

Abstract
The genetic cause of GAPO syndrome, a condition characterized by growth retardation, alopecia, pseudoanodontia, and progressive visual impairment, has not previously been identified. We studied four ethnically unrelated affected individuals and identified homozygous nonsense mutations (c.262C>T [p.Arg88*] and c.505C>T [p.Arg169*]) or splicing mutations (c.1435–12A>G [p.Gly479Phefs*119]) in ANTXR1, which encodes anthrax toxin receptor 1. The nonsense mutations predictably trigger nonsense-mediated mRNA decay, resulting in the loss of ANTXR1. The transcript with the splicing mutation theoretically encodes a truncated ANTXR1 containing a neopeptide composed of 118 unique amino acids in its C terminus. GAPO syndrome’s major phenotypic features, which include dental abnormalities and the accumulation of extracellular matrix, recapitulate those found in Antxr1-mutant mice and point toward an underlying defect in extracellular-matrix regulation. Thus, we propose that mutations affecting ANTXR1 function are responsible for this disease’s characteristic generalized defect in extracellular-matrix homeostasis.
Main Text
GAPO syndrome (MIM 230740) is the acronym for a complex disorder characterized by growth retardation, alopecia, pseudoanodontia, and, in many but not all cases, progressive optic atrophy.1 Although variations of these phenotypes have been associated with other syndromes, their combination is unique to individuals with GAPO syndrome, and more than 30 cases of various ethnic origins have been described.2–8 Most of the cases are from consanguineous parents, and inheritance patterns within these families have suggested that the disease is inherited as an autosomal-recessive trait. Altough affected individuals have no readily identifiable biochemical or endocrine abnormalities, histopathologic studies have revealed an abnormal accumulation of extracellular material,9,10 and clinical presentation has shown predominant involvement of connective tissue (fibroblasts, chondrocytes, and osteoblasts), venous malformations, and heart, lung, and ocular abnormalities. These clinicopathologic changes point to a generalized defect in extracellular-matrix homeostasis. However, prior research has been unable to identify the genetic roots or reveal the basic molecular mechanisms responsible for GAPO syndrome.
To identify the genetic defect in GAPO syndrome, we performed genomic analysis in four unrelated and ethnically diverse families (Figure 1). The study was approved by institutional review boards, and the investigations were performed according to the Declaration of Helsinki principles. Adults provided informed consent, and the affected child provided assent with parental consent. Consents to publish clinical photographs in scientific journals were also obtained.
Figure 1.

Family Pedigrees, Segregation of the ANTXR1 Mutations, and Facial Appearance of the Probands with GAPO Syndrome
(A) Pedigree of the Czech family, CZE1.
(B) Facial appearance of the Czech proband, II-1, after neurosurgery due to a posthemorrhagic malatic lesion in the frontal cortex.
(C) Pedigree of Egyptian family EGY1.
(D) Facial appearance of Egyptian proband V-3.
(E) Pedigree of Egyptian family EGY2. Note extensive consanguinity and similarly affected relatives.
(F) Facial appearance of Egyptian proband VI-3.
(G) Pedigree of the Sri Lankan family, SRI1.
(H) Facial appearance of the Sri Lankan proband, III-1.
Black symbols denote affected individuals, and open symbols denote unaffected parents and siblings. “NA” indicates that DNA was not available for the investigation. The diagonal slash denotes deceased individuals. The arrows indicate the probands displayed in the corresponding pictures.
We analyzed a previously reported Czech family trio11 (called CZE1) with one affected child (II-1 [Figures 1A and 1B]) who died from a heart attack at the age of 19 years, a previously reported Egyptian family (EGY1) with one affected child (V-3 [Figures 1C and 1D])10 who died from renal failure at the age of 12 years, and two recently identified cases in families from Egypt (EGY2) (VI-4 [Figures 1E and 1F]) and Sri Lanka (SRI1) (III-1 [Figures 1G and 1H]). All four cases demonstrated the major clinical hallmarks of GAPO syndrome as summarized in Table 1.
Table 1.
