

Abstract
Dementia disorders are characterized by clinicopathological criteria. Molecular understandings of these disorders, based on immunohistochemical studies, biochemical investigations, genetic approaches, and animal models have resulted in advances in diagnosis. Likewise translational research has allowed application of increasing basic scientific knowledge regarding neurodegeneration, to the rational development of new investigational therapies based on current understanding of disease pathogenesis. This review discusses application of translational research to both diagnosis and treatment of dementia disorders. The development of biomarkers has yielded imaging and biochemical methods that more assist in the diagnosis of neurodegenerative dementias, especially Alzheimer’s disease. New diagnostic criteria for disease are based on these molecular-based techniques. And these biomarkers are of potential use in monitoring disease activity during therapeutic trials. Translational investigations likewise have led towards new avenues in targeted dementia research. This is particularly so in the development and testing of disease-modifying treatments that might slow or deter progressive deterioration. Recent clinical trials have not been based on empiric trial of established drugs, but rather upon trial of drugs shown through culture and animal models to interfere with known elements of the pathogenetic cascade of Alzheimer disease.
Dementia is defined as a disorder manifest by loss of mental capacity affecting a person’s ability to function. Dementia affects over 6 million Americans today, most of whom are elderly. Dementing disorders were at one time viewed as of psychiatric origin in the younger population and of “senile” derivation - a consequence of aging - in the elderly. There was little hope in treating either of these. Translational research has emerged as a dominant driving force in both diagnostic and therapeutic advances in dementia. Multidisciplinary research efforts allow basic knowledge whether obtained in the laboratory through in vitro investigation, experimental animal science, or neuropathological studies to be applied to development of diagnostic techniques or disease-directed specific therapies. Translational research is bidirectional, with clinical information back-applied to basic studies, allowing further development of forward-applied methods.
Categorization of the dementias as clinicopathological disorders started in earnest in the early 20th century, with modern neuropathology, in conjunction with careful clinical studies. Neurodegenerative diseases include Alzheimer disease (AD), Lewy Body Dementia (LBD), Frontotemporal Dementia (FTD), Corticobasal Degeneration (CBD), Progressive Supranuclear Palsy (PSP), Vascular Dementia, Huntington Disease, and Creutzfeldt-Jakob disease. Clinical neuropathology, with classical staining techniques, did provide the founding basis for identifying these disorders. But increasing molecular understanding has sharpened the differentiation of these diseases. Use of immunochemical assays has allowed categorization of dementias by molecular typology (Table 1). This has been supplemented by identification of genes leading to specific dysfunctions. Alzheimer disease is most uniquely characterized biochemically by the accumulation of Aβ in plaques and vessels, with concomitant intracellular accretions of tau. Lewy Body Dementia shares with Parkinson’s disease the accumulation of abnormal alpha-synuclein aggregates. About half of frontotemporal dementia cases, together with PSP and CBD, may be grouped as disorders marked by deposits of abnormal tau. Other degenerative dementias are characterized by abnormalities of TDP-43, FUS, huntingtin, prion protein, and others (see Table 1). The identification of specific cellular proteins prominently involved in these neurodegenerative disorders has led to in vitro and rodent models probing disease pathways, and the promise of directed, rather than empiric, therapeutic trials.
Table 1.
Molecular Classification of Dementing Disorders.
| Clinical Disorder | Protein | Term | Genes |
|---|---|---|---|
| Alzheimer Disease | β-amyloid | β-amyloidopathy | PSEN1, PSEN2, APP, APOE, others |
| Progressive Supranuclear Palsy | Tau | Tauopathy | MAPT |
| Corticobasal Degeneration | Tau | Tauopathy | MAPT |
| Frontotemporal Dementia | Tau | Tauopathy | MAPT |
| Frontotemporal Dementia | TDP-43 | TDP-43-opathy | TARDP, PGN |
| Lewy Body Dementia | α-synuclein | α-synucleinopathy | GBA |
| Parkinson Disease Dementia | α-synuclein | α-synucleinopathy | SNCA, LRRK2, GBA |
| Creutzfeldt-Jakob Disease | prion protein | Prionopathy | PRNP |
| Huntington Disease | huntingtn | HTT |
Neurodegenerative dementing disorders are characterized by specific dominant abnormalities in particular proteins, with characteristic deposition of abnormal protein in neurons, glia, or brain extracellular space. In some disease there are co-pathologies. Alzheimer’s disease is marked by abnormal deposits of both β-amyloid and tau. Various genes have been shown to be involved in these disorders, either through mutations with autosomal dominant or recessive inheritance, or through risk factors of polymorphisms or mutations.
The dementing disorders of adults are age-dependent, with generally increasing incidence in later decades of life. The three most frequent pathologies are Alzheimer, Lewy Body, and Vascular disease abnormalities. Two or more of these pathologies commonly coexist in the same individual.1, 2 It is unclear whether this is simply coincidental, evidence of a shared diathesis, or indicative of interaction between the pathoetiologic cascades of these diseases. In addition to those millions of Americans with dementia, there are larger numbers of individuals who have cognitive impairment of insufficient severity, extent, or functional consequence, to meet criteria for dementia. This state is most often labeled “mild cognitive impairment” (MCI). Most of these persons are actually experiencing the earliest stages of one of the dementia disorders mentioned above, most commonly Alzheimer disease. There are also those persons who have no symptoms at all, but may be at risk of developing MCI, and dementia. These asymptomatic persons have been termed “pre-MCI”, and would only be identifiable by biomarkers suggesting that even without any symptoms, they do have the earliest pathological molecular, cellular, or radiological changes suggesting the beginning stages of a dementing disorder that has not become symptomatic. Recent consensus workgroups sponsored jointly by the National Institute on Aging of the US National Institutes of Health and the Alzheimer’s Association, have utilized the increasing body of knowledge on molecular disease markers to formulate important new criteria, not only for Alzheimer disease,3 but also for MCI,4 and for presymptomatic or preclinical disease.5 These criteria should allow for better definition of individuals who might be in the MCI or pre-clinical phases of dementia. The use of amyloid imaging to ascertain amyloid deposition in living persons, and the use of cerebrospinal examination to ascertain abnormalities in amyloid, tau, and phosphorylated tau are key techniques in the process. It is possible that the earlier identification of disease during prodromal periods may allow more effective therapeutic intervention, because the disease process could be prevented, interrupted earlier, or attacked before the onset of more irreversible changes such as neuronal cell loss. This review will principally focus on Alzheimer disease dementia, and its precursor stage MCI, since AD is responsible for more than 80% of cases of dementia in the elderly.
