

Abstract
Hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC) is a rare hereditary leukoencephalopathy that was originally identified by MRI pattern analysis, and it has thus far defied all attempts at identifying the causal mutation. Only 22 cases are published in the literature to date. We performed exome sequencing on five family trios, two family quartets, and three single probands, which revealed that all eleven H-ABC-diagnosed individuals carry the same de novo single-nucleotide TUBB4A mutation resulting in nonsynonymous change p.Asp249Asn. Detailed investigation of one of the family quartets with the singular finding of an H-ABC-affected sibling pair revealed maternal mosaicism for the mutation, suggesting that rare de novo mutations that are initially phenotypically neutral in a mosaic individual can be disease causing in the subsequent generation. Modeling of TUBB4A shows that the mutation creates a nonsynonymous change at a highly conserved asparagine that sits at the intradimer interface of α-tubulin and β-tubulin, and this change might affect tubulin dimerization, microtubule polymerization, or microtubule stability. Consistent with H-ABC’s clinical presentation, TUBB4A is highly expressed in neurons, and a recent report has shown that an N-terminal alteration is associated with a heritable dystonia. Together, these data demonstrate that a single de novo mutation in TUBB4A results in H-ABC.
Main Text
Hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC [MIM 612438]) is a rare leukodystrophy described by Van der Knaap et al. in 2002.1 To date, only 22 individual cases have been reported. It is characterized by variable onset (from infancy to childhood), developmental delay, extrapyramidal movement disorders (dystonia, choreoathetosis, rigidity, opisthotonus, and oculogyric crises), progressive spastic tetraplegia, ataxia, and, more rarely, seizures.1 MRI plays a fundamental role in the diagnostic work-up because neuroradiological findings are pathognomonic; diagnostic criteria include the combination of hypomyelination, cerebellar atrophy, and absence or disappearance of the putamen,1 all features that have been comprehensively confirmed in a recent autopsy case.2 The disease appears to be sporadic in nature given that there are no previously published sibling groups. The lack of familial groups has thus far foiled attempts at identifying a causative mutation or rare variant.
It has also been somewhat uncertain whether H-ABC represents a single disorder or the common neuroradiologic manifestations of a heterogeneous group of disorders. The reported finding of low levels of 5-methyltetrahydrofolate in the cerebrospinal fluid (CSF) of one affected individual resulted in a therapeutic trial with folinic acid, which led to an improvement.3,4 It was then suggested that H-ABC could be related to cerebral folate deficiency3,4 and, conversely, that because CSF analysis was normal in some affected individuals, H-ABC could be considered a symptomatic representation of heterogeneous disorders.5 Additionally, an H-ABC-affected individual with a partial response to L-dopa has been reported,6 and a 21-month-old boy with Down syndrome and a neuroradiologic presentation consistent with H-ABC has been described.7
The advent of exome-sequencing technology provides the opportunity to study individual H-ABC cases and to establish whether this disorder is indeed a single monogenic entity or a clustering of heterogeneous leukoencephalopathies. To understand the genetic underpinning of H-ABC, we recruited affected individuals and their family members via the Myelin Disorders Bioregistry Project or the Amsterdam Database of Unclassified Leukoencephalopathies with approval from the institutional review board at Children’s National Medical Center, the Baylor Neurogenetic Institute, VU University Medical Center, or the University of Queensland. Written informed consent was obtained for each study participant. Genomic DNA samples were collected from blood samples provided to the biorepositories, and in the case of family LD_0638, additional genomic DNA was isolated from buccal and saliva samples.
A total of 11 individuals from ten unrelated families fulfilling the MRI criteria for H-ABC diagnosis were included in this study (Figure 1 and Table 1). All affected individuals presented in infancy or early childhood with predominant motor dysfunction, which often manifested as delayed acquisition of milestones or unsteady walking. Most of these individuals had deterioration of motor skills, often marked initially by hemidystonia. Gait progressively deteriorated in all cases, and independent and even supported ambulation was lost over time. Language and cognitive development appeared relatively preserved, although over time, dysarthria increasingly impaired communication. Receptive language was often normal: many children functioned at age-appropriate levels.
Figure 1.
