

Abstract
Inherited white-matter disorders are a broad class of diseases for which treatment and classification are both challenging. Indeed, nearly half of the children presenting with a leukoencephalopathy remain without a specific diagnosis. Here, we report on the application of high-throughput genome and exome sequencing to a cohort of ten individuals with a leukoencephalopathy of unknown etiology and clinically characterized by hypomyelination with brain stem and spinal cord involvement and leg spasticity (HBSL), as well as the identification of compound-heterozygous and homozygous mutations in cytoplasmic aspartyl-tRNA synthetase (DARS). These mutations cause nonsynonymous changes to seven highly conserved amino acids, five of which are unchanged between yeast and man, in the DARS C-terminal lobe adjacent to, or within, the active-site pocket. Intriguingly, HBSL bears a striking resemblance to leukoencephalopathy with brain stem and spinal cord involvement and elevated lactate (LBSL), which is caused by mutations in the mitochondria-specific DARS2, suggesting that these two diseases might share a common underlying molecular pathology. These findings add to the growing body of evidence that mutations in tRNA synthetases can cause a broad range of neurologic disorders.
Main Text
As recognized more than a decade ago, nearly half of childhood leukoencephalopathies remain unclassified and are a diagnostic problem for pediatric neurologists and neuroradiologists.1 Many of these disorders are extremely rare, precluding conventional genetic linkage studies, but they are potentially amenable to whole-genome sequencing (WGS) or whole-exome sequencing (WES) analysis, particularly if unaffected family members are available and affected individuals can be nosologically clustered with MRI pattern recognition.1,2
The first child examined in this study, subject 1 (Figure 1), was diagnosed with a tethered cord as a newborn and subsequently developed global developmental delay and severe leg spasticity (for clinical details, see Table S1, available online); extensive white-matter abnormalities involved the supratentorial brain white matter, brain stem, and spinal cord (Figure 1 and Table S2). A comprehensive panel of imaging, metabolic, and genetic studies failed to yield a definitive diagnosis, and he was subsequently enrolled in an ongoing study investigating the etiology of unclassified neurological disorders at the Murdoch Children’s Research Institute (the study was approved by the Royal Children’s Hospital Research Ethics Committee [HREC reference number 28097], and informed consent was obtained from all participating individuals). WGS was performed on the affected proband and both parents (approved by the University of Queensland Ethics Office, projects 2012000375 and 2012000376). Sequencing reads were aligned to the reference human genome (UCSC Genome Browser hg19) with the Burrows-Wheeler Aligner,3 and downstream processing was carried out with a pipeline composed of the Genome Analysis Toolkit,4, 5 SAMtools,6 Picard (see Web Resources), Annovar,7 and a set of custom analysis and visualization tools. Average genome coverage for this family exceeded 38× (Table S3), which yielded more than 3.2 million variants per genome. A custom-developed differential family-trio analysis yielded a single likely candidate gene—DARS (MIM 603084, RefSeq accession number NM_001349.2), encoding an aspartyl-tRNA synthetase (AspRS). In this gene, we found a compound-heterozygous mutation, c.1099G>T (p.Asp376Tyr) (paternal allele) and c.821C>T (p.Ala274Val) (maternal allele) (see Figure 2 and Table 1), which was validated by Sanger sequencing. Intriguingly, we noted that recessive mutations in the DARS mitochondrial paralog, DARS2 (MIM 610956), had been previously associated with another white-matter disorder, leukoencephalopathy with brain stem and spinal cord involvement and elevated lactate (LBSL [MIM 611105]).8
Figure 1.
