

Abstract
Context:
The complexity of genetic testing in Kallmann syndrome (KS) is growing and costly. Thus, it is important to leverage the clinical evaluations of KS patients to prioritize genetic screening.
Objective:
The objective of the study was to determine which reproductive and nonreproductive phenotypes of KS subjects have implications for specific gene mutations.
Subjects:
Two hundred nineteen KS patients were studied: 151 with identified rare sequence variants (RSVs) in 8 genes known to cause KS (KAL1, NELF, CHD7, HS6ST1, FGF8/FGFR1, or PROK2/PROKR2) and 68 KS subjects who remain RSV negative for all 8 genes.
Main Outcome Measures:
Reproductive and nonreproductive phenotypes within each genetic group were measured.
Results:
Male KS subjects with KAL1 RSVs displayed the most severe reproductive phenotype with testicular volumes (TVs) at presentation of 1.5 ± 0.1 mL vs 3.7 ± 0.3 mL, P < .05 vs all non-KAL1 probands. In both sexes, synkinesia was enriched but not unique to patients with KAL1 RSVs compared with KAL1-negative probands (43% vs 12%; P < .05). Similarly, dental agenesis and digital bone abnormalities were enriched in patients with RSVs in the FGF8/FGFR1 signaling pathway compared with all other gene groups combined (39% vs 4% and 23% vs 0%; P < .05, respectively). Hearing loss marked the probands with CHD7 RSVs (40% vs 13% in non-CHD7 probands; P < .05). Renal agenesis and cleft lip/palate did not emerge as statistically significant phenotypic predictors.
Conclusions:
Certain clinical features in men and women are highly associated with genetic causes of KS. Synkinesia (KAL1), dental agenesis (FGF8/FGFR1), digital bony abnormalities (FGF8/FGFR1), and hearing loss (CHD7) can be useful for prioritizing genetic screening.
Kallmann syndrome (KS), ie, hypogonadotropic hypogonadism accompanied by anosmia, represents a unique form of isolated GnRH deficiency wherein the developing GnRH neurons along with the olfactory axons over which the GnRH neurons must traverse to inhabit their ultimate anatomic home within the hypothalamus, fail to migrate into the central nervous system (1–3). In the last 3 decades, mutations in several genes/pathways affecting GnRH neuronal migration (neurodevelopmental genes) have been identified in KS patients, namely the following: 1) KAL1 [Xp22.3]; 2) FGF8 [10q24.3] and FGFR1 [8p11.2-p11.1] (fibroblast growth factor [FGF] signaling pathway); 3) PROK2 [3p13] and PROKR2 [20p12.3] (PROK signaling pathway); 4) NELF [9q34.3] (Reference 4 and references within); 5) CHD7 [8q12.1-q12.2] (5, 6); 6) WDR11 [10q26.12] (7); 7) HS6ST1[2q14.3] (8); and 8) SEMA3A [7q21.11] (9).
This growing genetic and locus heterogeneity in KS pose considerable challenges to clinicians in the diagnostic workup of these patients, in their prioritization of genetic screening, and in providing optimal genetic counseling to patients and their families. Genetic information such as mode of inheritance, frequency of individual gene mutations, and regions of homozygosity in consanguineous/endogamous families can help target genetic screening in KS subjects. However, given the nature of the reproductive failure inherent to this condition, probands often present as sporadic cases (10) and/or the affected families are often too small to allow an accurate assessment of their inheritance pattern. In addition, although some genes show a clear pattern of inheritance (eg, KAL1: X linked) others show varied inheritance patterns with incomplete penetrance (CHD7, FGF8/FGFR1, PROK2/PROKR2). The above caveats emphasize the need for identifying those phenotypic features that can be also used to guide cost-effective prioritization of genetic testing.
Given the importance of these genes in the biology of other organ systems in addition to their critical role in the ontogeny of the GnRH neurons, it is conceivable that variations in the expression of each of these gene/pathways in other organ systems could result in unique clinical phenotypes that provide clues to their identity. For example, it has been reported that patients with KAL1 mutations have a severe reproductive phenotype, whereas those with FGFR1 mutations vary more widely in the severity of their GnRH deficiency (11, 12). Unilateral renal agenesis and synkinesia (mirror movements) have been suggested as unique phenotypes of patients with KAL1 gene defects (13–15), whereas skeletal anomalies, midline facial defects, and dental agenesis have been reported to be specific to FGF8/FGFR1 patients (16, 17). Likewise, obesity and sleeping disorders have been identified in some PROK2/PROKR2 patients and have been suggested to be phenotypic markers for mutations in this pathway (18). However, because of the rarity of KS, previous studies of this condition have typically involved relatively small cohorts of subjects who were not systematically phenotyped and in whom genetic testing was often limited.
To address this need, the present study performed a detailed comparative phenotypic evaluation in a large group of KS subjects harboring known rare sequence variations (RSVs) (genetic variants < 1% minor allele frequency in control populations) in 8 genes in 6 pathways [KAL1, the FGF (FGF8 and FGFR1), and PROK signaling pathways (PROK2 and PROKR2), CHD7, NELF, and HS6ST1]. We hypothesized that each of these 6 signaling pathways would exhibit specific reproductive and/or nonreproductive hallmarks that might permit some prioritization of genetic screening thus providing economically efficient guidance for genetic testing. We also performed a comparative phenotypic analysis in a cohort of KS patients exhibiting no demonstrable RSVs to contrast these groups and to identify those phenotypic features that might be specific to novel but as-yet-undiscovered KS genes and pathways.