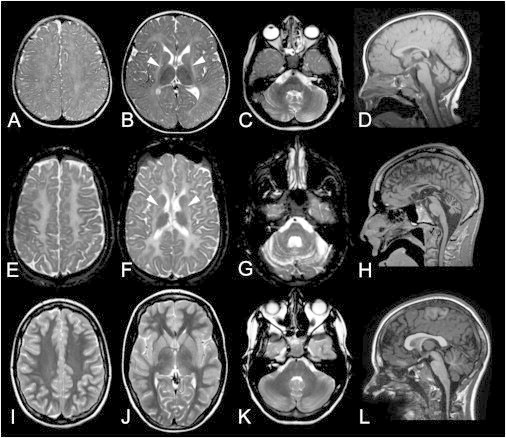
MRI Findings in H-ABC
Axial T2-weighted images (first three columns) and sagittal T1-weighted images (fourth column) of individual HA107 at the age of 3.5 years (A–D), individual HA27 at 21 years (E–H), and an unaffected individual at 14 years (I–L). We included the unaffected individual to demonstrate the low signal on T2-weighted images of normal myelinated white matter, the normal volume of the putamen, and the normal volume of the cerebellum. Note the relatively high T2 signal of the cerebral white matter in the two affected individuals (A, B, E, and F); this indicates lack of myelin. No putamen is visible in the affected individuals (B and F) (arrows are where the putamen should be). The cerebellar atrophy is already present in the younger individual (D) but is worse in the older individual (H).
Table 1.
Clinical Manifestation of Individuals with H-ABC
|
Individual |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| LD_0313.0 | LD_0345.0 | LD_0440.0 | LD_0605.0 | LD_0638.0A | LD_0638.0B | HA04 | HA07 | HA23 | HA27 | HA107 | |
| Gender | male | female | male | female | male | male | female | male | male | male | female |
| Ethnicity | white | Arab | white | Hispanic | Asian | Asian | white | white | white | white | white |
| Age at presentation | 1.5 years | 1 years | 3 years | 9 months | 2 years | 4.5 years | 1 years | 20 months | 6 months | 2.5 years | 2 years |
| Current age | 11 years | 7 years | 39 years | 30 years | 5 years | 8 years | 23 years | 21 years | 19 years | 29 years | 6 years |
| Presenting sign | mild gait instability; unclear speech | gait instability; delayed speech | motor | motor | mild gait instability; unclear speech | dystonia and dysarthria | delayed motor development; hypotonia | delayed motor development; spasticity | delayed motor development; hypotonia | progressive motor problems | delayed speech development; lack of motor developmental progress |
| Initial motor development | delayed; best skill was unsupported walking at 17 months; normal fine motor skills | delayed; best skill was the ability to take a few unsupported steps at 1 year | delayed | delayed | delayed; best skill was unsupported walking at 2 years | normal | delayed; able to stand at 12 months; walked with support at 24 months; walked a few steps without support at 33 months | normal; sat at 11 months; pulled to stand at 12 months; walked with support 18 months | delayed; sat at 18 months; walked with support at 3.5 years | normal; walked without support at 16 months; walked on toes | normal; walked without support at 13 months; wide-based gait |
| Onset of motor deterioration | 4.5 years; hemidystonia | 2 years | 3 years | 7 years | 4.5 years; hemidystonia | 4.5 years; hemidystonia | 7 years | 2 years; deterioration of hand function | 6 years | 2.5 years | 4 years |
| Loss of supported walking ability | 5 years | 2 years | 10 years | 10 years | supported walking at last examination at 5.5 years | supported walking at last examination at 7.5 years | 8 years | 9 years | ? | 14 years | 4 years |
| Spasticity | + | + | + | + | + | + | + | + | + | + | + |
| Ataxia | + | + | + | − | − | − | + | + | + | + | + |
| Tremor | − | − | + | + | − | + | + | + | + | + | + |
| Choreoathetosis | + | − | + | − | − | − | + | + | + | − | − |
| Dystonia | + | + | + | − | + | + | + | + | + | + | + |
| Rigidity | − | + | + | − | + | + | + | + | + | + | + |
| Dysarthria | + | + | + | + | + | + | + | + | + | + | + |
