

Abstract
Background:
Papillary thyroid carcinoma (PTC) shows high heritability, yet efforts to find predisposing genes have been largely negative.
Objectives:
The objective of this study was to identify susceptibility genes for PTC.
Methods:
A genome-wide linkage analysis was performed in 38 families. Targeted association study and screening were performed in 2 large cohorts of PTC patients and controls. Candidate DNA variants were tested in functional studies.
Results:
Linkage analysis and association studies identified the Slit-Robo Rho GTPase activating protein 1 gene (SRGAP1) in the linkage peak as a candidate gene. Two missense variants, Q149H and A275T, localized in the Fes/CIP4 homology domain segregated with the disease in 1 family each. One missense variant, R617C, located in the RhoGAP domain occurred in 1 family. Biochemical assays demonstrated that the ability to inactivate CDC42, a key function of SRGAP1, was severely impaired by the Q149H and R617C variants.
Conclusions:
Our findings suggest that SRGAP1 is a candidate gene in PTC susceptibility. SRGAP1 is likely a low-penetrant gene, possibly of a modifier type.
Papillary thyroid carcinoma (PTC) is the most common form of thyroid carcinoma accounting for 75%–85% of all thyroid cancer. The incidence of PTC appears to have increased in the United States and elsewhere in recent years (1, 2). It is estimated that 56 460 individuals in the United States will be diagnosed with thyroid cancer in 2012 (http://www.cancer.org/Research/CancerFactsFigures/index). Radiation is one well-known predisposing factor. The molecular pathogenesis of PTC is largely associated with point mutations in the BRAF or RAS family genes or RET/PTC gene rearrangements (3, 4).
Although PTC is mostly sporadic, approximately 5% is familial (5). A strong inherited genetic predisposition is suggested by case-control studies showing a 3- to 8-fold increase in risk in first-degree relatives (6–8). Over the past years, linkage studies have suggested several potential regions as harboring predisposing genes, including 1q21 (9, 10), 2q21 (11), 6q22 (10), 8p23 (12), 8q24 (13), and 19p13.2 (14). Although a few candidate predisposing genes have been proposed, decisive evidence implicating specific genes has not been forthcoming. The relative lack of success might be in part due to weaknesses in the design and execution of earlier experiments.
In a previous study involving linkage analysis of a large family with PTC and melanoma, we identified a candidate gene (a noncoding RNA gene named PTCSC1) in 8q24 (13). The successful use of linkage prompted us to undertake another linkage study in a series of 38 PTC families, many of which were recently acquired. We report here that genome-wide linkage analysis and the following association and functional studies point to the Slit-Robo Rho GTPase activating protein 1 gene (SRGAP1) as a candidate susceptibility gene in PTC.
Materials and Methods
Subjects
The studies were approved by the Institutional Review Boards at The Ohio State University Medical Center, M. Sklodowska Curie Memorial Cancer Center and Institute of Oncology, and the Medical University of Warsaw. All subjects gave written informed consent before participation.
Ohio families
A total of 38 families with at least 2 confirmed cases of nonmedullary thyroid cancer were recruited. The majority (32 of 38) had 3 or more affected individuals. Family history information, pathology reports confirming the diagnosis of thyroid cancer or thyroid disease, blood, and tissue samples were collected from consenting affected and key unaffected individuals. Of the 38 families, 36 were Caucasian non-Hispanic and 2 were Caucasian Hispanic. The pedigrees and clinical information are provided in Supplemental Figure 1 and Supplemental Table 1, published on The Endocrine Society's Journals Online web site at http://jcem.endojournals.org.
Sample sets of sporadic cases and controls from Ohio and Poland
Individuals with thyroid cancer from Ohio were histologically confirmed as having traditional PTC and follicular variant PTC. The control group comprised individuals without clinically diagnosed thyroid cancer from central Ohio who were randomly picked from a pool of controls for genetics projects. A set of randomly collected 429 cases and 571 controls (Ohio set 1) was used in genotyping single-nucleotide polymorphism (SNP) rs2168411. Another set of 742 cases and 828 controls (Ohio set 2) was used for screening the SRGAP1 variants (Supplemental Table 2). The individuals in these 2 sets overlapped. For Polish sporadic cases and controls, 2 sample sets were recruited; 1 set (906 cases and 866 controls) was from the Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology (Gliwice, Poland). This sample set was used in genotyping rs2168411 and screening the SRGAP1 variants. Another set (1738 cases and 1701 controls) was from the Medical University of Warsaw (Warsaw, Poland). This sample set was used for screening the variant R617C. The cases included traditional PTC and follicular variant PTC. Clinical information including age at onset, grade, and stage was obtained from medical records. The control groups comprised individuals without clinically diagnosed thyroid cancer. The age, gender, and race information and the results of statistical tests for sample sets used in the association study are provided in Supplemental Table 2.