Major advances in dementia over the past decade have transpired involving both diagnostic and therapeutic areas. These spring from increasingly diverse biochemical and microscopic techniques, genetic analyses, and ensuing cell culture, mice, or other model systems. Diagnostic accuracy has increased through translation of knowledge of molecular pathogenesis to development of biomarkers and genetic markers assisting in diagnosis, even prior to symptoms. Therapeutic trials have exploded, because the elucidation of the biochemical pathways, and ability to test interventions in model systems, has led to rationale drug trials to affect accumulating disease burden, rather than nonspecific or more arbitrarily chosen empiric trials of established substances. Here we will discuss firstly translational advances in diagnosis, then secondly, therapeutics.
Translational Research in Diagnosis of AD
Translational advances have spearheaded diagnostic understanding of dementia for the past century. It was the pioneering correlative clinical-pathological investigations of Alois Alzheimer, and Frederick Lewy, among others, who provided the basis of the definitions of the main degenerative dementias, based on their clinical symptoms and neuropathological findings. Since neuropathological descriptions typically are made only at autopsy, after death, diagnosis of dementia during life has traditionally been a clinical effort. Neurological and neuropsychological testing provide measures of an individual’s cognitive performance which can be judged normatively. However, the large variability in human performances, and the susceptibility of testing to cultural, educational, and achievement factors, have resulted in limitations of such testing to establish diagnosis. Particular problems include accurate early diagnosis of disease, correct antemortem diagnosis of established clinical disease (e.g. Alzheimer’s vs Lewy Body Dementia), and overlapping dementing disorders (e.g. combined Alzheimer’s and Lewy Body disorders). Thus there is a recognized need for tests that can provide “pathological” diagnosis during life. Traditional tissue methods such as biopsy are not likely to be widely applicable to disorders of brain function, both because of the surgical invasiveness of brain biopsy, and the large numbers of afflicted persons. Thus, translational research involving biomarkers (Table 2), either through imaging by MRI, SPECT, or PET, or through biochemical analysis of cerebrospinal fluid, body DNA, or blood serum or plasma, is now being used to develop newer improved diagnostic methods.
Table 2.
Translating Biology into Biomarkers of Possible Diagnostic Utility in Dementia.
| Magnetic Resonance Imaging (MRI) Structural Measures – |
| Global volume measures: whole brain, ventricular, gray matter, white matter, and cortical thickness measures |
| Regional measures including hippocampal volumes |
| Magnetic Resonance Imaging Functional Measures- |
| Arterial Spin Labeling (ASL) |
| BOLD imaging |
| Spectroscopy measures (e.g. NAA) |
| Nuclear Medicine Imaging Functional Measures |
| Single Photon Emission Computed Tomography (SPECT) – regional perfusion |
| Positron Emission Tomography (PET)- 18F-fluorodeoxyglucose regional metabolism |
| Nuclear Medicine Molecular Imaging |
| PET Amyloid imaging (e.g. 11C-PiB, 18F-florbetapir, 18F-florbetaben, 18F-flutemetamol) |
| PET transporter imaging (e.g. 14F-AV-144) |
| SPECT transporter imaging (e.g. 123I-ioflupane) |
| Cerebrospinal fluid analyses |
| β-amyloid-42 |
| tau |
| phosphorylated-tau |
| 14-3-3 protein |
| α-synuclein |
| Blood/plasma/serum analyses |
| DNA markers- genetic analyses |
| PS1, PS2, APP, APOE4, GBA, SNCA, GBA, LRRK2, others, |
| β-amyloid1-42, β-amyloid 1-40, others |
| Proteomics |
Brain atrophy is a feature of dementias
Neuropathological studies have shown that loss of neurons, synapses, and white matter, are accompaniments of dementia. These observations have translated into in vivo structural neuroimaging studies for diagnosis and followup of dementia. Structural imaging, originally computerized tomography (CT), then supplanted by magnetic resonance imaging (MRI) has shown value in measuring brain loss in dementia.6 Brains incur detectable volume losses due to synaptic and neuronal degenerative losses. These amount to 1-2 % change per year using various global measures of brain volume or ventricular volume, and somewhat greater volumetric changes in specific regional measures of hippocampal volume.7, 8 Large-scale detailed longitudinal data from many hundreds of subjects has emerged as a result of the Alzheimer’s Disease Neuroimaging Initiative (ADNI). These data have provided much better understanding of rates of structural atrophy, and how these relate to normal aging, conversion of MCI to AD, and progression of AD.9, 10 It is now commonplace for local radiologists to include the diagnostic possibility of Alzheimer disease upon reviewing images with marked mesiotemporal atrophy. For clinical trials, longitudinal measurements of brain volumes are now frequently incorporated because of their potential use in monitoring the effects of drug treatment.