Materials and Methods
Subjects
We studied 219 KS probands (ethnicity: 88% Caucasian; 3% African American; 6% Asian; and 3% mixed) selected from a cohort of 568 probands participating in a genetic study at the Reproductive Endocrine Unit of the Massachusetts General Hospital. The final assembled study population included 162 male KS probands (mean age ± SEM of assessment in our unit 25 ± 1.1 years) and 57 females (mean age at assessment in our unit: 29 ± 1.6 years) with the following diagnostic criteria: 1) a clinical diagnosis of KS characterized by absent or incomplete puberty by age 18 years, low sex steroid levels in the presence of low or normal gonadotropins, and anosmia or hyposmia; 2) otherwise normal anterior pituitary function biochemically: 3) with no demonstrable neuroanatomic or functional cause of hypogonadotropic hypogonadism (19); and 4) normal ferritin levels. Subjects included KS probands who were positive for an RSV (n = 151) in 1 of the 8 neurodevelopmental genes in 6 biological pathways and 68 consecutive KS probands who were negative for those genes/pathways. (see genetic groups below). Genetic variants identified in this study are termed as RSVs rather than mutations because the determination of an increasing number of these variants was based on statistical criteria because data on functional cellular consequence was not always available. An RSV was defined as follows: 1) a variant affecting splice junctions within 10 bp of coding sequence or if the variant was a protein-altering/protein-truncating nonsynonymous variant; and 2) present in less than 1% minor allele frequency (MAF) in the 98–262 control patients studied in our unit (see Supplemental Table 1, published on The Endocrine Society's Journals Online web site at http://jcem.endojournals.org) as well as less than 1% MAF in the National Heart, Lung, and Blood Institute Exome Sequencing Project [http://evs.gs.washington.edu/EVS/] consisting of 6503 individuals from Caucasian and African American ethnicity.
Genetic analysis
Genomic DNA was obtained from peripheral blood samples by standard phenol-chloroform extraction. Exonic and proximal intronic (≤15 bp from splice sites) DNA sequences of the following genes were amplified by PCR and determined by direct sequencing: KAL1 [online inheritance in man (OMIM) 308700], CHD7 (OMIM 608892), NELF (OMIM 608137), FGF8 (OMIM 600483), FGFR1 (OMIM 136350), PROK2 (OMIM 607002), PROKR2 (OMIM 607212), and HS6ST1 (OMIM 1 604846). The WDR11 and SEMA3A genes were not screened. The PCR primers and amplification conditions for each gene have been previously published (8, 20).
Genetic study groups
A total of 151 RSV positive Kallmann subjects were included in this study (116 males and 35 females) (see Figure 1 and Supplemental Table 2 for more details) of whom 109 underwent complete genetic screening for the 6 neurodevelopmental pathways (8 genes); 42 subjects had information missing on 1 or more neurodevelopmental genes (19 subjects had only 1 missing gene, 10 had 2 missing genes, and 13 had > 2 missing genes) (see Supplemental Table 3). Oligogenic probands were excluded unless they exhibited RSVs in both a ligand and receptor pair, ie, FGF8/FGFR1 or PROK2/PROKR2, to enable us to characterize phenotypic features associated with a specific signaling pathway. Therefore, as shown in Figure 1 and Supplemental Table 2, the final study group was as follows: 1) the KAL1 group: probands with an isolated RSV in KAL1; 2) the FGF signaling pathway group: probands with isolated RSVs in either FGF8 or FGFR1 and also subjects with FGF8 and FGFR1 RSVs in the same proband (1 digenic proband); 3) the PROK signaling pathway group: probands with isolated RSVs in either PROK2 or PROKR2 and also subjects with PROK2 and PROKR2 RSVs in the same proband (1 digenic proband); the 4) NELF group: probands with isolated RSV in NELF; the 5) HS6ST1 group: probands with isolated RSV in HS6ST1; the 6) CHD7 group: probands with isolated RSV in CHD7; and 7) the gene-negative group [n = 68 (46 males and 22 females)] consisted of probands without any KS-associated gene RSVs described above (8 neurodevelopmental genes). Phenotypic information on the 2 subjects with isolated NELF RSVs was incomplete, and hence, the NELF group was excluded from further comparative analysis.
Figure 1.

Number of KS probands with rare sequence variants in each of the neurodevelopmental genetic groups studied.
Clinical parameters
Phenotypic information was derived from clinical charts of patients, notes from referring physicians, and patient questionnaires. Phenotypes were aggregated and analyzed as described below.
Reproductive phenotypes
Neonatal markers and pubertal development.
The reproductive phenotypes of male subjects were classified as either absent or partial puberty as previously described (19). A history of cryptorchidism and micropenis at infancy (stretched penile length < 2.5 cm) (21, 22) were also collected; none of these infants had received sex steroid therapy. The data on the testicular volume (TV) (prepubertal TV < 4 mL and pubertal TV ≥ 4 mL) were included for analysis independent of prior androgen exposure. However, TV data from the patients who had previously received gonadotropins/GnRH were excluded. In the female subjects, partial puberty was defined as evidence of some degree of spontaneous breast development in the absence of sex steroid treatment (20).
Responses to exogenous pulsatile GnRH treatment.
In KS subjects treated with pulsatile GnRH therapy, we reviewed their response to physiological doses (2). In men, a positive outcome in response to long-term pulsatile GnRH (longer than 12 months) was defined as achieving normalized serum total T levels (>270 ng/dL), testicular growth, and development of sperm in the ejaculate (23). For women, a positive outcome was defined as evidence of ovulation by luteal phase progesterone levels during exogenous pulsatile GnRH therapy (24). In both sexes, a positive response was noted independent of dose and/or duration of therapy.
Spontaneous reversibility in men.
The definitive criterion for sustained reversal of KS was the ability to achieve a normal adult endogenous serum T level (>270 ng/dL) after the discontinuation of hormonal therapy (GnRH, gonadotropins, or androgens) as was previously reported (25). Reversibility was not assessed in the female subjects.