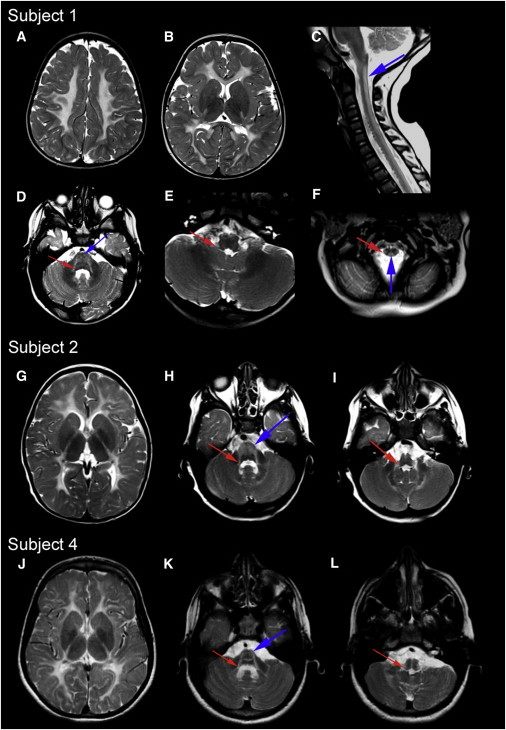
MRI Patterns Characteristic of HBSL
MRI images of subjects 1, 2 and 4: (A), (B), and (D)–(L) show axial T2-weighted images, and (C) shows a sagittal T2-weighted image. In (A), (G), and (J), note the diffuse T2 hyperintense signal of almost the entire supratentorial white matter. In subject 1, only the anterior section of the posterior limb of the internal capsule and a narrow band of the subcortical white matter in the occipital lobe are spared. In all subjects, the superior (D, H, and K; red arrows) and inferior (E, I, and L; red arrows) cerebellar peduncles and the anterior brainstem (D, H, and K; blue arrows) are affected. In the spinal cord, the dorsal columns (C and F; blue arrows) and the lateral corticospinal tracts (F, red arrow) are hyperintense. We note that in several individuals, the white-matter signal on the T2-weighted images was more elevated than usual for hypomyelinating leukoencephalopathies, similar to what has recently been described for another disease, hypomyelination with congenital cataracts (MIM 610532).2
Figure 2.
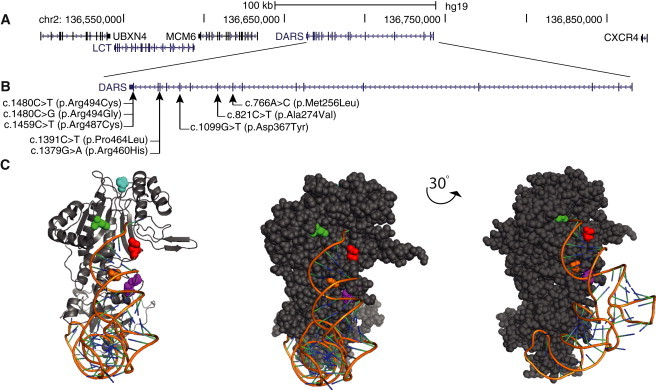
DARS Mutations in Individuals with HBSL
(A) Genomic organization of DARS in humans (UCSC Genome Browser hg19).
(B) Mutations and their positions within the DARS cDNA. Note that all identified mutations fall in the 3′ end of DARS, which encodes the active site of the AspRS. No mutations were detected within the hinge domain or the anticodon-binding domain.
(C) Five DARS mutations affect amino acids that are conserved in S. cerevisiae (Table 1), and the orientation of these amino acids within the protein can therefore be visualized with the crystal structure of the yeast DARS homolog, DSP1 (Protein Data Bank ID 1ASZ), in complex with tRNA-Asp. In this image, p.Ala274 (yeast p.Ala327) is shown in red, p.Asp367 (yeast p.Asp421) is shown in green, p.Pro464 (yeast p.Pro521) is shown in cyan, p.Arg487 (yeast p.Arg543) is shown in orange, and p.Arg494 (p.Arg554) is shown in magenta. Note that four of the affected amino acids sit within regions that stabilize the tRNA (orange ribbon) in complex with DSP1. Two alterations, p.Ala274Val and p.Asp367Tyr, sit in the active-site pocket and resulted from a compound-heterozygous mutation in a single subject (Table 1). An animation of this structure can be found online (DARS animation in Web Resources). Visualization of the predicted human DARS structure and the HBSL mutations can be found in Figure S1.