Neuropathological and clinical changes are not diffuse, but are variably distributed in different dementias
Functional measures of brain activity have been valuable in differential diagnosis. These include the nuclear medicine techniques of Single Photon Emission Computed Tomography (SPECT) and Positron Emission Tomography (PET). These methods have provided imaging of brain function through measurement of functional surrogate agents mapping blood perfusion and oxygen and glucose metabolism. These nuclear medicine studies provide “pattern” biomarkers allowing better distinction between disorders affecting primarily temporoparietal cortices (AD), parieto-occipital cortices (LBD), and frontotemporal regions (FTD).11 Alzheimer disease prominently affects temporal cortices, but also causes decreased activity in association cortices in frontal and parietal regions. Frontotemporal dementias typically are marked by decreased brain activity in frontal and anterior temporal regions. Lewy Body Dementia is often accompanied by significant decreases in brain activity in parieto-occipital regions, with lesser affliction of frontal and temporal regions. These patterns have had value for assisting in differential diagnosis of dementing disorders. However, there remains uncertainty regarding the interpretation and specificity of these imaging patterns.12 Functional MRI may provide even less invasive measures of brain function than nuclear medicine imaging.
Specific molecular and neurochemical changes mark each dementia, and in a revolutionary fashion, neuroimaging can identify such changes through use of specific ligands
In the research sphere, the use of Aβ-binding agents has provided the means to visualize early evidence of plaques in the living brain. The first agent to be widely used, the 11C radiolabeled agent BTA or PiB, has been handicapped by the need for a cyclotron near the PET imaging center, but 18F radiolabeled agents including florbetapir (AV-45), florbetaben (BAY-949172, or AV-1), and flumetamol have now achieved wide use in research studies. The national multicenter NIH-industry partnership funded ADNI studies have shown the ability even in different centers, using different imaging instruments, to reliably detect amyloid binding. A very high percentage of clinical Alzheimer Disease, and essentially 100% of pathologically-proven Alzheimer disease, shows amyloid binding through PET imaging techniques using these agents. Similarly, persons with MCI who progress to Alzheimer disease generally have evidence of amyloid deposition by amyloid PET imaging. However, amyloid-binding seen by PET also occurs in a moderate percentage (20-40%) of persons with normal cognition, depending on age.13, 14 The favored implication of this finding is that persons with such ligand binding are at the beginning, asymptomatic, stage of Alzheimer disease, that might become manifest at some later age. Imaging findings have now been incorporated into the new clinical criteria for Alzheimer disease, MCI, and presymptomatic AD.3-5 As clinical drug trials are aimed to interrupt Alzheimer disease at the earliest stage, trials may increasingly demand evidence of amyloid-binding by PET as an eligibility criteria to increase the homogeneity and accuracy of diagnosis. In one recent European trial which did not use amyloid-binding as an eligibility factor, nearly 20% of subjects entering the trial as clinically probable Alzheimer disease had no significant amyloid-binding,15 suggesting likely erroneous diagnosis of AD. Similar evidence for imperfect clinical diagnosis is present in the observational ADNI trial as well as neuropathological series. Ligand imaging may assist in diagnosis, but also is potentially useful in monitoring of treatment. Drug trials with an intravenously administered antibody specific for Aβ42, bapineuzumab, have shown in vivo reduction of brain amyloid levels in human research subjects 15. Nuclear medicine imaging techniques using PET or SPECT can also detect changes in nigrostriatal function seen in Parkinson Disease and Lewy Body Dementia. PET imaging with 18F-fluorodopa or with the investigational agent 14F-AV-144 can reveal presynaptic dopaminergic insufficiency. Likewise through the use of radiolabeled tropanes, which are taken up by the vesicular monoamine transporters, SPECT imaging can confirm clinically suspected dopaminergic deficits in Parkinson’s disease or Lewy Body Dementia, e.g. 123I-ioflupane (DATScan®), recently approved in the US.