Nonreproductive phenotypes
The 40-item University of Pennsylvania Smell Identification Test (Sensonics Inc, Haddon Heights, New Jersey) was used for an olfactory evaluation whenever possible. Self-reported anosmia was also included because this history has been validated as a reliable indicator of an objective olfactory deficit (26). Renal abnormalities ascertained by transabdominal ultrasound were recorded whenever available. Other nonreproductive phenotypes such as synkinesia, cleft lip and palate, dental agenesis, hearing loss, or skeletal anomalies (scoliosis/kyphosis, syndactyly, polydactyly, camptodactyly, clinodactyly, excessive joint mobility, short fourth metacarpal bones, foreshortened limb bones, and flat feet) were ascertained from clinical, evaluations, records, and questionnaire data. Probands specifically referred with an initial diagnosis of CHARGE syndrome (coloboma, heart defects, choanal atresia, retardation of growth, genital anomalies, and ear abnormalities) (27) were excluded from this study. Four nonreproductive CHARGE features (coloboma, heart defects, choanal atresia, and ear abnormalities) were retrieved from previously completed questionnaire data.
Statistical analysis
Data for quantitative features (body mass index and TV) are expressed as the mean ± SEM. To ascertain the enrichment of specific phenotypic features in each genetic group, the proportion of probands exhibiting the phenotype was compared between those who were RSV positive for a specific gene (eg, RSV positive for KAL1) vs all probands who were negative for the specific gene (eg, non-KAL1 probands). Therefore, a Fisher exact 2 × 2 test followed by a Bonferroni-Holms multiple comparison correction was used for each qualitative phenotype. For quantitative phenotypes, a similar approach was used applying a nonparametric Mann-Whitney test followed by Bonferroni-Holms correction. A Bonferroni-Holms corrected P < .05 was considered statistically significant. We also calculated the unadjusted odds ratio (OR) and 95% confidence intervals (CIs) for the association between select features previously reported to be clinically significant in specific KS genes (cleft lip/palate, synkinesia, hearing loss, syndactyly, renal agenesis, and dental agenesis). The data analysis of the OR calculation is shown in detail in Supplemental Table 4.
Results
Genetic groups
All comparative genetic groups are detailed in Figure 1 and Supplemental Table 2. Additional information on MAF, previous functional status, and predicted functional consequence by in silico programs for all RSVs included in this study is provided in Supplemental Table 1.
Reproductive phenotypes (Table 1 and Figure 1)
Table 1.
Prevalence of Reproductive Phenotypes Across Genetic Groups
| Reproductive Phenotypes | KAL1 | FGF8/FGFR1 | PROK2/PROKR2 | CHD7 | HS6ST1 | Gene-Negative Group |
|---|---|---|---|---|---|---|
| Testicular volume, mL, mean ± SEM, n | 1.5 ± 0.1 (18) |
3.6 ± 0.6 (26) |
3.0 ± 0.6 (17) |
1.7 ± 0.2 (9) |
6.8 ± 2.8 (3) |
4.3 ± 0.6 (38) |
| Cryptorchidism | 56% (15/27) |
51% (17/33) |
27% (6/22) |
82% (9/11) |
0 (0/4) |
41% (17/41) |
| Micropenis | 25% (7/28) |
32% (10/31) |
37% (6/16) |
36% (4/11) |
25% (1/4) |
25% (9/36) |
| Puberty, males | ||||||
| Absent | 96% (25/26) |
69% (20/29) |
77% (17/22) |
100% (11/11) |
33% (1/3) |
52% (23/44) |
| Partial | 4% (1/26) |
31% (9/29) |
23% (5/22) |
0% (0/11) |
67% (2/3) |
48% (21/44) |
| Puberty, females | ||||||
| Absent | 0% 0% |
50% (6/12) |
75% (4/5) |
78% (7/9) |
ND ND |
62% (10/16) |
| Partial | 100% (2/2) |
50% (6/12) |
25% (1/5) |
22% (2/9) |
ND ND |
38% (6/16) |
| Positive outcome to GnRH therapy, males | 50% (3/6) |
100% (5/5) |
100% (5/5) |
100% (1/1) |
ND ND |
78% (14/18) |
| Positive outcome to GnRH therapy, females | 100% (1/1) |
100% (2/2) |
ND ND |
100% (2/2) |
ND ND |
78% (7/9) |
| Male reversal | 0% (0/8) |
20% (2/10) |
0% (0/7) |
0% (0/3) |
50% (1/2) |
16% (6/38) |
Abbreviation: ND, no data available. For qualitative phenotypes, numerator in parentheses characterizes the number of probands that are positive to phenotypic feature. Denominator represents the total number of probands with available information.
Neonatal markers of GnRH deficiency
Both micropenis and cryptorchidism were present in similar frequency across all genetic groups.
Pubertal status at presentation
Nearly 96% of male KS subjects with KAL1 RSVs presented with absent puberty compared with 66% without KAL1 RSVs [P < .05, OR 13 (95% CI 1.7–99)]. The mean TV was prepubertal in the vast majority of KS patients across all RSV-positive groups (Figure 2A). However, when comparing the percent of males who harbored a specific gene RSV with those who were negative for the respective gene, KAL1 emerged as the group with the most severe pubertal presentation (TV: 1.5 ± 0.1 mL vs 3.7 ± 0.3 mL, KAL1 vs non-KAL1 probands, respectively, P < .05) (Figure 2B), with the other groups demonstrating a considerably more variable distribution. Among the remaining groups, the CHD7 RSV group showed a similar trend toward a severe prepubertal presentation with a mean TV of 1.7 ± 0.2 mL, although this did not reach statistical significance (P = .1). As previously reported, female KS subjects harbored variants in most of the neurodevelopmental genes (20). However, the severity of pubertal presentation in female KS subjects was not different between the genetic groups.
Figure 2.
