Syst Biol. 2005;152:214–220. doi: 10.1049/ip-syb:20050043. [DOI] [PubMed] [Google Scholar]
- 94.Timmer J, et al. Systems biology of mammalian cells: a report from the Freiburg conference. Bioessays. 2010;32:1099–1104. doi: 10.1002/bies.201000109. [DOI] [PubMed] [Google Scholar]
- 95.Vo TD, et al. Systems analysis of energy metabolism elucidates the affected respiratory chain complex in Leigh’s syndrome. Mol Genet Metab. 2007;91:15–22. doi: 10.1016/j.ymgme.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 96.Kim HU, et al. Genome-scale metabolic network analysis and drug targeting of multi-drug resistant pathogen Acinetobacter baumannii AYE. Mol Biosyst. 2010;6:339–348. doi: 10.1039/b916446d. [DOI] [PubMed] [Google Scholar]
- 97.Jamshidi N, Palsson BO. Investigating the metabolic capabilities of Mycobacterium tuberculosis H37Rv using the in silico strain iNJ661 and proposing alternative drug targets. BMC Syst Biol. 2007;1:26. doi: 10.1186/1752-0509-1-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Beste DJ, et al. GSMN-TB: a web-based genome-scale network model of Mycobacterium tuberculosis metabolism. Genome Biol. 2007;8:R89. doi: 10.1186/gb-2007-8-5-r89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fang X, et al. A systems biology framework for modeling metabolic enzyme inhibition of Mycobacterium tuberculosis. BMC Syst Biol. 2009;3:92. doi: 10.1186/1752-0509-3-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fang X, et al. Modeling synergistic drug inhibition of Mycobacterium tuberculosis growth in murine macrophages. Mol Biosyst. 2011;7:2622–2636. doi: 10.1039/c1mb05106g. [DOI] [PubMed] [Google Scholar]
- 101.Fang K, et al. Exploring the metabolic network of the epidemic pathogen Burkholderia cenocepacia J2315 via genome-scale reconstruction. BMC Syst Biol. 2011;5:83. doi: 10.1186/1752-0509-5-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Raghunathan A, et al. Systems approach to investigating host–pathogen interactions in infections with the biothreat agent Francisella. Constraints-based model of Francisella tularensis. BMC Syst Biol. 2010;4:118. doi: 10.1186/1752-0509-4-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chavali AK, et al. Systems analysis of metabolism in the pathogenic trypanosomatid Leishmania major. Mol Syst Biol. 2008;4:177. doi: 10.1038/msb.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chavali AK, et al. Metabolic network analysis predicts efficacy of FDA-approved drugs targeting the causative agent of a neglected tropical disease. BMC Syst Biol. 2012;6:27. doi: 10.1186/1752-0509-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kim HU, et al. Integrative genome-scale metabolic analysis of Vibrio vulnificus for drug targeting and discovery. Mol Syst Biol. 2011;7:460. doi: 10.1038/msb.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Oberhardt MA, et al. Genome-scale metabolic network analysis of the opportunistic pathogen Pseudomonas aeruginosa PAO1. J Bacteriol. 2008;190:2790–2803. doi: 10.1128/JB.01583-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee DS, et al. Comparative genome-scale metabolic reconstruction and flux balance analysis of multiple Staphylococcus aureus genomes identify novel antimicrobial drug targets. J Bacteriol. 2009;191:4015–4024. doi: 10.1128/JB.01743-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Raghunathan A, et al. Constraint-based analysis of metabolic capacity of Salmonella typhimurium during host–pathogen interaction. BMC Syst Biol. 2009;3:38. doi: 10.1186/1752-0509-3-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lo A, et al. Using a systems biology approach to explore hypotheses underlying clinical diversity of the renin angiotensin system and the response to antihypertensive therapies. In: Kimko H, Peck C, editors. Clinical Trial Simulations: Applications and Trends. Vol. 1. Springer; 2011. pp. 457–482. [Google Scholar]
- 110.Hallow K, et al. A systems modeling approach to understanding the mechanisms of renal protection observed in the avoid study: 5C.02. 20th European Meeting on Hypertension; 2010. p. E225. [Google Scholar]