Molecular brain changes are also discernible through analysis of cerebrospinal fluid (CSF)
Obtained through painless lumbar puncture, CSF contains brain proteins shed into the surrounding fluid that are increasingly the subject of analysis. AD shows certain hallmark changes in the CSF, with reduction of Aβ42. It has been proposed that Aβ42 is lower concentration in the CSF of AD patients due to deposition in the brain parenchyma,16 but Aβ42 is decreased in various other, non-AD disorders.17 An alternative explanation is that decreased Aβ42 relates to decreased brain synaptic activity. Tau protein is elevated in CSF of AD patients, but this is also non-specific. Increased CSF tau presumably reflects increased neurodegeneration, with release of this intraneuronal cytoskeletal protein. Increased CSF phosphorylated-tau (P-tau; particularly tau phosphorylated at the 181 position) is a more specific hallmark of AD, related to increased phosphorylation of tau in AD. Because each of these biomarkers in isolation is not highly specific, combinations of biomarkers as ratios of Aβ42:tau or other indices has had particular utility in diagnosis. CSF biomarkers have now been incorporated into the new clinical criteria for Alzheimer disease, MCI, and presymptomatic AD.3-5 Blood plasma or serum tests are increasingly sought, and might ultimately provide the least invasive measure, but to date there has been less clear success in measurements of blood, whether by measures of specific markers such as Aβ40 and Aβ42, or by changes in clusters of proteins as revealed by proteomic analysis.18 Like amyloid-imaging, CSF biomarker profiles can be used to select individuals with molecular characteristics of AD for clinical trials, and may be potentially used to monitor therapeutic efficacy. Decreased CSF levels of tau might be evidence of decreased degree of neurodegeneration, and preliminary data in some drug trials, e.g. bapineuzumab, has shown such change.19
Genetic factors were discovered in the epidemiologic search for risk factors for dementia
Early-onset AD can be caused by genetic mutations in APP, the gene coding for the β-amyloid precursor protein, or in PSEN1 or PSEN2, genes involved in the gamma-secretase-mediated cleavage of APP to Aβ42 (Table 1). Late-onset AD is associated with the presence of the APOE gene ε4 allele, which conveys significant risk for AD - about a 2-fold risk for one copy and more than 4-fold risk for two copies of this allele compared to individuals with no e4 alleles. Overall, in the US population in the 55-75 year age range, only about 1/3 of the general population has an ε4 allele, but about 2/3 of those with Alzheimer disease have one or more ε4 alleles. While the presence of an ε4 allele has not been proven to alter disease course or prognosis, it is increasingly clear that there may be differential sensitivity to drug therapy efficacy or side-effects in those with versus without an ε4 allele. Several studies have suggested an effect of ε4 allele on efficacy: e.g. on donepezil in MCI 20 although this may relate to diagnosis, on rosiglitazone in AD 21 although subsequent studies have not confirmed any effect of this drug, and on bapineuzumab in AD 19 although this may relate more to side-effects. Although definite effects of APOE are not proven in efficacy data, it is reasonably clear that APOE ε4 allele is a risk for the side-effect of amyloid reducing therapy originally termed “vasogenic edema”, and now termed ARIA (amyloid-related imaging abnormality) 22. There is a markedly increased risk of this sometimes symptomatic brain change in persons with APOE ε4 alleles. This observation is responsible for the protocol in which treatment is stratified by genetic background in the bapineuzumab phase 3 studies, in which individuals without ε4 alleles are eligible for higher doses of drug than those with ε4 alleles.
Recent genetic analytic studies have now resulted in an increasing family of genes for which polymorphisms might increase genetic risk of late-onset “sporadic” AD. These genes include members of protein processing, membrane or cholesterol, and immune/inflammatory pathways (Sorl1, CR1, PICALM, CLU, MS4A4, CD2AP, CD33, EPHA1).23-25 While variation in these genes appears to be only responsible for small fractions of the risk of AD, the genetic findings make it likely that newly identified pathways could be addressed in drug treatment strategies.
For Parkinson’s disease, there are likewise genes that are relatively determinative of inherited disease whether autosomal dominant (e.g. SNCA, LRRK2) or recessive (e.g. PARK2, PARK7, PINK1), and those that may convey risk (UCHL1, SNCAIP, and GBA). GBA may particularly provide risk of dementia in PD, or Lewy Body Dementia.26
For Frontotemporal Dementia, there are now several autosomal dominant genes known to convey risk of FTD including MAPT (tau), GRN, CHMP2B, VCP, and C9ORF72 (Tables 1 and 2).27, 28
Translational Research in Therapy for AD
Therapies for dementia may be divided into symptomatic therapies for secondary symptoms, disease-specific symptomatic therapies, and disease-modifying therapies. Secondary symptoms of dementia are clinically important, and include agitation, aggression, hallucinations, delusions, depression, and incontinence. While these symptoms can be refractory to present treatments, the neurotransmitter-based medications including neuroleptics, antidepressants, anxiolytics, and antispasmodic drugs have been valuable, although side-effects are sometimes limiting.
Symptomatic therapies for AD
These were first developed after the recognition of the particular cholinergic deficit in AD. Drugs causing increased brain acetylcholine, through acetylcholinesterase inhibition were developed. The first such drug was tacrine hydrochloride, which became available in 1993. Three further cholinesterase inhibitors have been developed successfully for clinical use. Donepezil HCl, galantamine HBr, and rivastigmine tartrate have been shown to be of modest symptomatic benefic in AD. Following the development of these drugs, the activity-dependent glutamatergic NMDA-receptor antagonist, memantine, was demonstrated to have modest symptomatic benefit in persons with moderate to severe AD.29 However, none of these five neurotransmitter-based drugs, labeled by the FDA for use in Alzheimer disease (four acetylcholinesterase inhibitors, and one NMDA glutamatergic receptor antagonist), provide more than a modest symptomatic benefit in afflicted patients. And these drugs do not appear to modify the molecular pathology or clinical course of patients with Alzheimer disease. Investigations are ongoing of other drugs that involve other neurotransmitter systems, as well the use of intranasal insulin.30
Disease-modifying therapies
Since established therapies do not actually modify disease course, the strongest focus of drug discovery for dementing disorders, are investigations into agents that might alter disease progression through affecting basic disease pathophysiology. A variety of empiric trials or existing medications or natural substances, including various vitamins, fish oils, botanical compounds (e.g. Gingko biloba), hormones (e.g. estrogen), HMGcoA reductase inhibitors, anti-inflammatory agents have been suggested to be possibly helpful, but subsequently proven to be inefficacious in the treatment of Alzheimer disease. Hence it is translational medical science that most likely will provide advances in treatment of Alzheimer disease and the other dementing disorders (Table 3).
Table 3.