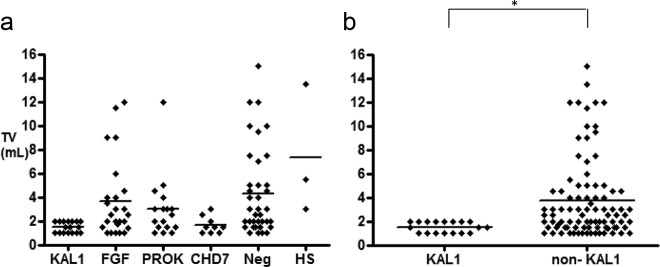
Pubertal severity at presentation in male KS probands. A, Individual values of TV at presentation in male probands within each gene/signaling group. Prepubertal TV less than 4 mL and pubertal TV of 4 mL or greater. KAL1, KAL1; FGF, FGF8/FGFR1 signaling pathway; PROK, PROK2/PROKR2 signaling pathway; CHD7, CHD7; Neg, gene–negative group; HS, HS6ST1. B, Comparison of individual TV at presentation between probands harboring KAL1 mutations (KAL1 group) vs probands who were negative for KAL1 mutations (non-KAL1 group). *P < .05
Response to exogenous pulsatile GnRH therapy
The response to exogenous GnRH therapy could be evaluated in 35 male patients. All PROK2/PROKR2 and FGF8/FGFR1 subjects responded positively to GnRH therapy as did the only CHD7 male who received pulsatile GnRH therapy. In contrast, both the KAL1 RSV group and the gene-negative group had a more variable response to GnRH (50% and 78% responding favorably to therapy, respectively). Fourteen female probands in this cohort underwent pulsatile GnRH therapy. Although the 5 RSV-positive females from the KAL1, FGF8/FGFR1, and CHD7 groups responded positively to GnRH therapy, 22% of gene-negative females had an unfavorable response. No statistical difference was observed between the groups in either sex for this phenotype.
Male reversibility
Long-term clinical follow-up was available in 68 male subjects. Three subjects (2 with FGFR1 heterozygous variants and 1 with a HS6ST1 heterozygous variant) underwent reversal. No KAL1 or PROK2/PROKR2 subjects showed evidence of reversal. In the gene-negative group, 16% (6 of 38) of subjects displayed a reversal. No significant difference in the occurrence of reversal was seen between the gene/pathway groups.
Nonreproductive phenotypes (Table 2 and Figure 3)
Table 2.
Prevalence of Nonreproductive Phenotypes Across Genetic Groups
| Non Reproductive Phenotypes | KAL1 | FGF8/FGFR1 | PROK2/PROKR2 | CHD7 | HS6ST1 | Gene-Negative Group |
|---|---|---|---|---|---|---|
| BMI, mean ± SEM (n) | 27 ± 1.5 (19) |
25.5 ± 0.9 (38) |
28 ± 2.2 (18) |
23.5 ± 1.3 (17) |
25 ± 3.8 (4) |
26 ± 0.7 (52) |
| Olfactory function | ||||||
| Hyposmic | 3% (1/38) |
30% (16/54) |
6% (2/31) |
18% (4/22) |
50% (2/4) |
34% (23/68) |
| Anosmic | 97% (37/38) |
70% (38/54) |
94% (29/31) |
82% (18/22) |
50% (2/4) |
66% (45/68) |
| Synkinesia | 43% (13/30) |
7% (3/44) |
26% (7/27) |
5% (1/20) |
0 (0/3) |
11% (6/53) |
| Dental agenesis | 0 (0/9) |
39% (13/33) |
0 (0/19) |
13% (2/15) |
0 (0/3) |
3% (1/36) |
| Sindactyly/polidactyly/camptodactyly | 0 (0/20) |
23% (8/35) |
0 (0/21) |
0 (0/20) |
0 (0/4) |
0 (0/55) |
| Hearing loss | 14% (3/21) |
16% (7/43) |
4% (1/24) |
40% (8/20) |
0 (0/4) |
11% (6/55) |
| Renal agenesis | 18% (3/17) |
0 (0/20) |
0 (0/16) |
0 (0/7) |
0 (0/4) |
17% (4/23) |
| Cleft lip/palate | 0 (0/30) |
11% (5/47) |
0 (0/26) |
9% (2/22) |
25% (1/4) |
6% (4/64) |
| Clinodactyly | 5% (1/20) |
9% (3/35) |
0 (0/21) |
5% (1/20) |
25% (1/4) |
4% (2/55) |
| Scoliosis/kyphosis | 10% (2/20) |
6% (2/35) |
10% (2/21) |
5% (1/20) |
0 (0/4) |
24% (13/55) |
| Excessive joint mobility | 10% (2/20) |
0 (0/35) |
10% (2/21) |
5% (1/20) |
0 (0/4) |
13% (7/55) |
| Short limb bones | 5% (1/20) |
11% (4/35) |
5% (1/21) |
10% (2/20) |
25% (1/4) |
2% (1/55) |
| Short fourth metatarsal | 10% (2/20) |
14% (5/35) |
5% (1/21) |
5% (1/20) |
0 (0/4) |
13% (7/55) |
| Flat feet | 5% (1/20) |
23% (8/35) |
29% (6/21) |
5% (1/20) |
50% (2/4) |
11% (6/55) |
| Congenital heart disease | 0 (0/9) |
0 (0/30) |
0 (0/14) |
6% (1/18) |
0 (0/3) |
0 (0/48) |
| External ear defect | 0 (0/10) |
0 (0/33) |
0 (0/17) |
0 (0/18) |
0 (0/4) |
0 (0/52) |
| Choanal atresia | 0 (0/11) |
0 (0/34) |
0 (0/16) |
5% (1/20) |
0 (0/4) |
0 (0/49) |
| Coloboma | 0 (0/11) |
3% (1/34) |
0 (0/16) |
0 (0/19) |
0 (0/4) |
2% (1/52) |
Abbreviation: BMI, body mass index. n indicates the total number of probands with measured BMI. For qualitative phenotypes, numerator in parentheses characterizes the number of probands that are positive to phenotypic feature. Denominator represents the total number of probands with available information.