Table 1.
DARS Mutations Associated with HBSL
| Subject | Country of Origin | Genomic Position (hg19) | cDNAaMutation | Exon | Protein Alteration | Inheritance | Frequencyb | dbSNP ID | Conservationc | Damage Predictiond |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Australia | chr2: 136,673,803 | c.1099G>T | 11 | p.Asp367Tyr | paternal | 0% | - | S. cerevisiae | damaging |
| chr2: 136,678,161 | c.821C>T | 10 | p.Ala274Val | maternal | 0.001% | rs112205661 | S. cerevisiae | damaging | ||
| 2,e 3,e 5, 8 | India and Pakistan | chr2: 136,680,399 | c.766A>C | 9 | p.Met256Leu | homozygous | 0% | - | C. elegans | damaging |
| 4 | India | chr2: 136,664,933 | c.1459C>T | 16 | p.Arg487Cys | homozygous | 0% | - | S. cerevisiae | damaging |
| 6,e 7e | USA | chr2: 136,668,744 | c.1379G>A | 15 | p.Arg460His | paternal | 0% | - | C. elegans | damaging |
| chr2: 136,664,912 | c.1480C>T | 16 | p.Arg494Gly | maternal | 0.001% | rs147077598 | S. cerevisiae | damaging | ||
| 9,e 10e | UK | chr2: 136,668,733 | c.1391C>T | 15 | p.Pro464Leu | ND | 0.0018%f | rs148806569 | C. elegans | damaging |
| chr2: 136,664,912 | c.1480C>T | 16 | p.Arg494Cys | ND | 0% | - | S. cerevisiae | damaging |
The following abbreviation is used: ND, not determined.
DARS RefSeq NM_001349.2.
Frequency of occurrence according to the ESP6500SI release of the NHBLI Exome Variant Server (see Web Resources).
Conservation was analyzed in 11 species, including human, chimp, rhesus, mouse, chicken, frog, zebrafish, tetraodon, Drosophila, C. elegans, and S. cerevisiae.
Please see Table S4 for damage scores from six damage prediction algorithms.
Sibling pairs are as follows: 2 and 3, 6 and 7, and 9 and 10.
Frequency specifically within the Africa-American (AA) ESP6500SI population.
Concurrently, as part of an ongoing study applying MRI pattern analysis2 to unclassified leukoencephalopathies at VU University Medical Center (the study was approved by the VUMC Medical Ethics Committee, and informed consent was obtained from all participating individuals), four subjects—one pair of brothers and two single unrelated individuals from consanguineous Indian families—with a previously undescribed disorder were identified. All had diffuse hypomyelination of the cerebral white matter and striking abnormalities of specific brain stem structures (Figure 1 and Table S2). In those subjects who had a spinal MRI, the dorsal columns also showed abnormal signal. Clinically, all subjects developed nystagmus in the first year of life and had severe leg spasticity and mild cerebellar dysfunction. For identification of the responsible mutations, WES was performed on one subject and his mother and on another unrelated affected subject. Using the bioinformatic methods outlined above and in Table S3, we identified two homozygous DARS mutations: subject 2 was homozygous for c.766A>C (p.Met256Leu), and subject 4 was homozygous for c.1459C>T (p.Arg487Cys) (Figures 1 and 2 and Table 1). Sanger sequencing confirmed these mutations and a c.766A>C (p.Met256Leu) homozygous mutation in subject 2’s affected sibling (subject 3; see Tables 1 and S1). Both sets of parents were confirmed to be heterozygous carriers.