| Oculomotor abnormalities | nystagmus; oculomotor apraxia; hypometric saccades | none | none | none | hypometric saccades | none | oculomotor apraxia | normal | normal | rotatory and pendular nystagmus | none |
| Vision | normal | normal | normal | normal | normal | normal | decreased | normal | normal | normal | normal |
| Hearing | normal | normal | normal | normal | normal | normal | normal | normal | normal | sensorineural deafness (homozygous otoa deletion); cochlear implant | normal |
| Cognitive decline | + | + | + | + | − | + | + | mild intellectual disability | mild intellectual disability | − | − |
| Language development | initially normal; loss of most spontaneous speech over time; normal receptive skills | delayed; best skill was five to ten intelligible words; normal receptive language skills | delayed; single words until age 6 years; better receptive skills | delayed; single words until age 7 years; better receptive skills | normal | initially normal, although great loss of intelligibility over time | initially normal; 2 word sentences at 2 years; increasing dysarthria | delayed; increasing dysarthria | spoke syllables; no active speech; uses computer-assisted communication | almost normal at 6 years; increasing dysarthria; loss of active speech at 12 years | no active speech; uses signs and computer-assisted communication |
| Epilepsy | − | − | − | − | − | − | − | − | − | − | + |
To identify the disease-causing mutation or variant, we performed exome sequencing on each of the 11 affected individuals, the unaffected parents in seven of the families, and one unaffected sibling. In brief, exomes were captured with the SeqCap EZ Human Exome Library v.3.0 and sequenced on an Illumina HiSeq 2000 with the 100 bp paired-end read-sequencing protocol at the Queensland Centre for Medical Genomics or the VU University Medical Center sequencing center. Reads were aligned to the reference human genome (UCSC Genome Browser hg19) with the Burrows-Wheeler Aligner (BWA),8 and downstream processing of sequence data was done with Picard v.1.8, SAMtools v.0.1.18,9 and the Genome Analysis Toolkit (GATK) v.2.2.8.10 Variants (SNPs and indels) were identified with GATK according to version four of the GATK Best Practice Variant Detection guide11 or Varscan v.2.2.5.12 Variants were annotated with the use of Annovar13 with UCSC Known Genes models, and known polymorphisms were identified with dbSNP135, 1000 Genomes (April 30, 2012, release), and the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP) Exome Variant Server (ESP6500 release), and minor allele frequencies were recorded from each data set. Subsequent analysis and identification of candidate variants was performed with an in-house workflow incorporating the annotated variant data and pedigree information.
We produced 8–30 Gb of sequence for each individual, yielding a mean depth of 65-fold coverage and an average of 95% of target bases sequenced at least 18 times (Table S1, available online). A single heterozygous de novo mutation in TUBB4A (MIM 602662; RefSeq accession number NM_006087) was identified in all affected individuals (Table 2; Sanger sequencing validation is shown in Figure S1) but was absent from all unaffected parents and siblings with the exception of the mother in family LD_0638 (more below). This mutation—c.745G>A (RefSeq NM_006087.2) (g.6495765C>T) (RefSeq NC_000019.9)—is not present in dbSNP135, the 1000 Genomes Project database, or the NHLBI Exome Variant Server, consistent with its presumed pathogenicity and the relative population incidence of H-ABC.
Table 2.