Figure 3.

Prevalence of nonreproductive phenotypes and their relative enrichment within specific genetic groups. Six panels show the relative enrichment of synkinesia (A), dental agenesis (B), hearing loss (C), syndactyly/camptodactyly/polydactyly (D), renal agenesis (E), and cleft lip/palate (F) within specific genetic groups. In each panel, 2 histograms are shown: the larger histogram shows the percentage of probands with RSVs in a specific gene compared with probands negative for the gene (eg, in panel A, KAL1 probands vs non-KAL1 probands and likewise in panels B–F). The unadjusted OR for the enrichment of the relevant phenotype within the specific genetic group is shown in the top right hand corner of the panel. The smaller histogram in each panel displays the absolute percentage of probands displaying the phenotype within each individual genetic group. CHD7, CHD7; FGF, FGF8/FGFR1 signaling pathway; KAL1, KAL1; Neg, gene-negative group; non-KAL1, all probands without KAL1 mutations; non-FGF, all probands without FGF8/FGFR1 mutations; non-CHD7, all probands without CHD7 mutations; PROK, PROK2/PROKR2 signaling pathway. *P < .05 for KAL1 vs non-KAL1 (A); FGF vs Non-FGF (B and D); CHD7 vs Non-CHD7 (C).
Body mass index
Body mass index was similar in all 6 groups, and the results are summarized in Table 2.
Synkinesia
Although synkinesia was seen across most groups, KAL1 RSV patients had a significantly higher prevalence of this phenotype compared with non-KAL1 probands [43% vs 12%, respectively; P < .05 (OR 5.9; 95% CI 2.4–14)] (Figure 3A). Interestingly, the PROK2/PROKR2 group also had a considerable prevalence of this feature, although this did not reach statistical significance compared with non-PROK2/PROKR2 probands.
Dental agenesis
Dental agenesis was detected in the FGF8/FGFR1, CHD7, and gene-negative groups, but only the FGF8/FGFR1 signaling group was significantly enriched for this feature [39% (FGF) vs 4% (non-FGF), P < .05 (OR 17; 95% CI 4.5–66)] (Figure 3B).
Hearing loss
Hearing loss was seen across all groups, but a significantly higher incidence in the CHD7 group was observed compared with non-CHD7 probands [40% (CHD7) vs 13% (non-CHD7); P < .05 (OR 4.5; 95% CI 1.6–12)] (Figure 3C).
Bone/skeletal phenotypes
Digital bone abnormalities (polydactyly or syndactyly or campylodactyly) were detected only in the FGF8/FGFR1 probands [23% (FGF) vs 0% (non-FGF); P < .05 (OR 74; CI 4–1331)] (Figure 3D). Other bone phenotypes such as clinodactyly, scoliosis or kyphosis, excessive joint mobility, short fourth metacarpal bones, foreshortened limb bones, and flat feet were distributed similarly across all groups.
Renal abnormalities
Unilateral renal agenesis was detected in the KAL1 group as well as the gene-negative group, affecting both kidneys equally. However, no significant difference between the genetic groups was seen for renal agenesis, including for the KAL1 gene, which has been previously associated with this phenotype [18% (KAL1) vs 17% (non-KAL1), P = .52 (OR 3.5; 95% CI 0.7–17)] (Figure 3E). The only other noteworthy renal finding was a double ureter seen in a KS male with a missense heterozygous FGFR1 variant.
Cleft lip/palate
Cleft lip/palate was detected in FGF8/FGFR1, CHD7, HS6ST1, and gene-negative groups but without significant differences between the groups including for the FGF8/FGFR1 probands [11% (FGF) vs 5% (non-FGF probands), P = .51 (OR 2.2; 95% CI 0.7–8)] (Figure 3F).
Additional CHARGE features
Only 1 of 22 probands (5%) harboring CHD7 variants had additional CHARGE features (choanal atresia and congenital heart disease). Coloboma was present in 2 KS females without CHD7 variants (1 with FGFR1 RSV and 1 in the gene-negative group).
Further considerations: gene-negative group
In both sexes, KS subjects in the gene-negative group displayed variable reproductive phenotypes. Their mean TV at diagnosis was in the early pubertal ranges (4.3 ± 0.6 mL), and 38% of KS females had undergone thelarche (Table 1). All other ascertained nonreproductive phenotypic features were also present in this group except syndactyly and some CHARGE features (Table 2 and Figure 3).
Discussion
This is the largest study to date to perform comparative phenotypic analyses in a genetically well-characterized KS cohort for identifying phenotypic features that might be utilized for prioritization of genetic screening. Severe pubertal reproductive deficits marked the male KS subjects with KAL1 RSVs, whereas most other genetic groups (barring CHD7) displayed more variable severity in their reproductive defects. Among nonreproductive phenotypes, dental agenesis, digital bony abnormalities (FGF8/FGFR1), synkinesia (KAL1), and hearing loss (CHD7) stood out as discriminatory phenotypic signatures (Figure 4). However, apart from the digital bone abnormalities that appeared unique to the FGF8/FGFR1 group, the other features crossed several groups of genes and biological pathways, suggesting potential biological interactions between these neurodevelopmental genes and pathways.
Figure 4.

Genetic signature(s) enriched within each phenotype expressed on the basis of strength of statistical association. Highly associated phenotypes represent features in which specific genetic mutations were enriched with statistical significance in this study (P < .05); Possibly associated phenotypes represent features in which no specific genes were enriched statistically in this study, but genes with either previously reported associations or with considerable prevalence of the relevant phenotypes are shown.