We then queried two biorepositories, the Centre for Children with White Matter Disorders and the Myelin Disorders Bioregistry Project (approved by the VUMC Medical Ethics Committee and the Children’s National Medical Center’s Ethics Program, respectively; informed consent was obtained from all participating individuals), and identified five more subjects who had the same MRI pattern and who were all confirmed to have mutations in DARS (Tables 1 and S1–S4). This included a five-member family that had been analyzed by ultra-high-coverage exome sequencing, which revealed a compound-heterozygous DARS mutation in two affected sisters (subjects 6 and 7; Tables 1 and S1–S4); this mutation was absent from their unaffected sibling. Taken together, these findings demonstrate that a previously uncharacterized leukoencephalopathy characterized by hypomyelination with brain stem and spinal cord abnormalities and leg spasticity (HBSL) is caused by recessive mutations in DARS.
To evaluate the potential effects of these DARS mutations, we assessed their relative incidence, conservation, and predicted pathogenicity (Table 1). Five were absent from SNPdb 135, the 1000 Genomes Project databases, and the National Heart, Lung, and Blood Institute (NHBLI) Exome Sequencing Project Exome Variant Server (ESP6500SI release), and the remaining three had minor allele frequencies of ∼0.001 (Table 1). All are conserved in the C. elegans DARS ortholog, drs-1, and five are conserved in the S. cerevisiae ortholog, DPS1. AspRS DARS is a fundamental component of the translational machinery, and a wide range of previous studies have shown that loss of AspRS activity in Drosophila and yeast is lethal (reviewed in Table S5) and that selective knockdown of AspRS in Drosophila neuroblasts leads to shorter lineages, alterations to cell shape, and defects in the ganglion mother cell.9 Consistent with these reports, we found that when assessed with a combination of six damage-prediction algorithms, all the HBSL DARS mutations were classified as pathogenic by at least two independent metrics (Table S4), strongly suggesting that these variants are the cause of HBSL.
The mutations identified in this study lie in the 3′ third of DARS, the region which corresponds to the C-terminal active-site domain of DARS. Two previous site-directed-mutagenesis studies in yeast have shown that alteration of any of the amino acids that reside within the active site’s hydrogen-bond network or the nearby side chains is detrimental to the enzyme10 and that any alteration in the C-terminal lobe of the protein leads to a minimum 3-fold reduction of enzyme activity.11 To investigate how the mutations identified in this study might affect DARS activity, we mapped each affected amino acid to a predicted human DARS crystal structure (Figure S1) and all the highly conserved amino acids to the yeast DPS1 crystal structure (Figure 2). Strikingly, we found that two mutations alter amino acids that sit within the active-site pocket (p.Ala274Val and p.Asp367Tyr from subject 1), three alter amino acids that are likely to destabilize the tRNA-protein interaction (p.Arg487Cys, p.Arg494Cys, and p.Arg494Gly), and the remaining mutations alter amino acids that are likely to be fundamental to chains supporting the active site (Figure 2 and Figure S1).
To gain further insight into the pathogenic role of these mutations in HBSL, we performed an investigation of DARS expression in humans and mice by using a curated set of publicly available data. Consistent with the fact that DARS is a core component of the translational machinery, we found that it is diffusely localized in cytoplasm and broadly expressed across tissue types (by both RNA-seq and array-based measures of expression), including the central nervous system (Figures S2 and S3). In mice, Dars shows specific expression in sagittal brain sections with staining in the hippocampus, the dentate gyrus, and the molecular layer of the cerebellum, which is strikingly similar to the expression patterns of glycyl-tRNA synthetase (Gars) and lysyl-tRNA synthetase (Kars), but shows little overlap with glial and oligodendrocyte markers (Figure S4). For example, Dars, Kars, and Gars are clearly expressed in the cerebellar molecular layer (which houses Purkinje neurons), whereas the oligodendrocyte markers Mog, CNPase, and Olig2 are highly expressed in the granular layer (Figure S4). DARS expression in the developing and adult human brain shows similar patterns—it is highly expressed in the ventricular and subventricular zones, including hippocampal subfields, the midlateral temporal cortex, and the frontal polar cortex (Figure S5). Likewise, DARS immunostaining of the cerebellum, cerebral cortex, hippocampus, and lateral ventricle all show preferential neuronal staining, and staining of peripheral neurons is evident in the colon (Figure S6). Overall, these data are consistent with HBSL’s clinical presentation.