Clinical Investigational Drug Studies in Alzheimer Disease
| INCREASE BETA-AMYLOID CLEARANCE |
| Active immunization – |
| AN-1792, ACC-001 (vanutide cridificar), CAD-106, UB-311, V-950 |
| Passive immunization- |
| Bapineuzumab (AAB-001), solanezumab (LY2062430), crenezumab (MABT-5102A), gantenerumab (R-1450), ponezumab (PF-04360365), GSK-933776, |
| Immunoglobulin therapy – IVIG (IGIV) |
| DECREASE BETA-AMYLOID PRODUCTION |
| Gamma secretase modulators – tarenflurbil (R-flurbiprofen) |
| Gamma-secretase inhibitors – semagacestat (LY450139), begacestat (GSI-953), BMS-708163, GSI-136, PF-3084014, MK0752 |
| Alpha-secretase enhancers – acitretin (etretinate, varenicline) |
| Beta-secretase inhibitors – CTS-21166 |
| DECREASE BETA-AMYLOID FIBRIL FORMATION OR AGGREGATION |
| tramiprosate |
| scyllo-inositol (D-005) |
| DETER TAU AGGREGATION |
| methylthioninium (methylene blue) |
| INHIBIT TAU PHOSPHORYLATION |
| GSK-3 inhibitors – Lithium, Valproic acid, |
| DETER NEURODEGENERATION |
| Davunetide (NAP, AL-108) |
| Nerve growth factor (NGF) |
| NGF-Adeno-associated Virus (AAV2-NGF) gene (CERE-110) |
| Brain-Derived Neurotrophic Factor (BDNF) |
| Exenatide |
Drugs in bold typeface are still under development in phase 2 or 3.
Pathological studies show that Alzheimer disease is marked by accumulation of beta-amyloid protein in the brain in the form of plaques, and hyperphosphorylated tau in the form of tangles. In vitro, animal model, and human genetic evidence all points to a centrality of beta-amyloid in the pathophysiological cascade causing Alzheimer disease.31 Thus many drugs in development have a mechanism of action based on either decreasing beta-amyloid production (through inhibition of beta-secretase or gamma secretase), increasing beta-amyloid clearance (through active or passive immunization), or decreasing beta-amyloid aggregation/fibrillization (Table 3).
Inhibitors of Beta-amyloid production in AD
Beta amyloid production can be inhibited by reduction of gamma-secretase or beta-secretase, or by enhancement of alpha-secretase (Figure 1). Tarenflurbil (Flurizan®), otherwise known as R-flurbiprofen, acts in vitro as a selective inhibitor of Aβ42 production both in vitro and in mouse models.32 This drug recently failed to show any efficacy or harm in a large phase 3 double-blind randomized placebo-controlled trial with over 2000 subjects.33 It is unclear if this was because of differing action in humans than mice, or whether there was simply insufficient dose or inadequate central nervous system penetration, or whether the putative mechanism of action did not occur in humans. Semagacesat (LY450139) is an inhibitor of gamma-secretase, with evidence of effect on amyloid production in vitro, in mice, and demonstrably in humans.34 But a large phase 3 double-blind randomized placebo-controlled trial was recently stopped upon interim review, because of evidence that subjects treated with drug were doing less well cognitively than those treated with placebo 35. The reasons for these unexpected findings are unclear, but might possibly relate to the changed balance of amyloid products, to insufficient overall inhibition of gamma-secretase, or to “off-target” effects of this inhibitor on other cellular processes. Other gamma-secretase inhibitors are under development, notably including BMS-708163, for which a phase 2 trial has been completed, and phase 3 development is underway. Inhibition of beta-secretase also results in decreased Aβ42 production in animal models 36, and inhibitors of this enzymatic activity are under development. Enhancement of alpha-secretase activity by etretinate has been reported, and limited clinical trials of this agent have started.
Figure 1. Hypothetical Working Model of Molecular and Clinical Features of Alzheimer Disease.
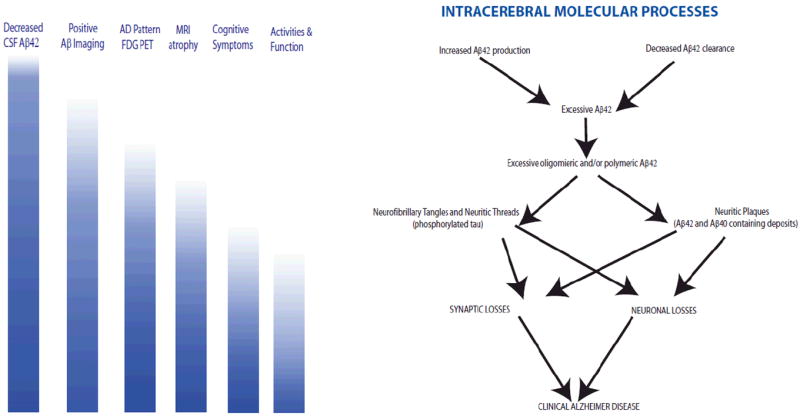
As depicted on the right side of the figure, the pathogenetic cascade of Alzheimer disease is hypothesized to initially involve either overproduction, or impaired clearance, of Aβ42. This peptide aggregates into oligomers and then polymeric deposits. Development of diffuse and neuritic plaques ensues, with concomitant depositions of hyperphosphorylated tau as neurofibrillary tangles and neuritic threads. Ultimately there are synaptic losses and neuronal losses. On the left side of the figure, in parallel to the cascade depicted on the right, are indicators of the degree to which clinical biomarkers may be present at different stages of the pathogenetic cascade: darker blue color indicates a higher likelihood of the listed biomarker being positive. Thus early aspects of β-amyloid dysregulation can be detected through decreased Aβ42 in the CSF, and through nuclear medicine-based amyloid imaging. Later stages of the cascade show detectable early brain functional changes through FDG-PET, subsequent brain atrophy, cognitive symptoms, and functional change.