Male KS subjects with KAL1 mutations have been extensively studied (11, 13, 14, 28–31). Consistent with those reports, this study confirms that males bearing KAL1 RSVs are completely anosmic, display a consistently severe reproductive phenotype characteristic of a highly penetrant Mendelian disorder, and rarely undergo a reversal of their condition (32). Synkinesia and unilateral renal agenesis have been previously reported to be highly specific to KAL1/X-linked KS pedigrees (13–15, 33). In this study, synkinesia was significantly enriched in the KAL1 group, suggesting that the presence of this feature could be useful for prioritizing this gene. However, renal agenesis, previously thought to be specific to KAL1 positive patients, was also detected in the gene-negative group, suggesting that the specificity of renal agenesis for the KAL1 pedigrees may be lower than previously reported.
In contrast to the KAL1 group, patients harboring RSVs in the FGF8/FGFR1 pathway displayed a considerably broader range of reproductive phenotypes including a reversal of their reproductive defect. These observations are in keeping with previous reports showing variable reproductive deficits in this group (11, 12, 33). Midline facial defects, dental agenesis, and skeletal anomalies have all been reported to be markers of mutations in the FGF8/FGFR1 pathway in KS (17, 33–36). In this study, we confirmed enrichment of dental agenesis in this group. Although skeletal anomalies as a whole did not differ between the groups, a detailed evaluation revealed that certain digital bony abnormalities (polydactyly, syndactyly, and camptodactyly) were unique to the FGF8/FGFR1 group, presumably reflecting the critical role of FGF signaling in limb formation (37). Furthermore, although the present study is underpowered to make inferences about HS6ST1 probands, the presence of cleft lip/palate in these subjects hints at its potential interaction with the FGF8/FGFR1 signaling cascade (8).
In our cohort of KS patients, RSVs in PROK2/PROKR2 were almost always found in the heterozygous state as previously reported by us and others (18, 38, 39). Although the precise mode of inheritance in these patients is still puzzling (40), recent evidence suggests that oligogenic inheritance may in part account for this observation (41, 42). Interestingly, recent evidence also suggests that some heterozygous variants in PROKR2 may act in a dominant-negative manner (43). The spectrum and severity of the reproductive phenotypes in this group in our study were similar to those reported in an independent group of monoallelic subjects with a similar spectrum of mutations (18). Although no specific nonreproductive feature was statistically enriched in the PROK2/PROKR2 group, a substantial prevalence of synkinesia was detected. This observation requires further validation in larger cohorts of patients.
Prior studies have described that CHD7 mutations are present in individuals with a pure isolated GnRH deficiency phenotype (5, 6). This study added some more systematic detailed reproductive phenotype in KS subjects harboring CHD7 variants. Hearing loss represented a significantly enriched nonreproductive feature in this group, which can be useful in prioritizing genetic screening in KS patients. Additional CHARGE syndrome-associated features were demonstrable only in 5% of CHD7 probands, and although other CHARGE-related features cannot be excluded without more extensive phenotypic evaluations (44), these observations confirm the previous observations that RSVs in CHD7 are important and are frequent causes of a pure reproductive phenotype (5, 6).
Evaluation of the gene-negative group allowed us to speculate on the potential nature of hitherto undiscovered KS genes. The reproductive phenotypes at presentation in this group tended to be milder with a reversal of reproductive deficit documented in some patients. However, a few subjects failed to respond favorably to GnRH treatment (23), suggesting that these as-yet-unknown genes may impact at all 3 levels of the hypothalamo-pituitary-gonadal axis. Among the nonreproductive features, there was some overlap with phenotypes enriched in the KAL1 group (renal agenesis, synkinesia), FGF8/FGFR1 group (dental agenesis, bone anomalies), and CHD7 group (iris coloboma) (16, 45). Although we cannot exclude mutations in the regulatory regions of KAL1, FGF8/FGFR1, or CHD7 or epigenetic changes in these genes, it is tempting to speculate that novel genes that may interact with existing pathways are awaiting elucidation. Therefore, weighing the known KS-associated genetic pathways in the bioinformatic analyses of next generation sequencing in these gene-naive patients will yield high dividends given the overlapping phenotypic features seen in this study.
This study has a few important limitations. Although it represents the largest KS population to be subjected to comprehensive genetic analysis of the 8 genes in these 6 neurodevelopmental pathway genes, only a small number of RSV positive probands were identified within each genetic group. In addition, phenotypic ascertainment was incomplete in some patients and consequently enrichment of some clinical features may have been missed due to lack of statistical power (eg, additional renal ultrasound in KS patients with KAL1 RSV may have revealed significance for renal agenesis). However, rigorous power calculations that are typically applied to common traits are not be readily applicable to rare Mendelian disorders such as KS. Also, due to the international referral nature of the study population, data on micropenis may have been underestimated due to the lack of ascertainment of the phenotype. Because of cost implications, genetic analysis was confined to the coding regions of the 8 KS genes, and thus, potential regulatory mutations cannot be excluded and mutations in newer KS genes such as WDR11 or SEMA3A were not ascertained. Although the RSV determinations in all genetic groups were based on strict minor allele frequency criteria (both our control population and the National Heart, Lung, and Blood Institute exome database), confirmatory functional data were available for only a subset of the discovered variants. Although in silico predictions of some variants were shown to be benign, the lack of specificity of the in silico programs and the rarity of these variants in extensive normative databases indicate that these variants must undergo further biological evaluation prior to discounting their pathogenicity.
In summary, this study will hopefully assist clinicians caring for KS patients to identify select phenotypic features (eg, synkinesia, dental agenesis, digital bony abnormalities, and hearing loss) that can aid in the prioritization of genetic screening in patients in need of a genetic diagnosis. Similarly, with the anticipated increase in accessibility to genetic testing, information from this study will allow targeted phenotyping in KS subjects with newly documented KS gene mutations.
Acknowledgments
We thank all our patients, their families, and the collaborating physicians from across the world who facilitated our recruitment efforts and volunteered their time and energy to enable this study. We also thank Dr Ye-Ming Chan for helpful discussions on this manuscript. Biostatistical analysis was conducted with support from the Harvard Clinical and Translational Science Center (National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health Award 8UL1TR000170-05 and financial contributions from Harvard University and its affiliated academic health care centers). The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic health care centers, or the National Institutes of Health.