H-ABC-Associated TUBB4A Genotypes
| Family | Individual | Affected | Genotype | Wild-Type Reads (G) | Alternative Reads (A) |
|---|---|---|---|---|---|
| LD_0313 | LD_0313.0A | yes | G/A | 132 | 118 |
| LD_0313.1 | no | G/G | 138 | 9 | |
| LD_0313.2 | no | G/G | 76 | 6 | |
| LD_0345 | LD_0345.0A | yes | G/A | 59 | 55 |
| LD_0345.1 | no | G/G | 55 | 5 | |
| LD_0345.2 | no | G/G | 105 | 2 | |
| LD_0440 | LD_0440.0A | yes | G/A | 114 | 81 |
| LD_0440.01 | no | G/G | 132 | 2 | |
| LD_0440.1 | no | G/G | 66 | 4 | |
| LD_0440.2 | no | G/G | 246 | 2 | |
| LD_0605 | LD_0605.0A | yes | G/A | 127 | 121 |
| LD_0605.1 | no | G/G | 173 | 7 | |
| LD_0605.2 | no | G/G | 144 | 4 | |
| LD_0638 | LD_0638.0A | yes | G/A | 124 | 124 |
| LD_0638.0B | yes | G/A | 140 | 110 | |
| LD_0638.1 | no | G/A | 193 | 54 | |
| LD_0638.2 | no | G/G | 241 | 2 | |
| HA04 | HA04 | yes | G/A | 43 | 40 |
| HA07 | HA07 | yes | G/A | 41 | 40 |
| HA23 | HA23 | yes | G/A | 46 | 35 |
| HA27 | HA27 | yes | G/A | 42 | 42 |
| HA28 | no | G/G | 50 | 1 | |
| HA29 | no | G/G | 50 | 0 | |
| HA107 | HA107 | yes | G/A | 51 | 65 |
| HA108 | no | G/G | 127 | 0 | |
| HA109 | no | G/G | 118 | 0 |
TUBB4A (also known as TUBB4 and TUBB5) is a member of the highly conserved β-tubulin protein family that forms heterodimers with α-tubulins and then in turn forms copolymers that assemble into microtubules, an essential component of the cytoskeleton. TUBB4A is primarily expressed in the nervous system,14–16 and its role in H-ABC is supported by several studies showing that neurological disorders characterized by abnormal neuronal migration, differentiation, and axon guidance and maintenance have been attributed to mutations in the α-tubulin- and β-tubulin-encoding genes TUBA1A (MIM 602529), TUBA8 (MIM 605742), TUBB2B (MIM 612850), and TUBB3 (MIM 602661).17,18 Indeed, an autosomal-dominant mutation (c.4C>G [p.Arg2Gly]) in TUBB4A was recently identified in an extended family affected by dystonia type 4 (DYT4 [MIM 128101]; Figure 2A).16 This disorder is characterized by a “whispering” dysphonia, generalized dystonia, and gait ataxia with onset in the second to fourth decade. In contrast to individuals with H-ABC, however, individuals with DYT4 are reported to have a normal MRI.20 Although H-ABC is a hypomyelinating leukoencephalopathy, it is distinguished from other disorders in this class by the presence of abnormalities of the deep gray nuclei, suggesting neuronal involvement. Further pathologic studies will be necessary to establish whether the H-ABC-related TUBB4A mutation results in cytoskeletal abnormalities in neurons and glia.
Figure 2.
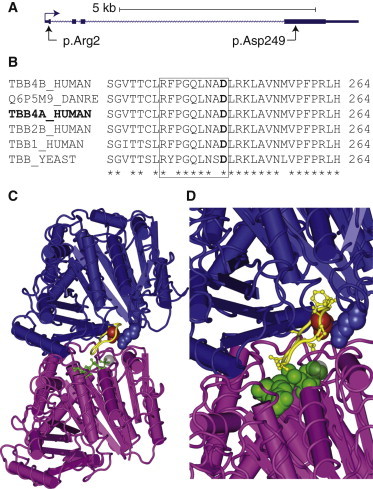
The Highly Conserved TUBB4A Residue Asp249 Sits at the Intradimer Interface of α-Tubulin and β-Tubulin
(A) The genomic structure of TUBB4A shows the region encoding Arg2 in exon 1 and the region encoding Asp249 in exon 4.
(B) Multiple-sequence alignment of a section of four human β-tubulins, a zebrafish β-tubulin, and the Saccharomyces cerevisiae β-tubulin. Residue Asp249 is marked in bold text, residues that form the T7 loop are boxed, and asterisks indicate invariant residues. Sequences are labeled with their Uniprot ID.
(C) Structure of bovine tubulin heterodimer.19 Guanosine triphosphate (GTP; green) can be seen bound at the interface of α-tubulin (magenta) and β-tubulin (blue). Residue Asp249 is shown as red spheres, its interacting partner Arg2 is shown as blue spheres, and the remainder of the T7 loop is shown in yellow.
(D) A magnified view of the intradimer interface demonstrates the roll of Asp249 in coordinating the interaction between the T7 loop and the α-tubulin-bound GTP.