It is becoming increasingly apparent that mutations in genes encoding both cytoplasmic and mitochondrial tRNA synthetases (ARSs) can cause a wide range of neurological and multisystem disorders (reviewed in Table 2). For example, leukoencephalopathy with thalamus and brain stem involvement and high lactate (LTBL) is caused by mutations in EARS29 (MIM 612799), encoding a mitochondrial tRNA synthetase, and several forms of Charcot-Marie-Tooth disease (CMT [MIM 613287, 601472, 608323, and 613641]), a peripheral neuropathy, are caused by damaging mutations in AARS (MIM 601065), GARS (MIM 600287), YARS (MIM 603623), and KARS (MIM 601421) (Table 2), encoding cytoplasmic aminoacyl-tRNA synthetases. Similarly, recessive mutations in DARS2, encoding the mitochondria-specific aspartyl-tRNA synthetase, cause LBSL.8
Table 2.
Disease-Associated Genes Encoding tRNA Synthetases
| Gene | Gene MIM | Cognate Amino Acid | Localization | Disease | Hallmarks | Phenotype MIM | Inheritance | Reference |
|---|---|---|---|---|---|---|---|---|
| AARS | 601065 | alanine | C | CMT 2N | axonal neuropathy | 613287 | AD | Latour et al.12 |
| AARS2 | 612035 | alanine | M | combined oxidative phosphorylation deficiency 8 | muscular hypotonia, fatal hypertrophic cardiac myopathy | 614096 | AR | Gotz et al.13 |
| DARS | 603084 | aspartate | C | HBSL | leukoencephalopathy (hypomyelination) with brain stem and spinal cord involvement | - | AR | this study |
| DARS2 | 610956 | aspartate | M | LBSL | leukoencephalopathy with brain stem and spinal cord involvement and elevated lactate | 611105 | AR | Scheper et al.8 |
| EARS2 | 612799 | glutamine | M | LTBL | leukoencephalopathy with thalamic and brain stem involvement and elevated lactate | 614924 | AR | Steenweg et al.14 |
| FARS2 | 611592 | phenylalanine | M | combined oxidative phosphorylation deficiency 14 | neonatal encephalopathy with seizures and liver involvement | 614946 | AR | Elo et al.15 and Shamseldin et al.16 |
| GARS | 600287 | glycine | C | CMT 2D; distal hereditary motor neuropaty type V | axonal neuropathy | 601472 and 600794 | AD | Antonellis et al.17 |
| HARS | 142810 | histidine | C | Usher syndrome type 3B | retinitis pigmentosa, sensorineural deafness, ataxia | 614504 | AR | Puffenberger et al.18 |
| HARS2 | 600783 | histidine | M | Perrault syndrome 2 | sensorineural deafness, primary amenorrhea, ovarian dysgenesis | 614926 | AR | Pierce et al.19 |
| KARS | 601421 | lysine | C | recessive intermediate CMT | neuropathy with axonal and demyelinating features | 613641 | AR | McLaughlin et al.20 |
| LARS2 | 604544 | leucine | M | Perrault syndrome | sensorineural deafness, premature ovarian failure | - | AR | Pierce et al.21 |
| MARS2 | 609728 | methionine | M | spastic ataxia 3 | autosomal-recessive spastic ataxia with leukoencephalopathy (arsal) | 611390 | AR | Bayat et al.22 |
| RARS2 | 611524 | arginine | M | pontocerebellar hypoplasia type 6 | pontocerebellar and cerebellar atrophy, epilepsy | 611523 | AR | Edvardson et al.23 |
| SARS2 | 612804 | serine | M | hyperuricemia, pulmonary hypertension, renal failure, and alkalosis | renal failure in infancy, metabolic alkalosis, pulmonary hypertension | 613845 | AR | Belostotsky et al.24 |
| YARS | 603623 | tyrosine | C | dominant intermediate CMT | neuropathy with axonal and demyelinating features | 608323 | AD | Jordanova et al.25 |
| YARS2 | 610957 | tyrosine | M | mitochondrial myopathy | sideroblastic anemia and myopathy with exercise intolerance | 613561 | AR | Riley et al.26 |
Abbreviations are as follows: C, cytoplasm; M, mitochondria; AD, autosomal dominant; and AR, autosomal recessive.