Main Clinical Findings in the Four Studied Individuals with GAPO Syndrome
| Features |
Cases |
|||
|---|---|---|---|---|
| V-3 from EGY1 | VI-4 from EGY2 | II-1 from CZE1 | III-1 from SRI1 | |
| General | ||||
| Age at evaluation (years) | 3 | 10 | 18 | 4 |
| Gender | male | male | male | male |
| Parental consanguinity (first or second cousins) | + | + | − | + |
| Family history of similarly affected case | − | + | − | − |
| Height | –2 SDs | –3.7 SDs | –4 SDs | –4 SDs |
| Head circumference | –2 SDs | +2 SDs | +1 SD | –2 SDs |
| Bone age | delayed | mild delay | normal for age | delayed |
| Craniofacial | ||||
| Plagiocephaly | + | + | + | − |
| Frontal bossing | + | + | + | + |
| Broad forehead | + | + | + | + |
| Enlarged persistent anterior fontanel | + | + | + | + |
| Widely spaced eyes | + | + | + | + |
| Epicantus | + | + | − | + |
| Depressed nasal bridge | + | + | + | + |
| Short nose | + | + | + | + |
| Long philtrum | + | + | + | + |
| Thick and anteverted nares | + | + | + | + |
| Thick lower lip | + | + | + | + |
| Micrognathia | + | + | + | + |
| Pseudoanodontia | + | + | + | + |
| Skin and Hair | ||||
| Sparse scalp hair (alopecia) | + | + | + | + |
| Scalp pigmented with scars and papules | − | + | − | − |
| Sparse eyebrows and eyelashes | + | + | + | + |
| Ophthalmologic | ||||
| Megalocornea | + | + | + | NR |
| Nystagmus | + | − | − | + |
| Esotropia | + | − | + | − |
| Shallow anterior chamber | − | + | + | NR |
| Bilateral engorged tortuous retinal vessels | − | + | + | + |
| Bilateral optic atrophy | + | − | + | + |
| VEP (abnormal pattern) | + | − | + | NR |
| Other | ||||
| Umbilical hernia | + | + | + | − |
| Hyperextensible joints | + | − | − | + |
| Mild webbing between fingers | − | + | − | − |
| Facial nerve palsy | + | − | − | − |
| MRI brain changes | bilateral high signal of deep white matter at deep parietal and occipital region and around the optic nerve | ND | ND | ND |
Abbreviations are as follows: VEP, visual-evoked potential; NR, not recorded; and ND, not done.
Participants provided venous blood samples, and genomic DNA was isolated with standard technology. We first genotyped genomic DNA from all three Czech family members (i.e., both unaffected parents and the affected child) by using Affymetrix GeneChip Mapping 6.0 Arrays. We used data from both SNP and copy-number probes and identified in all three individuals copy-number alterations relative to a built-in reference as previously described.12 In our analysis of the Czech proband, II-1, no rare or potentially disease-causing deletion or amplification larger than 10 Kb was revealed to be compatible with an expected autosomal-recessive inheritance model. Because ∼0.85% of the proband genome was found to be autozygous, we estimated that the parents might be fifth-degree relatives. Accordingly, when we used the Affymetrix Genotyping Console Software version 4.1 algorithm to compare values from the user’s sample set and SNP-specific distributions derived from a reference set of 200 ethnically diverse individuals,12 we identified in the proband sample two extended autozygous regions on chromosome 2 (chr2: 60,738,227–74,103,186) and chromosome 4 (chr4: 20,458,688–32,646,855), and they contained 114 and 29 genes, respectively (Figure S1A, available online).