Therapies to increase beta-amyloid clearance in AD
Active immunization
Injection with beta amyloid peptide is efficacious in mouse models of Alzheimer disease. Aβ deposits in the brain are reduced,37 and memory function is improved in maze tasks.38 The first such drug tried in humans was AN-1792, for which a phase 2 trial was halted prematurely due to the occurrence of meningoencephalitis, a symptomatic in about 5% of treated patients. Limited clinical data obtained from this study did not show efficacy,39 although a follow-up study did seem to show possible evidence suggestive of efficacy.40 Postmortem studies, of some immunized individuals, showed apparent evidence that immunization may have had the intended effect in those treated. Individuals with serological antibody response did show successful clearance of amyloid,41 and immunized individuals may have shown beneficial changes in abnormal neurites.42 The possibility that amyloid clearance was successful but was still accompanied by lack of clinical improvement or clinical deterioration has raised the issue as to whether Aβ-removing strategies will indeed be successful.41 However, with such small numbers, lack of clarity as to the influence of subclinical side-effects in treated patients, and lag from treatment to autopsy, it is not possible to make any conclusions at this time. It is likely that the severe inflammatory symptoms from AN-1792 related significantly to the immunological adjuvant. Experimental laboratory studies have led to introduction of other forms of Aβ immunization, including different haptens and adjuvants, such as vanutide cridificar (ACC-001), and CAD-106 are now in phase 2 trials. Such trials, as well as the passive immunization trials, should result in clearer data as to whether amyloid reduction strategies will be effective in stabilizing, slowing, or less likely reversing, AD clinical symptoms.
Passive Immunization
Passive immunization with antibodies to beta amyloid peptide have demonstrated efficacy in mouse models of Alzheimer disease. Bapineuzumab (AAB-001) is a humanized mouse monoclonal antibody to the N-terminal portion of beta-amyloid. It has undergone phase 2 trial, and is now undergoing two large multicenter phase 3 trials, with a total of over 4000 patients being studied. While the phase 2 trial did not meet efficacy endpoints 19, there was clear evidence of increased side-effects, including vasogenic edema of the brain in apolipoprotein e4 carriers 19, 22, and suggestive evidence of benefit from active treatment, particularly in non-carriers of the apolipoprotein ε4 allele.19 The phase 3 trial is ongoing in both carriers (at lower dose) and non-carriers of the e4 allele. A different humanized mouse monoclonal antibody, to the mid-portion of beta-amyloid, solanezumab (LY2062430), is also undergoing phase 3 trials, after phase 2 trials and cerebrospinal fluid testing showed encouraging biomarker changes.34, 43 This antibody may act more to clear soluble beta-amyloid, although it may secondarily cause plaque clearance. Additional antibodies are also in phase 1 or phase 2 trials (Table 3).
Nonspecific immunoglobulin therapy and other immunization strategies
The success in mice of specific anti-Aβ passive immunization prompted small human phase 1 and phase 2 trials of nonspecific human immunoglobulin (IGIV) therapy.44 Biochemical experiments have suggested that human intravenous immunoglobulin contains some polyclonal anti-beta amyloid antibody at low levels,44 and this would be one putative mechanism of action of human immunoglobulin preparations, if they indeed were shown to have beneficial effects in AD. Alternative mechanisms of action include modifying the immune response. A phase 3 trial of IGIV is ongoing in mild-to-moderate AD. An alternative to immunization with protein or passive immunoglobulin administration has been the concept of DNA vaccination,45 in which injection of DNA in mice has led to decreased beta-amyloid in the brain.
Therapies to inhibit beta-amyloid aggregation in AD
Antifibrillation/anti-aggregation agents
In vitro and animal model experiments have suggested that treatment with agents that might prevent aggregation of beta-amyloid might have efficacy for AD. The first such agent tested in a phase 3 trial, tramiprosate, also known as homotaurine, failed to show any efficacy in cognitive tests, or first-order biomarker tests. The developer of that drug has discontinued development of the molecule as a drug, but has recharacterized it as a “medical food” (Vivimind®) marketed in Canada. However, evidence is lacking that it might benefit AD. Another agent that appears to show promising preclinical results is ELN D005, also known as scyllo-inositol, which has now completed phase 2 testing, and will apparently proceed to phase 3 testing shortly.
Therapies to prevent neuronal degeneration in AD
Agents that might inhibit tau dysfunction, or support neuronal integrity
Given the prominence of neurofibrillary degeneration in AD, it is reasonable to speculate that agents that might prevent the formation of the phosphorylated tau that aggregates into neurofibrillary tangle in the brain of persons with AD. In vitro and animal experiments have suggested that a variety of agents might deter formation of tangles. Methylene blue (methylthioninium) was tried in a small phase 2 study, but showed no clear efficacy. Taxol has been suggested to be of potential utility on the basis of animal studies, but its toxicity will likely preclude human trials. Davunetide, also known as AL-108, has been proposed, based on animal experimentation, to be of potential utility in AD, and a phase 1 trial has been performed. Growth factors might improve neuronal survival. Injection of nerve growth factor (NGF) gene delivery into the brain,46 has been tried in a limited number of persons in a phase 1, proof of concept experiment. Since then injections of DNA coding for Brain-derived neurotrophic factor (BDNF) has been the subject of phase 1 experiments.47 Neuronal degeneration is marked by a number of final processes including cell membrane breakdown, calcium influxes, caspase activations, and other cell-death pathways. Research continues on developing agents that might intervene beneficially at these more terminal steps of nervous system injury that occur in common in ischemic and neurodegenerative disease.