Genotyping
SNP genotyping of genomic DNA from blood using Affymetrix GeneChip Human Mapping Nsp 250K arrays (Santa Clara, California) was performed as described (13). SNP genotype calls were made with Genechip genotyping analysis software (GTYPE) 4.0 (Affymetrix) with default parameters or using the BRLMM program from Affymetrix. The SNP call rate was greater than 92% with a P = .3. The Mendelian error rate was below 0.2% and errors were removed before analysis.
In addition, 8 microsatellite markers in 12q14 were genotyped. The PCR primer sequences are available upon request. The PCR assays were performed according to the standard PCR protocol except that 1 PCR primer for each marker was labeled with a fluorescent dye (HEX, FAM, or TET). The allele analysis was performed by using an ABI 3730 DNA analyzer (Life Technologies Corp, Grand Island, New York).
For the association studies, a total of 168 tag SNPs spanning the 12q14 linkage region (∼3.2 Mb) were picked using Tagger software (http://www.broadinstitute.org/mpg/tagger/). SNP genotyping was performed using the Sequenom MassARRAY platform (San Diego, California) (15), SNaPshot assay (Life Technologies Corp, Grand Island, New York), or real-time PCR allelic discrimination assay (Life Technologies). Association analyses were performed using GeneSpring GT2 software (Agilent Technologies, Santa Clara, California).
Statistical analysis
Genome-wide posterior probability of linkage (PPL) analyses were conducted using the software package KELVIN (16), which implements the PPL class of models for measuring the strength of genetic evidence (16). The PPL is on the probability scale and indicates the probability that a disease-related gene is located at a given genetic position. The genetic map was based on that located on the Internet (http://compgen.rutgers.edu/mapopmat) (17) (release October 2006).
DNA resequencing and screening for SRGAP1 mutations
Blood genomic DNA from one PTC patient from each of 21 families (Supplemental Figure 1A) was used for Sanger resequencing using a PE3730 DNA analyzer. The PCR primers and PCR conditions are available upon request. Three SRGAP1 mutations were screened for in blood genomic DNA from patients with sporadic PTC and control individuals. A SNaPshot assay (Life Technologies) was performed as described. PCRs for amplifying the SRGAP1 exons 4, 7, and 16 were performed and followed by a single nucleotide extension reaction. The primer sequences for PCR and extension for detecting each of the mutations are listed in Supplemental Table 3.
DNA constructs, cell culture, and transfection
Cloned Homo sapiens full-length SRGAP1 in pCMV MYC-DDK expression vector was purchased from OriGene (Rockville, Maryland). Mutations in SRGAP1 were generated by the QuickChange method (Stratagene, La Jolla, California). Cell division cycle 42 (CDC42) cDNA was cloned into the pCMVSPORT6 expression vector (Open Biosystem, Huntsville, Alabama) by using primers incorporating NotI and SalI restriction sites. PCR primers used for making all mutations and cloning are available upon request. DNA constructs expressing the full-length rat ROBO1 protein in a hemagglutinin (HA)-tagged form and expressing the full-length H sapiens slit homolog 2 (Drosophila) (SLIT2) protein in a HIS/MYC-tagged form were generously provided by Ramesh Ganju (The Ohio State University, Columbus, Ohio). The SW1736 cell line was obtained from Rebecca Schweppe (University of Colorado Cancer Center, Denver, Colorado) (18). The batch used in this study was thawed from an original frozen stock. The SW1736 cell line was cultured with RPMI 1640 medium supplemented with 20% fetal bovine serum. The human embryonic kidney (HEK) 293 cell line was maintained in DMEM with 10% fetal bovine serum and 1% penicillin-streptomycin at 37°C in 5% CO2. For transient expression of proteins, cells were transfected using LipofectAMINE 2000 (Life Technologies) as detailed in the figure legends. SLIT2 was obtained from the supernatants of SLIT2-transfected HEK 293 cells. The conditioned media were collected and used for treatment or concentrated using Centricon Plus-20 filters (Millipore, Billerica, Massachusetts) for SLIT2 expression analysis by Western blotting using an anti-MYC antibody.
CDC42 activation assay
CDC42 activation assay kit was used (Cell Biolabs, San Diego, California). Cell lysates were incubated with p21-activated kinase-1-glutathione-S-transferase (GST) agarose beads for 60 minutes at 4°C according to the protocols of Cell Biolabs. GTP bound CDC42 was analyzed by Western blotting using an anti-CDC42 antibody (Cell Biolabs). Input control was analyzed using an anti-HA (Cell Signaling Technology, Danvers, Massachusetts) anti-MYC antibody (Santa Cruz Biotechnology, Santa Cruz, California).