The TUBB4A c.745G>A mutation (RefSeq NM_006087.2) identified in this study causes a nonsynonymous change to an aspartic acid (p.Asp249Asn) (Figure 2A) that is highly conserved in all β-tubulins spanning from yeast to primates (Figure 2B),19 and it is predicted to be “probably damaging” (score 1.0) by PolyPhen-2 and “damaging” (score 0.001) by SIFT.21,22 Asp249 is located within the TUBB4A T7 loop, which interacts with the guanosine triphosphate (GTP) nucleotide bound to the N-site of the α-tubulin and is important for the longitudinal interaction between tubulins (Figure 2C).19 Additionally, Asp249 forms a salt bridge with the β-tubulin N-terminal residue Arg2 (altered in DYT4), an interaction that is likely to be important for positioning of T7-loop residues that interact with the α-tubulin-bound GTP (Figure 2D). We predict that disruption of the interaction between Asp249 and Arg2 might lead to inefficient dimerization, reduced microtubule polymerization, or reduced microtubule stability.
Intriguingly, this alteration has been identified in other β-tubulin isotypes or subfamily members in other animal species. For example, a dominant p.Asp249Asn substitution has been described in a hematopoietic-specific β-tubulin isotype in Cavalier King Charles spaniels, where it is responsible for the inherited disease macrothrombocytopenia.23 In Caenorhabditis elegans, a c.745G>A (p.Asp249Asn) mutation in the β-tubulin-encoding gene mec-7 leads to a dominant loss-of-touch-sensitivity phenotype caused by disrupted migration of the specialized touch receptor neuron cells.24 It is likely that the H-ABC-associated TUBB4A mutation might similarly disrupt neuronal growth or axonal function.
Given the severity of H-ABC and its seemingly sporadic presentation, it had been previously proposed that this disease was likely to be caused by a de novo mutation.2 In our family cohort study, however, one family (LD_0638) includes siblings diagnosed with H-ABC. To resolve this apparent incongruity, we performed a detailed investigation of the exome-sequencing data from this family quartet. It revealed that, as expected, the father (LD_0638.2) is homozygous for the reference base and that both affected children (LD_0638.0A and LD_0638.0B) are heterozygous for the TUBB4A mutation. The initial SNP call for the mother (LD_0638.1), however, was annotated as “heterozygous,” despite the fact that she is asymptomatic, suggesting either that we were incorrect about the pathogenicity of the TUBB4A mutation or that some other subtle genetic process was confounding our results. Detailed inspection of the sequence data derived from the mother revealed 193 reads that supported the wild-type allele, whereas only 54 reads supported the mutant allele. This ratio of 3.6:1 is substantially higher than the ratios observed in any of the 11 heterozygous H-ABC-affected individuals in this study (1.0:1–1.4:1; Table 2). Given the high read depth for all individuals at this locus (minimum of 50-fold coverage), these results suggest that individual LD_0638.1 could be mosaic for the c.745G>A variant.
To confirm mosaicism, we collected additional DNA samples from saliva and buccal cells of individuals LD_0638.1 and LD_0638.2. A 216 nt genomic fragment centered on the c.745G>A mutation was amplified with primers 5′-CAACGAGGCACTCTACGACA-3′ and 5′-CTGGTCAGGGGTGCGAAG-3′, and 1 ng of each PCR product was prepared for sequencing with the Nextera XT Library Preparation Kit. Sequencing of the pooled libraries was completed according to the manufacturer’s recommendations with the MiSEQ v.2 instrument and the MiSeq Reagent Kit, which generated paired 150 bp reads. Reads were aligned to the reference human genome (UCSC Genome Browser hg19) with the BWA tool with default parameters.8 We obtained more than 1,000,000-fold coverage over the TUBB4A de novo mutation site in each sample tested (Table 3). The results showed that in the asymptomatic mother (LD_0638.1), the c.745G>A allele was present in 25% of reads from blood, 29% of reads from buccal cell DNA, and 26% of reads from salivary DNA, suggesting a level of mosaicism between 50% and 58% (Table 3). These results suggest that rare de novo mutations that are initially phenotypically neutral in a mosaic individual can be disease causing in the subsequent generation if they are inherited.
Table 3.