Non-AD dementias
The biological underpinnings of DLB, FTD, PSP, CBD, and CJD, are reasonably described, but the pathogenetic cascade is less well understood than for AD. Thus translational advances in diagnosis and therapy are in less advanced stages for these non-AD dementias. For FTD and related disorders, possible medications including possibly tau-disruptive, or neuronal supportive, medications for the tau forms of this disorder are under consideration, including AL-108 (mentioned above) which is being tried in a phase 2 study of progressive supranuclear palsy. Likewise memantine, which has neuroprotective effects in vitro, is being tried in a phase 2 study of FTD. Quinacrine, has shown efficacy in vitro in deterring prion protein aggregation, leading to trials of this agent has been in CJD; these limited trials to date have been without success.48, 49 Even though the understanding of the molecular cascades of AD is limited, it is nonetheless more advanced than the understanding of the molecular pathological underpinnings of these other neurodegenerative disorders. Thus, translational investigations leading to potential diagnostic or therapeutic methodology for the non-AD degenerative disorders are lagging behind those for AD.
Acknowledgments
Supported by funding from the National Institutes of Health/ National Institute on Aging (grant P50AG08702), the Alzheimer’s Association (IIRG 08-92010), the Alzheimer’s Disease Discovery Foundation, the Panasci Fund for Lewy Body Research, and the Taub Institute for Research.
Footnotes
DISCLOSURE
The author has received personal compensation for consulting activities with Bayer, Biogen IDEC, Dainippon, Johnson and Johnson, and Pfizer pharmaceuticals. He has also received personal compensation for editorial activities for Archives of Neurology. He has funding for research activities from Bayer, Elan, Genentech, Johnson and Johnson, Lilly, and Pfizer pharmaceutical companies.
References
- 1.White L. Brain lesions at autopsy in older Japanese-American men as related to cognitive impairment and dementia in the final years of life: a summary report from the Honolulu-Asia aging study. J Alzheimers Dis. 2009;18(3):713–725. doi: 10.3233/JAD-2009-1178. [DOI] [PubMed] [Google Scholar]
- 2.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol. 2009 Aug;66(2):200–208. doi: 10.1002/ana.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011 May;7(3):263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011 May;7(3):270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011 May;7(3):280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sabuncu MR, Desikan RS, Sepulcre J, et al. The Dynamics of Cortical and Hippocampal Atrophy in Alzheimer Disease. Arch Neurol. 2011 Aug;68(8):1040–1048. doi: 10.1001/archneurol.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barnes J, Bartlett JW, van de Pol LA, et al. A meta-analysis of hippocampal atrophy rates in Alzheimer’s disease. Neurobiol Aging. 2009 Nov;30(11):1711–1723. doi: 10.1016/j.neurobiolaging.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scher AI, Xu Y, Korf ES, et al. Hippocampal morphometry in population-based incident Alzheimer’s disease and vascular dementia: the HAAS. J Neurol Neurosurg Psychiatry. 2011 Apr;82(4):373–376. doi: 10.1136/jnnp.2008.165902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ewers M, Sperling RA, Klunk WE, Weiner MW, Hampel H. Neuroimaging markers for the prediction and early diagnosis of Alzheimer’s disease dementia. Trends Neurosci. 2011 Aug;34(8):430–442. doi: 10.1016/j.tins.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vemuri P, Wiste HJ, Weigand SD, et al. Serial MRI and CSF biomarkers in normal aging, MCI, and AD. Neurology. 2010 Jul 13;75(2):143–151. doi: 10.1212/WNL.0b013e3181e7ca82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen K, Ayutyanont N, Langbaum JB, et al. Characterizing Alzheimer’s disease using a hypometabolic convergence index. Neuroimage. 2011 May 1;56(1):52–60. doi: 10.1016/j.neuroimage.2011.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Womack KB, Diaz-Arrastia R, Aizenstein HJ, et al. Temporoparietal hypometabolism in frontotemporal lobar degeneration and associated imaging diagnostic errors. Arch Neurol. 2011 Mar;68(3):329–337. doi: 10.1001/archneurol.2010.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sojkova J, Driscoll I, Iacono D, et al. In vivo fibrillar beta-amyloid detected using [11C]PiB positron emission tomography and neuropathologic assessment in older adults. Arch Neurol. 2011 Feb;68(2):232–240. doi: 10.1001/archneurol.2010.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aizenstein HJ, Nebes RD, Saxton JA, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008 Nov;65(11):1509–1517. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rinne JO, Brooks DJ, Rossor MN, et al. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010 Apr;9(4):363–372. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- 16.Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006 Mar;59(3):512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 17.Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010 Mar;6(3):131–144. doi: 10.1038/nrneurol.2010.4. [DOI] [PubMed] [Google Scholar]
- 18.Ray S, Britschgi M, Herbert C, et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat Med. 2007 Nov;13(11):1359–1362. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- 19.Salloway S, Sperling R, Gilman S, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009 Dec 15;73(24):2061–2070. doi: 10.1212/WNL.0b013e3181c67808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petersen RC, Thomas RG, Grundman M, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005 Jun 9;352(23):2379–2388. doi: 10.1056/NEJMoa050151. [DOI] [PubMed] [Google Scholar]
- 21.Risner ME, Saunders AM, Altman JF, et al. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 2006 Jul-Aug;6(4):246–254. doi: 10.1038/sj.tpj.6500369. [DOI] [PubMed] [Google Scholar]