This work was supported by National Institutes of Health Grants U54 HD028138 and R01 HD15788 (to W.F.C.), R01 HD056264 (to N.P.), and R01 HD42708 (to J.E.H.).
Disclosure Summary: W.F.C. is a consultant for Quest and Athena Diagnostics. The remaining authors have nothing to declare.
For editorial see page 1860
- CHARGE
- syndrome consisting of coloboma, heart defects, choanal atresia, retardation of growth, genital anomalies, and ear abnormalities
- CI
- confidence interval
- FGF
- fibroblast growth factor
- KS
- Kallmann syndrome
- MAF
- minor allele frequency
- OR
- odds ratio
- OMIM
- online inheritance in man
- RSV
- rare sequence variation
- TV
- testicular volume.
References
- 1. Seminara SB, Hayes FJ, Crowley WF., Jr Gonadotropin-releasing hormone deficiency in the human (idiopathic hypogonadotropic hypogonadism and Kallmann's syndrome): pathophysiological and genetic considerations. Endocr Rev. 1998;19:521–539 [DOI] [PubMed] [Google Scholar]
- 2. Hoffman AR, Crowley WF., Jr Induction of puberty in men by long-term pulsatile administration of low-dose gonadotropin-releasing hormone. N Engl J Med. 1982;307:1237–1241 [DOI] [PubMed] [Google Scholar]
- 3. Schwanzel-Fukuda M, Pfaff DW. The migration of luteinizing hormone-releasing hormone (LHRH) neurons from the medial olfactory placode into the medial basal forebrain. Experientia. 1990;46:956–962 [DOI] [PubMed] [Google Scholar]
- 4. Balasubramanian R, Dwyer A, Seminara SB, Pitteloud N, Kaiser UB, Crowley WF., Jr Human GnRH deficiency: a unique disease model to unravel the ontogeny of GnRH neurons. Neuroendocrinology. 2010;92:81–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim HG, Kurth I, Lan F, et al. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 2008;83:511–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jongmans MC, van Ravenswaaij-Arts CM, Pitteloud N, et al. CHD7 mutations in patients initially diagnosed with Kallmann syndrome—the clinical overlap with CHARGE syndrome. Clin Genet. 2009;75:65–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim HG, Ahn JW, Kurth I, et al. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 2011;87:465–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tornberg J, Sykiotis GP, Keefe K, et al. Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism. Proc Natl Acad Sci USA. 2011;108:11524–11529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Young J, Metay C, Bouligand J, et al. SEMA3A deletion in a family with Kallmann syndrome validates the role of semaphorin 3A in human puberty and olfactory system development. Hum Reprod. 2012;27:1460–1465 [DOI] [PubMed] [Google Scholar]
- 10. Oliveira LM, Seminara SB, Beranova M, et al. The importance of autosomal genes in Kallmann syndrome: genotype-phenotype correlations and neuroendocrine characteristics. J Clin Endocrinol Metab. 2001;86:1532–1538 [DOI] [PubMed] [Google Scholar]
- 11. Salenave S, Chanson P, Bry H, et al. Kallmann's syndrome: a comparison of the reproductive phenotypes in men carrying KAL1 and FGFR1/KAL2 mutations. J Clin Endocrinol Metab. 2008;93:758–763 [DOI] [PubMed] [Google Scholar]
- 12. Pitteloud N, Meysing A, Quinton R, et al. Mutations in fibroblast growth factor receptor 1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. Mol Cell Endocrinol. 2006;254–255:60–69 [DOI] [PubMed] [Google Scholar]
- 13. Quinton R, Duke VM, Robertson A, et al. Idiopathic gonadotrophin deficiency: genetic questions addressed through phenotypic characterization. Clin Endocrinol (Oxf). 2001;55:163–174 [DOI] [PubMed] [Google Scholar]
- 14. Kirk JM, Grant DB, Besser GM, et al. Unilateral renal aplasia in X-linked Kallmann's syndrome. Clin Genet. 1994;46:260–262 [DOI] [PubMed] [Google Scholar]
- 15. Quinton R, Duke VM, de Zoysa PA, et al. The neuroradiology of Kallmann's syndrome: a genotypic and phenotypic analysis. J Clin Endocrinol Metab. 1996;81:3010–3017 [DOI] [PubMed] [Google Scholar]
- 16. Dode C, Levilliers J, Dupont JM, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33:463–465 [DOI] [PubMed] [Google Scholar]
- 17. Zenaty D, Bretones P, Lambe C, et al. Paediatric phenotype of Kallmann syndrome due to mutations of fibroblast growth factor receptor 1 (FGFR1). Mol Cell Endocrinol. 2006;254–255:78–83 [DOI] [PubMed] [Google Scholar]
- 18. Sarfati J, Guiochon-Mantel A, Rondard P, et al. A comparative phenotypic study of Kallmann syndrome patients carrying monoallelic and biallelic mutations in the prokineticin 2 or prokineticin receptor 2 genes. J Clin Endocrinol Metab. 2010;95:659–669 [DOI] [PubMed] [Google Scholar]
- 19. Pitteloud N, Hayes FJ, Boepple PA, et al. The role of prior pubertal development, biochemical markers of testicular maturation, and genetics in elucidating the phenotypic heterogeneity of idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2002;87:152–160 [DOI] [PubMed] [Google Scholar]
- 20. Shaw ND, Seminara SB, Welt CK, et al. Expanding the phenotype and genotype of female GnRH deficiency. J Clin Endocrinol Metab. 2011;96:E566–E576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Feldman KW, Smith DW. Fetal phallic growth and penile standards for newborn male infants. J Pediatr. 1975;86:395–398 [DOI] [PubMed] [Google Scholar]