Results
Genome-wide linkage scan
We performed a genome-wide linkage analysis in 38 families, some of which were analyzed previously (13) (Supplemental Figure 1). The linkage data set included genotypes from a total of 176 samples (113 affected with PTC), 12 obligate carriers, and 51 unaffected individuals. Genome-wide posterior PPL analysis with all families produced a PPL score of 30% in 12q14, which was by far the highest peak in the genome-wide scan with this method (Figure 1). Nonparametric linkage (NPL) using MERLIN produced a maximum NPL score of 2.8 and limit of detection score of 1.6 in 12q14 (Supplemental Figure 2). The linkage peaks obtained by the 2 methods coincided. The linkage interval was about 3.2 Mb, from SNP rs717271 to rs7957712. The previously observed peak in chromosome 8q24 (13) is lower in the present set of families (PPL 0.45%; NPL Z-score 0.98), likely due to the chance inclusion of more 8q24-related families in the previous experiment than the present one. Families included in the previous linkage and families being compatible with 8q24 linkage are indicated in Supplemental Figure 1.
Figure 1.
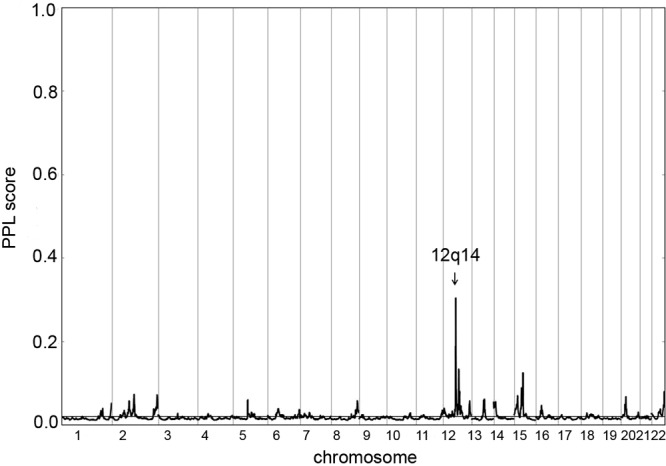
Plot of the genome-wide linkage scan with posterior PPL from 38 families. The x-axis shows chromosomal location; the y-axis shows PPL score. The 12q14 locus is indicated by an arrow.
Of the 38 families, 19 showed individual maximum NPL Z-scores greater than 1.0 in 12q14, which suggested possible linkage. Two additional smaller families with NPL Z-scores between 0.5 and 1.0 were also included in the following analyses. To fine map the 12q14 locus, we genotyped 8 microsatellite markers in 21 families (Supplemental Figure 1A). Linkage and haplotype analyses with the microsatellite markers confirmed that 21 of 38 families (55%) were compatible with linkage to 12q14. In each of the 21 families, a haplotype cosegregated with the disease phenotype (PTC individuals) as illustrated in 4 families (Figure 2). The 12q14 related families (n = 21) included 3 small families with only 2 members affected (number 2, number 5, and number 9). These families except one (number 16) were Caucasian non-Hispanic (Supplemental Figure 1). The clinical information (mean age at diagnosis, race, ethnicity, and histological subtype) of these 21 families were similar as the other 17 families without 12q14 linkage.
Figure 2.

Haplotypes and the missense mutations in 4 PTC families. A unique haplotype (boxed) cosegregates with the PTC phenotype in each family. Solid circles or squares, Individuals affected with PTC. Hatched symbols, Individuals affected with benign thyroid disease (benign nodule or goiter). DNA samples of individual 2 in family 20 and individuals 2 and 3 in family 13 were not available for haplotype analysis.
Association study
Linkage data suggested that the 12q14 locus might account for more than half of the families studied, which implied that the putative causal variant(s) or mutation(s) might be common in sporadic PTC and widespread in the population. We conducted an association study targeting the 12q14 locus. In the first round of association analyses, we genotyped 123 tag SNPs spanning the linkage peak region (∼3.2 Mb) in an Ohio cohort (sporadic PTC, n = 268; controls, n = 421). We then selected 18 SNPs with P ≤ .05 (χ2 test) and genotyped them in another Ohio cohort (sporadic PTC, n = 289; controls, n = 252). Overall, only SNP rs2168411 showed an association with PTC (Table 1). Among 429 cases and 571 controls, the G allele was more frequently detected in cases than in controls [P = .014; odds ratio (OR) 1.26; 95% confidence interval (CI) 1.05–1.52] (Table 1). To validate this association result, we genotyped SNP rs2168411 in a Polish cohort (cases, n = 906; controls, n = 866). As in the Ohio cohort, SNP rs2168411 showed an association with PTC in the Polish population (Table 1) with a P = .014 [OR 1.19; (95% CI, 1.04–1.36]. Combining all the data, SNP rs2168411 associated with PTC with a combined OR of 1.21 (95% CI 1.08–1.35), P = .0008.