To directly identify potential disease-causing mutations, we sequenced and analyzed the exomes of all three individuals from the Czech family as previously described.13 In the resulting data set, we searched for variants that were either private or present in the internal exome database or in the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server with allele frequencies lower than 0.1% and whose genotypes were compatible with an expected autosomal-recessive model of the disease. This analysis revealed 121 candidate variants in proband II-1. However, the only relevant variant compatible with a recessive disorder was a homozygous nonsense mutation (c.505C>T [p.Arg169*]) in ANTXR1, encoding anthrax toxin receptor 1, also known as tumor endothelial marker 8 (TEM8) (RefSeq accession number NM_032208.2) (Table S1 and S2). This mutation is localized in one of the extended homozygous regions identified in the proband’s genome and was inherited from both parents, who are heterozygous carriers. We confirmed the presence of the c.505C>T mutation in the parents and in the proband’s genomic DNA by Sanger sequencing (Figure S2A). The identified mutation was not reported in dbSNP, 1000 Genomes, the Exome Variant Server, or an internal exome database (>120 exomes). It was absent in an additional 200 control samples that we analyzed with an XhoI-based restriction assay of the corresponding PCR-amplified genomic DNA fragments. To confirm the recurrence of ANTXR1 mutations in another affected family, we sequenced ANTXR1 genomic DNA of the proband (VI-4) from family EGY2 and identified a homozygous nonsense mutation (c.262C>T [p.Arg88*]) (Figure S2B), which was also localized in an apparently autozygous region (according to the homozygous genotypes for common SNPs present across the analyzed ANTXR1 genomic sequence and quantitative-PCR results verifying the presence of both mutated alleles; Figure S3) and was not reported in dbSNP, 1000 Genomes, the Exome Variant Server, an internal exome database, or 200 control samples analyzed with a BsaJI-based restriction assay performed on PCR-amplified genomic DNA fragments.
In parallel, DNA samples from two other cases (V-3 from family EGY1 and III-1 from family SRI1) were independently analyzed by exome sequencing performed essentially as above and as described previously.14–17 As in a previous study,14 autozygous regions were identified directly from the exome data of both samples. Strikingly, this resulted in two large overlapping regions of homozygosity on chromosome 2; the total overlap was a ∼27 Mb region (chromosome 2: 43–70 Mb) containing 144 genes (Figures S1B and S1C). The only gene harboring private or rare homozygous coding or splice-site variants within this overlapping region was ANTXR1. For the EGY1 case (V-3), we identified the same nonsense mutation (c.262C>T [p.Arg88*]) as for the EGY2 case (VI-4) (Figure S2C), whereas in the SRI1 case (III-1), we identified a substitution, c.1435–12A>G (Figure S2D). This latter variant is predicted by ESE finder18,19 to generate an alternative strong splice acceptor site 11 nucleotides upstream of the last exon (Figure S4), and this would theoretically result in a frameshift of the complete reading frame of the last exon and proteosynthesis of a truncated ANTXR1 containing a neopeptide composed of 118 unique amino acids in its C terminus (p.Gly479Phefs*119) (Figure S5).
Two of the affected probands, V-3 and VI-4, from Egyptian families EGY1 and EGY2, respectively, harbor an identical c.262C>T [p.Arg88*] mutation. To determine whether these two probands might be distantly related and share a mutated chromosomal segment from a common ancestor, we examined ANTXR1 intragenic SNP haplotypes obtained by exome sequencing (for V-3 from EGY1) and Sanger sequencing (for VI-4 from EGY2). This revealed that the c.262C>T mutations are present on two distinct haplotypes, indicating that these mutations most likely developed independently or that these families share a very old ancestral allele (Figure S6). The c.262C nucleotide belongs to a CpG doublet, making deamination of the cytosine a possible explanation for the recurrence of the mutation.