Amplicon Sequencing of Family LD_0638 Shows that LD_0638.1 Is Mosaic for the TUBB4A c.745G>A Variant
| Individual | Affected | DNA Source | Wild-Type Reads (G) | Alternative Reads (A) | Percentage of Alternative Reads |
|---|---|---|---|---|---|
| LD_0638.1 | no | blood | 1,339,079 | 446,652 | 25% |
| buccal | 1,023,413 | 426,821 | 29% | ||
| saliva | 1,003,089 | 354,859 | 26% | ||
| LD_0638.2 | no | blood | 1,386,048 | 1580 | 0% |
| buccal | 1,071,589 | 701 | 0% | ||
| saliva | 991,880 | 1,011 | 0% | ||
| LD_0638.0A | yes | blood | 838,773 | 785,684 | 48% |
| LD_0638.0B | yes | blood | 680,222 | 637,585 | 48% |
Individuals with H-ABC have cerebellar atrophy, basal ganglia degeneration with a predilection for the putamen, and a striking lack of cerebral myelin development (hypomyelination). Intriguingly, expression data of TUBB4A in normal human brain samples suggest that it has its highest expression in the cerebellum, putamen, and supratentorial white matter.16 Although individuals with DYT4 share many phenotypic characteristics (including dysphonia, dystonia, and ataxia) with those with H-ABC, MRI features such as hypomyelination and disappearance of the putamen over time are not reported. The p.Arg2Gly alteration causing DYT4 is within the MREI (Met-Arg-Glu-Ile) autoregulatory motif of β-tubulin proteins; this motif is responsible for regulating the abundance of β-tubulins and their encoding mRNA in the cell, which might be a partial explanation for the different presentations of H-ABC and DYT4. All individuals who have the H-ABC phenotype and who have been tested thus far demonstrate a single mutation, but the finding of TUBB4A mutations in individuals with DYT4 suggests that other mutations in this gene could result in a phenotype of primary dystonia with or without involvement of the cerebral white matter.
We hypothesize that the single de novo TUBB4A mutation identified in individuals with H-ABC affects gene function in a dominant-negative fashion and leads to the loss of, or inefficient, dimerization of microtubules. This prediction, however, requires further validation in models of disease and in human material. Given TUBB4A expression in neuronal cells and previous pathologic descriptions,2 we also hypothesize that the H-ABC-related TUBB4A mutation results in a primary disturbance of neurons and the secondary involvement of glial cells. The finding of TUBB4A mutations provides further insight into the complex interplay among cellular cytoskeleton, function, and glial-neuronal interactions.
Acknowledgments
This study was supported by the National Institutes of Health Intramural Program, the Myelin Disorders Bioregistry Project, ZonMw TOP grant 91211005, the Institute for Molecular Bioscience Core Sequencing facility, the NeCTAR Genomics Virtual Lab (G.V.L.), and a University of Queensland Foundation Research Excellence Award. A.V. is supported by K08NS060695. R.J.T. is supported by an Australian Research Council Discovery Early Career Research Award and is a consultant to Isis Pharmaceuticals.
Contributor Information
Ryan J. Taft, Email: r.taft@uq.edu.au.
Adeline Vanderver, Email: avanderv@childrensnational.org.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, http://genome.ucsc.edu
VarScan, http://varscan.sourceforge.net
References
- 1.van der Knaap M.S., Naidu S., Pouwels P.J., Bonavita S., van Coster R., Lagae L., Sperner J., Surtees R., Schiffmann R., Valk J. New syndrome characterized by hypomyelination with atrophy of the basal ganglia and cerebellum. AJNR Am. J. Neuroradiol. 2002;23:1466–1474. [PMC free article] [PubMed] [Google Scholar]
- 2.van der Knaap M.S., Linnankivi T., Paetau A., Feigenbaum A., Wakusawa K., Haginoya K., Köhler W., Henneke M., Dinopoulos A., Grattan-Smith P. Hypomyelination with atrophy of the basal ganglia and cerebellum: follow-up and pathology. Neurology. 2007;69:166–171. doi: 10.1212/01.wnl.0000265592.74483.a6. [DOI] [PubMed] [Google Scholar]