- 22. Bin-Abbas B, Conte FA, Grumbach MM, Kaplan SL. Congenital hypogonadotropic hypogonadism and micropenis: effect of testosterone treatment on adult penile size why sex reversal is not indicated. J Pediatr. 1999;134:579–583 [DOI] [PubMed] [Google Scholar]
- 23. Sykiotis GP, Hoang XH, Avbelj M, et al. Congenital idiopathic hypogonadotropic hypogonadism: evidence of defects in the hypothalamus, pituitary, and testes. J Clin Endocrinol Metab. 2010;95:3019–3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martin K, Santoro N, Hall J, Filicori M, Wierman M, Crowley WF., Jr Clinical review 15: management of ovulatory disorders with pulsatile gonadotropin-releasing hormone. J Clin Endocrinol Metab. 1990;71:1081A–1081G [DOI] [PubMed] [Google Scholar]
- 25. Raivio T, Falardeau J, Dwyer A, et al. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med. 2007;357:863–873 [DOI] [PubMed] [Google Scholar]
- 26. Lewkowitz-Shpuntoff HM, Hughes VA, Plummer L, et al. Olfactory phenotypic spectrum in idiopathic hypogonadotropic hypogonadism: pathophysiological and genetic implications. J Clin Endocrinol Metab. 2012;97:E136–E144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Verloes A. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A. 2005;133A:306–308 [DOI] [PubMed] [Google Scholar]
- 28. Bhagavath B, Podolsky RH, Ozata M, et al. Clinical and molecular characterization of a large sample of patients with hypogonadotropic hypogonadism. Fertil Steril. 2006;85:706–713 [DOI] [PubMed] [Google Scholar]
- 29. Versiani BR, Trarbach E, Koenigkam-Santos M, et al. Clinical assessment and molecular analysis of GnRHR and KAL1 genes in males with idiopathic hypogonadotrophic hypogonadism. Clin Endocrinol (Oxf). 2007;66:173–179 [DOI] [PubMed] [Google Scholar]
- 30. Izumi Y, Tatsumi K, Okamoto S, et al. Analysis of the KAL1 gene in 19 Japanese patients with Kallmann syndrome. Endocr J. 2001;48:143–149 [DOI] [PubMed] [Google Scholar]
- 31. Massin N, Pecheux C, Eloit C, et al. X chromosome-linked Kallmann syndrome: clinical heterogeneity in three siblings carrying an intragenic deletion of the KAL-1 gene. J Clin Endocrinol Metab. 2003;88:2003–2008 [DOI] [PubMed] [Google Scholar]
- 32. Ribeiro RS, Vieira TC, Abucham J. Reversible Kallmann syndrome: report of the first case with a KAL1 mutation and literature review. Eur J Endocrinol. 2007;156:285–290 [DOI] [PubMed] [Google Scholar]
- 33. Sato N, Katsumata N, Kagami M, et al. Clinical assessment and mutation analysis of Kallmann syndrome 1 (KAL1) and fibroblast growth factor receptor 1 (FGFR1, or KAL2) in five families and 18 sporadic patients. J Clin Endocrinol Metab. 2004;89:1079–1088 [DOI] [PubMed] [Google Scholar]
- 34. Bailleul-Forestier I, Gros C, Zenaty D, Bennaceur S, Leger J, de Roux N. Dental agenesis in Kallmann syndrome individuals with FGFR1 mutations. Int J Paediatr Dent. 2010;20:305–312 [DOI] [PubMed] [Google Scholar]
- 35. Trarbach EB, Costa EM, Versiani B, et al. Novel fibroblast growth factor receptor 1 mutations in patients with congenital hypogonadotropic hypogonadism with and without anosmia. J Clin Endocrinol Metab. 2006;91:4006–4012 [DOI] [PubMed] [Google Scholar]
- 36. Falardeau J, Chung WC, Beenken A, et al. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest. 2008;118:2822–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Coumoul X, Deng CX. Roles of FGF receptors in mammalian development and congenital diseases. Birth Defects Res C Embryo Today. 2003;69:286–304 [DOI] [PubMed] [Google Scholar]
- 38. Abreu AP, Trarbach EB, de Castro M, et al. Loss-of-function mutations in the genes encoding prokineticin-2 or prokineticin receptor-2 cause autosomal recessive Kallmann syndrome. J Clin Endocrinol Metab. 2008;93:4113–4118 [DOI] [PubMed] [Google Scholar]
- 39. Cole LW, Sidis Y, Zhang C, et al. Mutations in prokineticin 2 and prokineticin receptor 2 genes in human gonadotrophin-releasing hormone deficiency: molecular genetics and clinical spectrum. J Clin Endocrinol Metab. 2008;93:3551–3559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Balasubramanian R, Plummer L, Sidis Y, et al. The puzzles of the prokineticin 2 pathway in human reproduction. Mol Cell Endocrinol. 2011;346:44–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dode C, Teixeira L, Levilliers J, et al. Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet. 2006;2:e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sykiotis GP, Plummer L, Hughes VA, et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci USA. 2010;107:15140–15144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Abreu AP, Noel SD, Xu S, Carroll RS, Latronico AC, Kaiser UB. Evidence of the importance of the first intracellular loop of prokineticin receptor 2 in receptor function. Mol Endocrinol. 2012;26:1417–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bergman JE, de Ronde W, Jongmans MC, et al. The results of CHD7 analysis in clinically well-characterized patients with Kallmann syndrome. J Clin Endocrinol Metab. 2012;97:E858–E862 [DOI] [PubMed] [Google Scholar]
- 45. Laitinen EM, Vaaralahti K, Tommiska J, et al. Incidence, phenotypic features and molecular genetics of Kallmann syndrome in Finland. Orphanet J Rare Dis. 2010;6:41. [DOI] [PMC free article] [PubMed] [Google Scholar]