Table 1.
Association Results of SNP rs2168411 in Ohio and Polish Cohorts
| Sample | Case number | Genotype |
Allele Frequency |
OR (95% CI)a | P Valuea | |||
|---|---|---|---|---|---|---|---|---|
| GG | GC | CC | G | C | ||||
| Ohio | ||||||||
| PTC | 429 | 71 | 205 | 153 | 0.404 | 0.596 | 1.26 (1.05, 1.52) | .014 |
| Control | 571 | 77 | 249 | 245 | 0.353 | 0.647 | ||
| Polish | ||||||||
| PTC | 906 | 145 | 405 | 356 | 0.384 | 0.616 | 1.19 (1.04, 1.36) | .014 |
| Control | 866 | 98 | 400 | 368 | 0.344 | 0.656 | ||
| Combinedb | ||||||||
| PTC | 0.394 | 0.606 | 1.21 (1.08, 1.35) | .0008 | ||||
| Control | 0.349 | 0.652 | ||||||
The allelic OR with 95% CI, and P values, obtained by applying logistic regression adjusting for age and gender.
For the combined study populations, the reported frequencies are the average, unweighted frequencies of the individual populations, and the OR and the P value were estimated using the Mantel-Haenszel model.
Germline mutations/variations in the SRGAP1 gene
SNP rs2168411 is located in intron 4 of SRGAP1. We resequenced all exons and exon-intron boundaries of SRGAP1 in 1 patient from each of the 21 families that were compatible with linkage to 12q14. Six germline single-nucleotide changes in the SRGAP1 coding region were found, 1 synonymous and 5 nonsynonymous (Table 2). There were also several previously reported polymorphisms in the 3′ untranslated region (UTR) (data not shown). The synonymous change SRGAP1-c.2274T>C (S758S), the numbering according to SRGAP1 (NM_020762.2 and NP_065813.1), was found in 7 families and was reported in the single-nucleotide polymorphism database (dbSNP) database with a heterozygosity frequency of 0.424.
Table 2.
DNA Variations in the SRGAP1 Coding Region in 21 PTC Families
| Nucleotide Change | Amino Acid Change | dbSNP ID | Affected Family | PolyPhen Prediction |
SIFT Prediction |
Minor Allele Frequencya | ||
|---|---|---|---|---|---|---|---|---|
| PSIC SD | Predicted Impact | Tolerance Score | Predicted Impact | |||||
| c.447A>C | Q149H | 1 family (number 2) | 1.004 | Possibly damaging | 0.22 | Tolerant | ||
| c.823G>A | A275T | 1 family (number 3) | 1.598 | Possibly damaging | 0.57 | Tolerant | ||
| c.1534G>A | V512I | rs74691643 | 1 family (number 4) | 0.117 | Benign | 0.7 | Tolerant | 0.013 |
| c.1849C>T | R617C | rs114817817 | 1 family (number 20) | 3.311 | Probably damaging | 0.01 | Intolerant | 0.002 |
| c.2274T>C | S758S | rs789722 | 7 families | n/ab | n/a | n/a | n/a | 0.424 |
| c.2624A>G | H875R | rs61754221 | 1 family (number 13) | 1.175 | Benign | 0.64 | Tolerant | 0.009 |
Abbreviation: n/a, not applicable.
Available at: http://evs.gs.washington.edu/EVS/.
For the 5 missense variants, PolyPhen-2 (19) predicted 2 variants as possibly damaging; one was in exon 4 (c.447A>C) with an amino acid change from glutamine to histidine (Q149H) and another one in exon7 (c.823G>A) with an amino acid change from alanine to threonine (A275T). One variant was predicted as probably damaging; it was in exon16 (c.1849C>T) with an amino acid change from arginine to cysteine (R617C). The R617C was reported with a minor allele frequency of 0.002 in a North American Caucasian population (Exome Variant Server, National Heart, Lung, and Blood Institute Exome Sequencing Project, Seattle, Washington (http://evs.gs.washington.edu/EVS/; accessed July 2012). Two variants (V512I and H875R) were predicted as benign; however, the H875R was predicted with a PolyPhen PISC score of 1.175, implying borderline functional impact. We also performed computer analysis using the SIFT algorithm (20); only the R617C was predicted as intolerant (Table 2).