- 22.Sperling RA, Jack CR, Jr, Black SE, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011 Jul;7(4):367–385. doi: 10.1016/j.jalz.2011.05.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reitz C, Cheng R, Rogaeva E, et al. Meta-analysis of the association between variants in SORL1 and Alzheimer disease. Arch Neurol. 2011 Jan;68(1):99–106. doi: 10.1001/archneurol.2010.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011 May;43(5):436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jun G, Naj AC, Beecham GW, et al. Meta-analysis confirms CR1, CLU, and PICALM as alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch Neurol. 2010 Dec;67(12):1473–1484. doi: 10.1001/archneurol.2010.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shulman JM, De Jager PL, Feany MB. Parkinson’s disease: genetics and pathogenesis. Annu Rev Pathol. 2011 Feb 28;6:193–222. doi: 10.1146/annurev-pathol-011110-130242. [DOI] [PubMed] [Google Scholar]
- 27.Rohrer JD, Warren JD. Phenotypic signatures of genetic frontotemporal dementia. Curr Opin Neurol. 2011 Oct 7; doi: 10.1097/WCO.0b013e32834cd442. [DOI] [PubMed] [Google Scholar]
- 28.Borroni B, Pilotto A, Bianchi M, Gilberti N, Padovani A. Genetic Contributors to Frontotemporal Lobar Degeneration: Beyond Monogenic Disease. Mini Rev Med Chem. 2011 Jul 15; doi: 10.2174/138955711797068517. [DOI] [PubMed] [Google Scholar]
- 29.Herrmann N, Li A, Lanctot K. Memantine in dementia: a review of the current evidence. Expert Opin Pharmacother. 2011 Apr;12(5):787–800. doi: 10.1517/14656566.2011.558006. [DOI] [PubMed] [Google Scholar]
- 30.Craft S, Baker LD, Montine TJ, et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment. A pilot clinical trial. Arch Neurol. 2011 doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Selkoe DJ. Biochemistry and molecular biology of amyloid beta-protein and the mechanism of Alzheimer’s disease. Handb Clin Neurol. 2008;89:245–260. doi: 10.1016/S0072-9752(07)01223-7. [DOI] [PubMed] [Google Scholar]
- 32.Kukar TL, Ladd TB, Bann MA, et al. Substrate-targeting gamma-secretase modulators. Nature. 2008 Jun 12;453(7197):925–929. doi: 10.1038/nature07055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Green RC, Schneider LS, Amato DA, et al. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA. 2009 Dec 16;302(23):2557–2564. doi: 10.1001/jama.2009.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siemers ER, Friedrich S, Dean RA, et al. Safety and changes in plasma and cerebrospinal fluid amyloid beta after a single administration of an amyloid beta monoclonal antibody in subjects with Alzheimer disease. Clin Neuropharmacol. 2010 Mar-Apr;33(2):67–73. doi: 10.1097/WNF.0b013e3181cb577a. [DOI] [PubMed] [Google Scholar]
- 35.Siemers E, Henley D, Sundell K, et al. Evaluating semagacestat, a gammasecretase inhibitor, in a phase III trial. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association. 2011;7(4, Supplement):S484–S485. [Google Scholar]
- 36.Citron M. Beta-secretase inhibition for the treatment of Alzheimer’s disease--promise and challenge. Trends Pharmacol Sci. 2004 Feb;25(2):92–97. doi: 10.1016/j.tips.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 37.Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999 Jul 8;400(6740):173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 38.Wilcock DM, Rojiani A, Rosenthal A, et al. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004 Dec 8;1(1):24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gilman S, Koller M, Black RS, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005 May 10;64(9):1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 40.Vellas B, Black R, Thal LJ, et al. Long-term follow-up of patients immunized with AN1792: reduced functional decline in antibody responders. Curr Alzheimer Res. 2009 Apr;6(2):144–151. doi: 10.2174/156720509787602852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holmes C, Boche D, Wilkinson D, et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008 Jul 19;372(9634):216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 42.Serrano-Pozo A, William CM, Ferrer I, et al. Beneficial effect of human anti-amyloid-beta active immunization on neurite morphology and tau pathology. Brain. 2010 May;133(Pt 5):1312–1327. doi: 10.1093/brain/awq056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siemers ER, Quinn JF, Kaye J, et al. Effects of a gamma-secretase inhibitor in a randomized study of patients with Alzheimer disease. Neurology. 2006 Feb 28;66(4):602–604. doi: 10.1212/01.WNL.0000198762.41312.E1. [DOI] [PubMed] [Google Scholar]
- 44.Relkin NR, Szabo P, Adamiak B, et al. 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol Aging. 2009 Nov;30(11):1728–1736. doi: 10.1016/j.neurobiolaging.2007.12.021. [DOI] [PubMed] [Google Scholar]
- 45.Lambracht-Washington D, Qu BX, Fu M, et al. DNA immunization against amyloid beta 42 has high potential as safe therapy for Alzheimer’s disease as it diminishes antigen-specific Th1 and Th17 cell proliferation. Cell Mol Neurobiol. 2011 Aug;31(6):867–874. doi: 10.1007/s10571-011-9680-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tuszynski MH. Nerve growth factor gene delivery: animal models to clinical trials. Dev Neurobiol. 2007 Aug;67(9):1204–1215. doi: 10.1002/dneu.20510. [DOI] [PubMed] [Google Scholar]
- 47.Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov. 2011 Mar;10(3):209–219. doi: 10.1038/nrd3366. [DOI] [PubMed] [Google Scholar]
- 48.Collinge J, Gorham M, Hudson F, et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol. 2009 Apr;8(4):334–344. doi: 10.1016/S1474-4422(09)70049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geschwind MD. Clinical trials for prion disease: difficult challenges, but hope for the future. Lancet Neurol. 2009 Apr;8(4):304–306. doi: 10.1016/S1474-4422(09)70050-X. [DOI] [PMC free article] [PubMed] [Google Scholar]