- 111.Defranoux NA, et al. In silico modeling and simulation of bone biology: a proposal. J Bone Miner Res. 2005;20:1079–1084. doi: 10.1359/JBMR.050401. [DOI] [PubMed] [Google Scholar]
- 112.Hansen RN, et al. EULAR Annual European Congress of Rheumatology. London, UK: 2011. May 25–28, In silico target evaluation of NKG2D for treatment of rheumatoid arthritis. [Google Scholar]
- 113.Chang S, et al. Mathematical model predicting outcomes of sepsis patients treated with xigris: enhance trial. Shock. 2006;25(S16):70–71. [Google Scholar]
- 114.Sarkar J, et al. Mathematical modeling of community-acquired pneumonia patients. Crit Care. 2009;13(S4):P49. [Google Scholar]
- 115.Marathe D, et al. Modeling of severe sepsis patients with community acquired pneumonia. Shock. 2010;33(S1):72–73. [Google Scholar]
- 116.Keating SM, et al. SBMLToolbox: an SBML toolbox for MATLAB users. Bioinformatics. 2006;22:1275–1277. doi: 10.1093/bioinformatics/btl111. [DOI] [PubMed] [Google Scholar]
- 117.Schellenberger J, et al. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat Protoc. 2011;6:1290–1307. doi: 10.1038/nprot.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ho R, et al. Chronic inflammation in asthma airway remodeling. 15th International Conference of the Inflammation Research Association; Chantilly, USA. September 21–24.2008. [Google Scholar]
- 119.Han TH, et al. Alteration of glucose and insulin regulatory networks in Type 2 diabetes mellitus. 17th Annual Meeting of the Population Approach Group in Europe; Marseille, France. June 18–20.2008. [Google Scholar]
- 120.Baillie R, et al. Cambridge Health Institute Discovery on Target. Boston, USA: 2010. Nov 2–4, Modeling glucose metabolism in diabetes. [Google Scholar]
- 121.Ghosh A, et al. Cambridge Health Institute Discovery on Target. Boston, USA: 2010. Nov 2–4, A systems approach to accelerating the pharmaceutical industry pipeline: competitive preclinical and clinical modeling in diabetes drug development. [Google Scholar]
- 122.Tess D, et al. Impact of modeling on GPR119 agonist development. American Conference on Pharmacometrics; San Diego, USA. April 3–6.2011. [Google Scholar]
- 123.Tess D, Baillie R. Biorbis World PK/PD Summit. Boston, USA: 2011. Apr 26–29, Creating and using a physiological model to support development of a GPR119 agonist diabetes therapy. [Google Scholar]
- 124.Akesson M, et al. Integration of gene expression data into genome-scale metabolic models. Metab Eng. 2004;6:285–293. doi: 10.1016/j.ymben.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 125.Zur H, et al. iMAT: an integrative metabolic analysis tool. Bioinformatics. 2010;26:3140–3142. doi: 10.1093/bioinformatics/btq602. [DOI] [PubMed] [Google Scholar]
- 126.Becker SA, Palsson BO. Context-specific metabolic networks are consistent with experiments. PLoS Comput Biol. 2008;4:E1000082. doi: 10.1371/journal.pcbi.1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Colijn C, et al. Interpreting expression data with metabolic flux models: predicting Mycobacterium tuberculosis mycolic acid production. PLoS Comput Biol. 2009;5:E1000489. doi: 10.1371/journal.pcbi.1000489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yizhak K, et al. Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics. 2010;26:i255–i260. doi: 10.1093/bioinformatics/btq183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.van Berlo RJP, et al. Predicting metabolic fluxes using gene expression differences as constraints. IEEE/ACM Trans Comput Biol Bioinform. 2011;8:206–216. doi: 10.1109/TCBB.2009.55. [DOI] [PubMed] [Google Scholar]
- 130.Jensen PA, Papin JA. Functional integration of a metabolic network model and expression data without arbitrary thresholding. Bioinformatics. 2011;27:541–547. doi: 10.1093/bioinformatics/btq702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jensen PA, et al. TIGER: toolbox for integrating genome-scale metabolic models, expression data, and transcriptional regulatory networks. BMC Syst Biol. 2011;5:147. doi: 10.1186/1752-0509-5-147. [DOI] [PMC free article] [PubMed] [Google Scholar]