Each of the 5 missense variants was found in only 1 family each (Table 2). We sequenced DNAs from all available additional family members (affected and nonaffected) in each family for the unique variant and found that 4 variants (Q149H, A275T, R617C, and H875R) cosegregated with the PTC phenotype in the relevant family and were inherited with the risk haplotype (Figure 2). Because the V512I was consistently predicted as benign and not shared by the affected individuals in family 4 (Supplemental Figure 3), we did not include the V512I in the further studies. Clinical information regarding the affected individuals in the 4 families shown in Figure 2 is provided in Supplemental Table 4. Paraffin thyroid tissue blocks from 3 patients (family 2-III-1, family 13-II-3, and family 20-II-2) were available. We analyzed the loss of heterozygosity by studying paired nontumor and tumor DNA samples from these patients using a SNaPshot assay (Life Technologies) as illustrated in Supplemental Figure 4. At least 3 of 6 SNP markers were informative for each sample. No loss of heterozygosity was observed (Supplemental Table 5).
To assess the frequency of the 4 missense variants in sporadic PTC cases and healthy controls, we screened sample sets from the Ohio and Polish populations. The Q149H and A275T variants were detected neither in 367 cases and 552 controls from Ohio or in 432 cases and 424 controls from Poland. Variant R617C was detected in 4 of 742 sporadic PTC cases from Ohio (0.54%) but in none of the 828 controls. In the 2 Polish sample sets combined, the R617C variant was found in 7 of 2644 PTC cases (0.26%) and 7 of 2567 controls (0.27%). All the individuals with the R617C variant were heterozygous. Variant H875R was detected in 4 of 367 (11%) Ohio sporadic PTC cases (minor allele frequency 0.005) and in 8 of 463 (17%) Ohio controls (minor allele frequency, 0.009). The frequency of H875R did not show any significant difference between the cases and controls (χ2 test, P = .637).
Missense variants affect SRGAP1 function
SRGAP1 regulates the small G-protein CDC42 in a Slit-Robo-dependent manner in neurons and affects cell motility (21). The Q149H and A275T changes are located in a Fes/CIP4 homology (FCH) domain, the R617C is located in the GAP domain and the H875R is located at the C-terminal end of the SRGAP1 protein (Figure 3A). To evaluate the linkage disequilibrium of these variants, SRGAP1 haplotypes were analyzed using HapMap data (Supplemental Figure 5). Incomplete linkage disequilibrium of the A275T, R617C, H875R, and SNP rs2168411 does occur. The amino acids in these positions in wild-type SRGAP1 are highly conserved in other SRGAP family members as well as in other species (Supplemental Figure 6). The Q149H and R617C mutations were also predicted to alter the protein structure of the FCH and GAP domains, respectively (Supplemental Figure 7).
Figure 3.
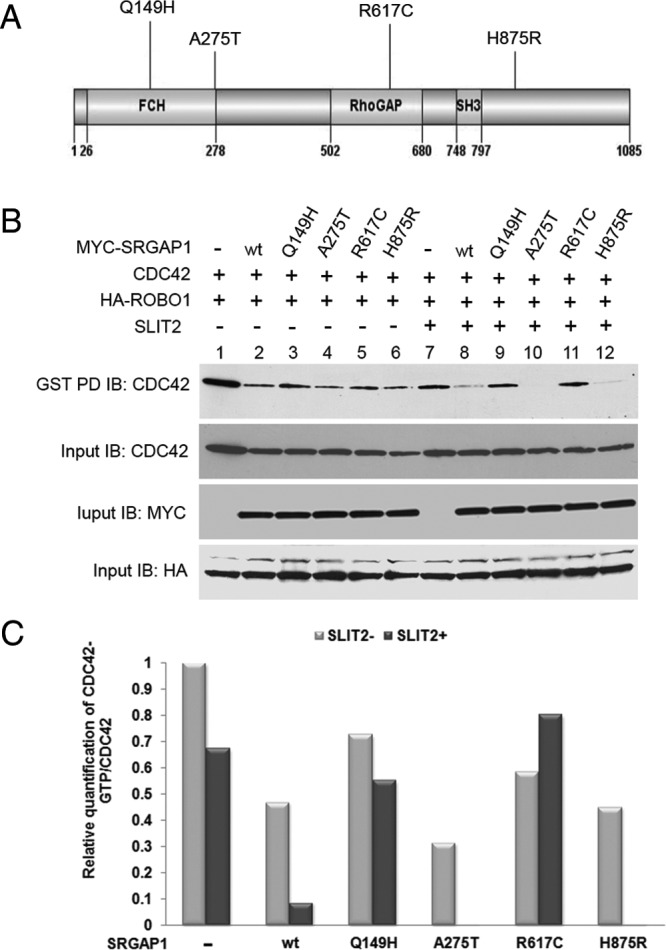
Regulation of CDC42 by SRGAP1 and its mutants. A, Schematic drawing indicating the mutation positions in the SRGAP1 protein. The domain regions are shown based on National Center for Biotechnology Information reference sequence NP_065813.1. B, SW1736 cells were cotransfected with HA-ROBO1 and with either empty-MYC vector or MYC-SRGAP1 wild-type (wt) or mutants (Q149H, A275T, R619C). After 48 hours, cells were either left untreated (−) or were treated with conditioned medium from HEK 293 cells overexpressing HIS-SLIT2 (+). GST fusion proteins of PBD-conjugated beads were used to pull down GTP-bound forms of the CDC42 GTPase. After GST pulldown (PD), CDC42 GTPase was detected using anti-CDC42 antibodies. Whole-cell lysates were also immunoblotted with anti-MYC, anti-HA, or anti-CDC42 antibody (input controls). C, Data from the experiments above were quantified using ImageJ software (http://rsb.info.nih.gov/ij/).