ANTXR1, also called TEM8, was initially identified as one of the tumor endothelial markers (TEMs) that displays elevated protein levels during tumor angiogenesis.20,21 Soon after its discovery, it was independently identified as the anthrax toxin receptor (ATR).22 Several variants of human ANTXR1 have been proposed to exist on the basis of the identification of rare alternative mRNA splice variants (Figure 2A). The biosynthesis of all known variants is driven by a common signal peptide (amino acids 1–27) and proceeds by cotranslational translocation in the endoplasmic reticulum. The full-length ANTXR1 variant v1, (RefSeq NM_032208.2) is by far the most prevalent transcript found in databases of cDNA and expressed sequence tags. It encodes a single-pass type 1 transmembrane glycoprotein that has a molecular weight of approximately 85 kDa and that is composed of a predicted N-terminal extracellular sequence (amino acids 28–322) containing a von Willebrand type A domain (amino acids 44–215), a transmembrane domain (amino acids 322–342), and large cytoplasmic domain (amino acids 343–564) (isoform 1). Variant v2 (RefSeq NM_053034.2) encodes protein isoform 2, which is structurally similar to variant 1 but contains a much shorter cytoplasmic domain (amino acids 343–368).22 Variant v3 (RefSeq NM_018153.3) encodes protein isoform 3, which does not contain the transmembrane or cytoplasmic domains and is predicted to be secreted.23 Two other transcript variants have recently been identified: v4 (GenBank accession number JX424838.1), potentially encoding membrane-bound protein isoform 4, which, compared to isoform 1, lacks 36 aa residues in its cytoplasmic domain, and v5 (GenBank JX424839.1), potentially encoding secreted protein isoform 5, which, compared to isoform 3, has an alternative C-terminal sequence.24 Because detecting cDNA for alternative splice variants v2–v5 is difficult in that it requires as many as 60 cycles of nested PCR24 and because similar conserved variants in other species have not yet been described, it is currently unclear whether they represent transcriptional noise caused by inappropriate splicing events and whether the encoded protein isoforms are produced at sufficient endogenous levels needed to impact biological function. However, the full-length ANTXR1 isoform 1 has been shown to promote interaction between cells and various components of the extracellular matrix,25,26 link extracellular ligands to the actin cytoskeleton,25,27 and regulate cell spreading.28–30
Figure 2.
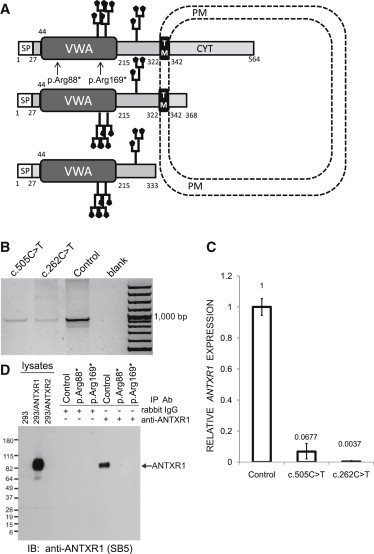
Effects of the Identified ANTXR1 Mutations
(A) A schematic representation of ANTXR1 shows the protein structure, cellular topology, and location of the p.Arg88* and p.Arg169* substitutions. Two potential N-glycosylation sites are depicted. The numbers denote amino acid residues defining the boundaries of predicted ANTXR1 domains. Only ANTXR1 isoforms 1, 2, and 3 are depicted. Abbreviations are as follows: SP, signal peptide; VWA, Von Willebrand factor type A domain; TM, transmembrane domain; CYT, cytoplasmic domain; and PM, plasma membrane.
(B) ANTXR1 cDNA analysis. Total RNA was isolated from pellets from a cultured skin fibroblast cell line with the use of TRIZOL solution (Invitrogen). RNA concentrations were determined spectrophotometrically at A260 nm by NanoDrop (NanoDrop Technologies), and RNA quality was verified with an Agilent 2100 bioanalyser, RNA Lab-on-a-Chip (Agilent Technologies). The first-strand cDNA synthesis was carried out with an oligo-dT primer and SuperScript III Reverse Transcriptase (Life Technologies). ANTXR1 cDNA was PCR amplified from the synthesized first-strand cDNA with oligonucleotide primers designed to span and amplify all three ANTXR1 variants in parallel (Table S2). Lanes 1 and 2 show reduced amounts of RT-PCR products from cases with p.Arg169* and p.Arg88* substitutions, and lane 3 shows the cDNA amount obtained under identical conditions from a control cell line, C. Lane 4 is a negative control. Lane 5 is a 100 bp DNA ladder.