- 3.Mercimek-Mahmutoglu S., Stockler-Ipsiroglu S. Cerebral folate deficiency and folinic acid treatment in hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC) syndrome. Tohoku J. Exp. Med. 2007;211:95–96. doi: 10.1620/tjem.211.95. author reply 97. [DOI] [PubMed] [Google Scholar]
- 4.Mercimek-Mahmutoglu S., van der Knaap M.S., Baric I., Prayer D., Stoeckler-Ipsiroglu S. Hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC). Report of a new case. Neuropediatrics. 2005;36:223–226. doi: 10.1055/s-2005-865715. [DOI] [PubMed] [Google Scholar]
- 5.Wakusawa K., Uematsu M., Tsuchiya S., Haginoya K., Blau N. The cerebrospinal fluid level of 5-methylterahydrofolate in a Japanese boy with hypomyelination with atrophy of the basal ganglia and cerebellum. Tohoku J. Exp. Med. 2007;213:373. doi: 10.1620/tjem.213.373. [DOI] [PubMed] [Google Scholar]
- 6.Wakusawa K., Haginoya K., Kitamura T., Togashi N., Ishitobi M., Yokoyama H., Higano S., Onuma A., Nara T., Iinuma K. Effective treatment with levodopa and carbidopa for hypomyelination with atrophy of the basal ganglia and cerebellum. Tohoku J. Exp. Med. 2006;209:163–167. doi: 10.1620/tjem.209.163. [DOI] [PubMed] [Google Scholar]
- 7.Narumi Y., Shiihara T., Yoshihashi H., Sakazume S., van der Knaap M.S., Nishimura-Tadaki A., Matsumoto N., Fukushima Y. Hypomyelination with atrophy of the basal ganglia and cerebellum in an infant with Down syndrome. Clin. Dysmorphol. 2011;20:166–167. doi: 10.1097/MCD.0b013e32834659a8. [DOI] [PubMed] [Google Scholar]
- 8.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cerritelli S.M., Crouch R.J. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 2009;276:1494–1505. doi: 10.1111/j.1742-4658.2009.06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koboldt D.C., Chen K., Wylie T., Larson D.E., McLellan M.D., Mardis E.R., Weinstock G.M., Wilson R.K., Ding L. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25:2283–2285. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee M.G., Loomis C., Cowan N.J. Sequence of an expressed human beta-tubulin gene containing ten Alu family members. Nucleic Acids Res. 1984;12:5823–5836. doi: 10.1093/nar/12.14.5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Su A.I., Wiltshire T., Batalov S., Lapp H., Ching K.A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hersheson J., Mencacci N.E., Davis M., Macdonald N., Trabzuni D., Ryten M., Pittman A., Paudel R., Kara E., Fawcett K. Mutations in the autoregulatory domain of β-tubulin 4a cause hereditary dystonia. Ann. Neurol. 2012 doi: 10.1002/ana.23832. Published online December 13, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tischfield M.A., Cederquist G.Y., Gupta M.L., Jr., Engle E.C. Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr. Opin. Genet. Dev. 2011;21:286–294. doi: 10.1016/j.gde.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cushion T.D., Dobyns W.B., Mullins J.G., Stoodley N., Chung S.K., Fry A.E., Hehr U., Gunny R., Aylsworth A.S., Prabhakar P. Overlapping cortical malformations and mutations in TUBB2B and TUBA1A. Brain. 2013;136:536–548. doi: 10.1093/brain/aws338. [DOI] [PubMed] [Google Scholar]
- 19.Löwe J., Li H., Downing K.H., Nogales E. Refined structure of α β-tubulin at 3.5 A resolution. J. Mol. Biol. 2001;313:1045–1057. doi: 10.1006/jmbi.2001.5077. [DOI] [PubMed] [Google Scholar]
- 20.Wilcox R.A., Winkler S., Lohmann K., Klein C. Whispering dysphonia in an Australian family (DYT4): a clinical and genetic reappraisal. Mov. Disord. 2011;26:2404–2408. doi: 10.1002/mds.23866. [DOI] [PubMed] [Google Scholar]
- 21.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 23.Davis B., Toivio-Kinnucan M., Schuller S., Boudreaux M.K. Mutation in beta1-tubulin correlates with macrothrombocytopenia in Cavalier King Charles Spaniels. J. Vet. Intern. Med. 2008;22:540–545. doi: 10.1111/j.1939-1676.2008.0085.x. [DOI] [PubMed] [Google Scholar]
- 24.Savage C., Xue Y., Mitani S., Hall D., Zakhary R., Chalfie M. Mutations in the Caenorhabditis elegans beta-tubulin gene mec-7: effects on microtubule assembly and stability and on tubulin autoregulation. J. Cell Sci. 1994;107:2165–2175. doi: 10.1242/jcs.107.8.2165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.