To test the potential impact of the SRGAP1 variants, expression constructs encoding the wild-type and the 4 mutant proteins were generated and each separately transfected in combination with ROBO1 and CDC42 expression vectors into the thyroid cancer cell line SW1736 (Figure 3B). Although all the proteins had some basal CDC42-GAP activity (Figure 3B, lanes 2–6), the addition of the ROBO1 ligand SLIT-2 to the transfected cells selectively enhanced the CDC42-GAP activity of wild-type and the A275T and H875R proteins; the active CDC42 levels were reduced (Figure 3B, lanes 8, 10, and 12). In contrast, CDC42-GAP activity was severely reduced by the Q149H and R617C constructs; the active CDC42 levels were similar to the lane without SRGAP1 (Figure 3B, lanes 7, 9, and 11). These observations were confirmed in the HEK 293 cells using the same experimental approaches (Supplemental Figure 8, A and B). The coimmunoprecipitation assay indicated that wild-type SRGAP1, and all 4 mutant SRGAP1 showed similar binding with ROBO1 in HEK 293 cells, implying that the 4 mutations probably did not affect the interaction between SRGAP1 and ROBO1 (Supplemental Figure 8C).
Discussion
By genome-wide linkage analysis of PTC families, we first disclosed a susceptibility locus in chromosome 12q14 that had not been reported before. This finding, together with reports of other PTC loci, supports the concept of genetic heterogeneity in PTC (5, 9, 11–13). Previously we reported the 8q24 locus through the linkage analysis with 26 families (13). In this study, 9 of 38 families were compatible with both 8q24 and 12q14 loci. It is not uncommon that multiple linkage loci are present in 1 cancer family. The 8q24 and 12q14 (and other undisclosed) loci may jointly contribute to predisposition to PTC. Linkage analysis in this study included 6 small families with only 2 members affected. Despite concerns that these families might unduly skew the results, the chromosome 12 linkage plots in the presence and absence of these 6 families (Supplemental Figure 2) were very similar. The SRGAP Q149H missense mutation was found in 1 of the small families (Family 2). Association studies targeted to 12q14 in cohorts of sporadic PTC cases/controls pinpointed a SNP in SRGAP1. The association was modest (OR 1.2) with a P = .0008, implying that this SNP was unlikely to be causative. Indeed, Sanger DNA resequencing revealed 4 germline missense variants in PTC families that appeared to display at least medium-high penetrance based on the present limited data. This scenario fits the common disease, rare allele concept (22, 23).
The fact that variants A275T and Q149H found in just 1 family each did cosegregate with the PTC phenotype yet were not found in some 800 sporadic cases suggests that they are extreme examples of rare or ultrarare variants contributing to the genetic predisposition. For the R617C and H875R variants, the data suggest a different scenario. The R617 and H875R were found in 1 family each in which they cosegregated with disease. However, in Ohio the R617C was also seen in 4 of 742 sporadic cases but in none of the 828 controls, suggesting a role as a low-penetrance mutation. Remarkably in Poland R617C occurred in 7 of 2644 cases and 7 of 2567 controls. This apparent contradiction may well be a clue to the nature of this variation. One explanation is that the phenotype arises only or preferably in individuals who harbor a second hit that could be another germline change and that this putative variant has different allele frequencies in Ohio vs Poland. It could also be an environmental factor that affects populations differently. The H875R was seen in Ohio sporadic PTC and controls with a similar frequency, implying a weak or nonfunctional variant. We note that of the 21 families whose linkage results were compatible with linkage to 12q14, only a few are accounted for by the variants in SRGAP1 that we describe. This is best explained by assuming that other variants may be present in the SRGAP1 gene or its vicinity, acting in cis on SRGAP1. In the public database, numerous SNPs occur in the genomic region of SRGAP1. In the 21 familial PTC patients, we identified numerous previously reported and unreported SNPs, some of them located in the promoter region and in the 3′UTR. Potential microRNA binding sites were found in the 3′ UTR. Functional investigation of these DNA polymorphisms is warranted.