Intriguingly, despite the fact that DARS and DARS2 have mutually exclusive subcellular localizations (Table 2), LBSL and HBSL are strikingly similar in their presentation. For example, they both manifest with abnormalities in the same central nervous system structures, including the superior and inferior cerebellar peduncles, the medial lemniscus and pyramidal tracts in the brain stem and the dorsal columns, and the lateral corticospinal tracts in the spinal cord—structures that are rarely affected in other leukoencephalopathies and the combination of which appears to be unique to HBSL and LBSL. The similarity in MRI abnormalities is such that one of the individuals (subject 7) had been initially classified as “variant LBSL” (Figure S7). Consistent with other reports,10 we find it unlikely that HBSL and LBSL are caused by amino acid mischarging alone (although this might be an important factor). Indeed, recent work has demonstrated that mutations in DARS2 decrease catalytic activity by impairing dimer formation or by reducing protein expression27, 28 but that activity of the respiratory chain is normal in individuals with LBSL.8 Many ARSs are known to have alternative functions beyond translation,29 including AspRS, which is involved in asparagine biosynthesis.30 This suggests that the disease etiology and progression of HBSL and LBSL, and possibly other ARS-associated illnesses, might be driven by processes and biological relationships that are not yet fully understood.
Acknowledgments
We are grateful for the support and cooperation of the subjects with hypomyelination with brain stem and spinal cord involvement and leg spasticity, as well as their families. We wish to thank Illumina for their reagent contributions and continuous support of this project. We also thank Dennis Gascoigne and Michael Pheasant for their early support of this work and data-analysis advice, as well as David Amor of the Victorian Clinical Genetics Services. We are indebted to Emiel Polder and Nienke Postma for their help with DARS sequencing and to Marianna Bugiani for discussions on DARS expression. R.J.T. is supported by an Australian Research Council Discovery Early Career Research Award and is a consultant to Isis Pharmaceuticals. A.V. is supported by a grant from the National Institute of Neurologic Disorders and Stroke, National Institutes of Health (1K08NS060695) and by the Myelin Disorders Bioregistry Project. This work was supported by the Mission Massimo Foundation, the Victorian State Government Operational Infrastructure Support Program, the Institute for Molecular Bioscience funds, the NeCTAR Genomics Virtual Lab, a University of Queensland Foundation Research Excellence Award, and ZonMw TOP grant 91211005.
Published: May 2, 2013
Footnotes
Supplemental Data include seven figures and five tables and can be found with this article online at http://www.cell.com/AJHG.
Contributor Information
Ryan J. Taft, Email: r.taft@imb.uq.edu.au.