The results of biochemical analyses demonstrated that 2 of the 4 germline variants found in thyroid cancer cases decrease the GAP activity of SRGAP1. The R167C mutation lies directly within the GAP domain, whereas the Q149H mutation lies in the FCH domain. The FCH domain is involved in protein-protein interactions and thus could affect the recruitment of cofactors necessary for SRGAP1 function (24, 25). The rho GTPases are implicated in almost every fundamental cellular process (26). Several rho GAP domain-containing proteins have been shown to be cancer related (27, 28). For example, the deleted in liver cancer 1 (DLC1) gene encodes rho GTPase-activating protein and plays tumor suppressor roles (28). Germline missense and nonsense mutations in the DLC1 gene were detected in colon and prostate cancer patients. Our findings again emphasize the importance of rho GTPase domains in tumorigenesis.
We speculate that at least 2 missense variants in SRGAP1 (Q149H and R617C) could be loss-of-function-type changes affecting CDC42 activity. CDC42 acts as a signal transduction convergence point in intracellular signaling networks, mediates multiple signaling pathways, and plays a role in tumorigenesis (29, 30). Functional analysis using microarrays suggested that the activation of the CDC42/p21-activated kinase signaling plays a role in invasive PTC (31). The well-defined function of CDC42 in cell proliferation, survival, and migration provides an attractive molecular explanation for the genetic data. Taken together, we propose SRGAP1 as a potential low-penetrant susceptibility gene in PTC.
Acknowledgments
We thank Dr Ramesh Ganju for generously providing the Robo-1 and Slit-2 expression constructs and Dr Rebecca Schweppe for the cell line SW1736. We also thank Jan Lockman for technical support; the Ohio State University Comprehensive Cancer Center Microarray Shared Resource for the SNP genotyping; the Ohio State University Comprehensive Cancer Center Nucleic Acid Shared Resource for the SNP and microsatellite marker genotyping and sequencing, and the Biomedical Genomics Core of The Research Institute at Nationwide Children's Hospital (Columbus, Ohio) for targeted deep sequencing. Tissue samples were provided by the Cooperative Human Tissue Network, which is funded by the National Cancer Institute.
This work was supported by National Cancer Institute Grants P30CA16058 and P01CA124570 and Grants N N402 193740 and N N519 579938 from the Polish National Science Center.
Disclosure Summary: M.R. has previously been on a clinical advisory board for Veracyte. The other authors have no potential conflicts of interest.
Footnotes
- CDC42
- cell division cycle 42
- CI
- confidence interval
- FCH
- Fes/CIP4 homology
- GST
- glutathione-S-transferase
- HA
- hemagglutinin
- HEK
- human embryonic kidney
- NPL
- nonparametric linkage
- OR
- odds ratio
- PPL
- posterior probability of linkage
- PTC
- papillary thyroid carcinoma
- SLIT2
- slit homolog 2 (Drosophila)
- SNP
- single-nucleotide polymorphism
- SRGAP1
- Slit-Robo Rho GTPase activating protein 1 gene
- UTR
- untranslated region.
References
- 1. Yu GP, Li JC, Branovan D, McCormick S, Schantz SP. Thyroid cancer incidence and survival in the national cancer institute surveillance, epidemiology, and end results race/ethnicity groups. Thyroid. 2010;20:465–473 [DOI] [PubMed] [Google Scholar]
- 2. Albores-Saavedra J, Henson DE, Glazer E, Schwartz AM. Changing patterns in the incidence and survival of thyroid cancer with follicular phenotype—papillary, follicular, and anaplastic: a morphological and epidemiological study. Endocr Pathol. 2007;18:1–7 [DOI] [PubMed] [Google Scholar]
- 3. Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003;63:1454–1457 [PubMed] [Google Scholar]
- 4. Ciampi R, Nikiforov YE. RET/PTC rearrangements and BRAF mutations in thyroid tumorigenesis. Endocrinology. 2007;148:936–941 [DOI] [PubMed] [Google Scholar]
- 5. Vriens MR, Suh I, Moses W, Kebebew E. Clinical features and genetic predisposition to hereditary nonmedullary thyroid cancer. Thyroid. 2009;19:1343–1349 [DOI] [PubMed] [Google Scholar]
- 6. Goldgar DE, Easton DF, Cannon-Albright LA, Skolnick MH. Systematic population-based assessment of cancer risk in first-degree relatives of cancer probands. J Natl Cancer Inst. 1994;86:1600–1608 [DOI] [PubMed] [Google Scholar]