Nicole I. Wolf, Email: n.wolf@vumc.nl.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
Burrows-Wheeler Aligner, http://bio-bwa.sourceforge.net/
DARS animation, http://goo.gl/nwj60
Genome Analysis Toolkit, http://www.broadinstitute.org/gatk/
NHBLI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Picard, http://picard.sourceforge.net/
SAMtools, http://samtools.sourceforge.net/
UCSC Genome Browser, http://genome.ucsc.edu/
Supplemental Data
References
- 1.van der Knaap M.S., Breiter S.N., Naidu S., Hart A.A., Valk J. Defining and categorizing leukoencephalopathies of unknown origin: MR imaging approach. Radiology. 1999;213:121–133. doi: 10.1148/radiology.213.1.r99se01121. [DOI] [PubMed] [Google Scholar]
- 2.Steenweg M.E., Vanderver A., Blaser S., Bizzi A., de Koning T.J., Mancini G.M.S., van Wieringen W.N., Barkhof F., Wolf N.I., van der Knaap M.S. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain. 2010;133:2971–2982. doi: 10.1093/brain/awq257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M., et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scheper G.C., van der Klok T., van Andel R.J., van Berkel C.G.M., Sissler M., Smet J., Muravina T.I., Serkov S.V., Uziel G., Bugiani M., et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat. Genet. 2007;39:534–539. doi: 10.1038/ng2013. [DOI] [PubMed] [Google Scholar]
- 9.Neumüller R.A., Richter C., Fischer A., Novatchkova M., Neumüller K.G., Knoblich J.A. Genome-wide analysis of self-renewal in Drosophila neural stem cells by transgenic RNAi. Cell Stem Cell. 2011;8:580–593. doi: 10.1016/j.stem.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cavarelli J., Eriani G., Rees B., Ruff M., Boeglin M., Mitschler A., Martin F., Gangloff J., Thierry J.C., Moras D. The active site of yeast aspartyl-tRNA synthetase: structural and functional aspects of the aminoacylation reaction. EMBO J. 1994;13:327–337. doi: 10.1002/j.1460-2075.1994.tb06265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prevost G., Eriani G., Kern D., Dirheimer G., Gangloff J. Study of the arrangement of the functional domains along the yeast cytoplasmic aspartyl-tRNA synthetase. Eur. J. Biochem. 1989;180:351–358. doi: 10.1111/j.1432-1033.1989.tb14655.x. [DOI] [PubMed] [Google Scholar]
- 12.Latour P., Thauvin-Robinet C., Baudelet-Méry C., Soichot P., Cusin V., Faivre L., Locatelli M.-C., Mayençon M., Sarcey A., Broussolle E., et al. A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 2010;86:77–82. doi: 10.1016/j.ajhg.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Götz A., Tyynismaa H., Euro L., Ellonen P., Hyötyläinen T., Ojala T., Hämäläinen R.H., Tommiska J., Raivio T., Oresic M., et al. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am. J. Hum. Genet. 2011;88:635–642. doi: 10.1016/j.ajhg.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steenweg M.E., Ghezzi D., Haack T., Abbink T.E.M., Martinelli D., van Berkel C.G.M., Bley A., Diogo L., Grillo E., Te Water Naudé J., et al. Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’ caused by EARS2 mutations. Brain. 2012;135:1387–1394. doi: 10.1093/brain/aws070. [DOI] [PubMed] [Google Scholar]
- 15.Elo J.M., Yadavalli S.S., Euro L., Isohanni P., Götz A., Carroll C.J., Valanne L., Alkuraya F.S., Uusimaa J., Paetau A., et al. Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum. Mol. Genet. 2012;21:4521–4529. doi: 10.1093/hmg/dds294. [DOI] [PubMed] [Google Scholar]
- 16.Shamseldin H.E., Alshammari M., Al-Sheddi T., Salih M.A., Alkhalidi H., Kentab A., Repetto G.M., Hashem M., Alkuraya F.S. Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J. Med. Genet. 2012;49:234–241. doi: 10.1136/jmedgenet-2012-100836. [DOI] [PubMed] [Google Scholar]
- 17.Antonellis A., Ellsworth R.E., Sambuughin N., Puls I., Abel A., Lee-Lin S.-Q., Jordanova A., Kremensky I., Christodoulou K., Middleton L.T., et al. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet. 2003;72:1293–1299. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Puffenberger E.G., Jinks R.N., Sougnez C., Cibulskis K., Willert R.A., Achilly N.P., Cassidy R.P., Fiorentini C.J., Heiken K.F., Lawrence J.J., et al. Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS ONE. 2012;7:e28936. doi: 10.1371/journal.pone.0028936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pierce S.B., Chisholm K.M., Lynch E.D., Lee M.K., Walsh T., Opitz J.M., Li W., Klevit R.E., King M.-C. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc. Natl. Acad. Sci. USA. 2011;108:6543–6548. doi: 10.1073/pnas.1103471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McLaughlin H.M., Sakaguchi R., Giblin W., Wilson T.E., Biesecker L., Lupski J.R., Talbot K., Vance J.M., Züchner S., Lee Y.C., et al. NISC Comparative Sequencing Program A recurrent loss-of-function alanyl-tRNA synthetase (AARS) mutation in patients with Charcot-Marie-Tooth disease type 2N (CMT2N) Hum. Mutat. 2012;33:244–253. doi: 10.1002/humu.21635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pierce S.B., Gersak K., Michaelson-Cohen R., Walsh T., Lee M.K., Malach D., Klevit R.E., King M.-C., Levy-Lahad E. Mutations in LARS2, Encoding Mitochondrial Leucyl-tRNA Synthetase, Lead to Premature Ovarian Failure and Hearing Loss in Perrault Syndrome. Am. J. Hum. Genet. 2013;92:614–620. doi: 10.1016/j.ajhg.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bayat V., Thiffault I., Jaiswal M., Tétreault M., Donti T., Sasarman F., Bernard G., Demers-Lamarche J., Dicaire M.-J., Mathieu J., et al. Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol. 2012;10:e1001288. doi: 10.1371/journal.pbio.1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edvardson S., Shaag A., Kolesnikova O., Gomori J.M., Tarassov I., Einbinder T., Saada A., Elpeleg O. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Belostotsky R., Ben-Shalom E., Rinat C., Becker-Cohen R., Feinstein S., Zeligson S., Segel R., Elpeleg O., Nassar S., Frishberg Y. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am. J. Hum. Genet. 2011;88:193–200. doi: 10.1016/j.ajhg.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jordanova A., Irobi J., Thomas F.P., Van Dijck P., Meerschaert K., Dewil M., Dierick I., Jacobs A., De Vriendt E., Guergueltcheva V., et al. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet. 2006;38:197–202. doi: 10.1038/ng1727. [DOI] [PubMed] [Google Scholar]
- 26.Riley L.G., Cooper S., Hickey P., Rudinger-Thirion J., McKenzie M., Compton A., Lim S.C., Thorburn D., Ryan M.T., Giegé R., et al. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia—MLASA syndrome. Am. J. Hum. Genet. 2010;87:52–59. doi: 10.1016/j.ajhg.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Berge L., Dooves S., van Berkel C.G.M., Polder E., van der Knaap M.S., Scheper G.C. Leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation is associated with cell-type-dependent splicing of mtAspRS mRNA. Biochem. J. 2012;441:955–962. doi: 10.1042/BJ20110795. [DOI] [PubMed] [Google Scholar]
- 28.van Berge L., Kevenaar J., Polder E., Gaudry A., Florentz C., Sissler M., van der Knaap M.S., Scheper G.C. Pathogenic mutations causing LBSL affect mitochondrial aspartyl-tRNA synthetase in diverse ways. Biochem. J. 2013;450:345–350. doi: 10.1042/BJ20121564. [DOI] [PubMed] [Google Scholar]
- 29.Hurto R.L. Unexpected functions of tRNA and tRNA processing enzymes. Adv. Exp. Med. Biol. 2011;722:137–155. doi: 10.1007/978-1-4614-0332-6_9. [DOI] [PubMed] [Google Scholar]
- 30.Klipcan L., Safro M. Amino acid biogenesis, evolution of the genetic code and aminoacyl-tRNA synthetases. J. Theor. Biol. 2004;228:389–396. doi: 10.1016/j.jtbi.2004.01.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.