- 7. Czene K, Lichtenstein P, Hemminki K. Environmental and heritable causes of cancer among 9.6 million individuals in the Swedish family-cancer database. Int J Cancer. 2002;99:260–266 [DOI] [PubMed] [Google Scholar]
- 8. Dong C, Hemminki K. Modification of cancer risks in offspring by sibling and parental cancers from 2,112,616 nuclear families. Int J Cancer. 2001;92:144–150 [PubMed] [Google Scholar]
- 9. Malchoff CD, Sarfarazi M, Tendler B, et al. Papillary thyroid carcinoma associated with papillary renal neoplasia: genetic linkage analysis of a distinct heritable tumor syndrome. J Clin Endocrinol Metab. 2000;85:1758–1764 [DOI] [PubMed] [Google Scholar]
- 10. Suh I, Filetti S, Vriens MR, et al. Distinct loci on chromosome 1q21 and 6q22 predispose to familial nonmedullary thyroid cancer: A SNP array-based linkage analysis of 38 families. Surgery. 2009;146:1073–1080 [DOI] [PubMed] [Google Scholar]
- 11. McKay JD, Lesueur F, Jonard L, et al. Localization of a susceptibility gene for familial nonmedullary thyroid carcinoma to chromosome 2q21. Am J Hum Genet. 2001;69:440–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cavaco BM, Batista PF, Sobrinho LG, Leite V. Mapping a new familial thyroid epithelial neoplasia susceptibility locus to chromosome 8p23.1-p22 by high-density SNP genome-wide linkage analysis. J Clin Endocrinol Metab 2008;93(11):4426–4430 [DOI] [PubMed] [Google Scholar]
- 13. He H, Nagy R, Liyanarachchi S, et al. A susceptibility locus for papillary thyroid carcinoma on chromosome 8q24. Cancer Res. 2009;69:625–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Canzian F, Amati P, Harach HR, et al. A gene predisposing to familial thyroid tumors with cell oxyphilia maps to chromosome 19p13.2. Am J Hum Genet. 1998;63:1743–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gabriel S, Ziaugra L, Tabbaa D. SNP genotyping using the Sequenom MassARRAY iPLEX platform. Curr Prot Hum Genet. 2009;60:2.12.1–2.12.16 [DOI] [PubMed] [Google Scholar]
- 16. Vieland VJ, Huang Y, Seok SC, et al. KELVIN: a software package for rigorous measurement of statistical evidence in human genetics. Hum Hered. 2011;72:276–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matise TC, Chen F, Chen W, et al. A second-generation combined linkage physical map of the human genome. Genome Res. 2007;17:1783–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schweppe RE, Klopper JP, Korch C, et al. Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. J Clin Endocrinol Metab. 2008;93:4331–4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protocols. 2009;4:1073–1081 [DOI] [PubMed] [Google Scholar]
- 21. Wong K, Ren XR, Huang YZ, et al. Signal transduction in neuronal migration: roles of GTPase activating proteins and the small GTPase Cdc42 in the Slit-Robo pathway. Cell. 2001;107:209–221 [DOI] [PubMed] [Google Scholar]
- 22. Meindl A, Hellebrand H, Wiek C, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet. 2010;42(5):410–414 [DOI] [PubMed] [Google Scholar]
- 23. Schork NJ, Murray SS, Frazer KA, Topol EJ. Common vs. rare allele hypotheses for complex diseases. Curr Opin Genet Dev. 2009;19:212–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ahmed S, Bu W, Lee RT, Maurer-Stroh S, Goh WI. F-BAR domain proteins: Families and function. Commun Integr Biol. 2010;3:116–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ghose A, Van Vactor D. GAPs in Slit-Robo signaling. Bioessays. 2002;24:401–404 [DOI] [PubMed] [Google Scholar]
- 26. Moon SY, Zheng Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003;13:13–22 [DOI] [PubMed] [Google Scholar]
- 27. Kandpal RP. Rho GTPase activating proteins in cancer phenotypes. Curr Protein Pept Sci. 2006;7:355–365 [DOI] [PubMed] [Google Scholar]
- 28. Kim TY, Vigil D, Der CJ, Juliano RL. Role of DLC-1, a tumor suppressor protein with RhoGAP activity, in regulation of the cytoskeleton and cell motility. Cancer Metastasis Rev. 2009;28:77–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Etienne-Manneville S. Cdc42—the centre of polarity. J Cell Sci. 2004;117:1291–1300 [DOI] [PubMed] [Google Scholar]
- 30. Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett. 2008;582:2093–2101 [DOI] [PubMed] [Google Scholar]
- 31. Vasko V, Espinosa A, Scouten W, et al. Gene expression and functional evidence of epithelial-to-mesenchymal transition in papillary thyroid cancer invasion. Proc Natl Acad Sci USA. 2007;104:2803–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]