(C) Relative expression levels of ANTXR1 mRNA amounts normalized to glyceraldehydes-3-phosphate dehydrogenase (GAPDH) mRNA amounts in skin fibroblasts. Quantitative PCR was carried out on a StepOne Plus Real Time System (Applied Biosystems). The reactions were carried out in a 96-well plate in a 20 μl reaction volume containing 10 μl 2 × Maxima SYBR Green qPCR Master Mix (Thermo Scientific), 0.2 μM forward and reverse primer, and 5 ng cDNA. Data were analyzed by StepOne Software v.2.0. The comparative Ct (ΔΔ Ct) method was used for normalizing target-gene mRNA to GAPDH mRNA. The relative amounts of the ANTXR1cDNA were significantly reduced in cases compared to control samples. The means ± SD of three experiments performed in triplicate are shown.
(D) Immunoblot (IB) analysis of immunoprecipitated (IP) total-protein extracts showing absence of ANTXR1 in cultured skin fibroblasts from cases with p.Arg88* and p.Arg169* substitutions. Cultured cells were lysed in TNT lysis buffer (50 mM Tris [pH 7.5], 75 mM NaCl, and 1% Triton X-100 plus complete protease inhibitor cocktail [Roche]) and clarified by centrifugation. Protein extracts were quantified with a BCA assay (Pierce), normalized, and immunoprecipitated with a rabbit monoclonal ANTXR1 antibody (clone 37). This rabbit monoclonal antibody was produced as part of a collaboration between Epitomics and the National Cancer Institute and will be described in more detail elsewhere. After immunoprecipitation using protein A agarose, protein extracts were separated by SDS-PAGE, transferred to a PDVF membrane (Millipore), and detected by immunoblotting with SB5 mouse monoclonal ANTXR1 antibodies followed by HRP-conjugated anti-mouse or anti-rabbit F(ab’)2 secondary antibodies (Jackson). Chemiluminescence was visualized with the ECL plus system (Amersham) according to the supplier’s instructions. Lysates of 293 cells stably transfected with an empty vector (293), 293 cells stably expressing human ANTXR1 (293/ANTXR1), and 293 cells stably expressing ANTXR2 (293/ANTXR2) were used as negative, positive, and specificity controls, respectively. Equal protein amounts in the original lysates were immunoprecipitated in parallel with either control rabbit nonspecific IgG antibodies or rabbit ANTXR1 antibodies.
In three of the four GAPO cases, the identified mutations introduce premature stop codons in ANTXR1 mRNA, and in the fourth case (III-1 in SRI1), the mutation most likely results in a loss-of-function allele. From cases II-1 (CZE1) and VI-4 (EGY2), we studied skin fibroblast cell lines harboring the ANTXR1 mutations encoding p.Arg169* and p.Arg88*. To characterize the molecular consequences of the identified mutations on ANTXR1 mRNA expression, splicing, and stability, we isolated total RNA from two cases and control skin fibroblasts and performed RT-PCR and quantitative-PCR analyses. In fibroblasts from affected individuals, we found a single PCR product comparable in size to a control specimen (Figure 2B). Sanger sequencing demonstrated that the obtained PCR products corresponded to cDNA of the major transcript variant v1 of ANTXR1 and independently confirmed the presence of the premature-stop-codon-encoding mutations previously identified in corresponding genomic DNA in affected samples (data not shown). The amounts of ANTXR1 cDNA were significantly reduced in GAPO compared to control samples, most likely through partial degradation of mutated transcripts via nonsense-mediated mRNA decay (NMD) (Figure 2C). Next, we examined whether any mRNAs might have escaped NMD and translated into any form of ANTXR1. We immunoprecipitated ANTXR1 from skin fibroblast lysates and culture media by using a rabbit monoclonal ANTXR1 antibody recognizing an epitope between amino acid residues 28 and 81 of ANTXR1 (A.C. and B.S.C., unpublished data) and then performed immunoblotting for ANTXR1. For immunodetection of ANTXR1, we used either SB5 mouse monoclonal antibodies recognizing the extracellular region of ANTXR1 (amino acid residues 82–145)25,27,31 or the rabbit monoclonal antibody. This analysis revealed the presence of the full-length ANTXR isoform 1 in control fibroblasts but a complete loss of ANTXR1 in both affected fibroblasts (Figure 2D and data not shown). Specific chemiluminiscence signals indicating the presence of the secreted ANTXR1 isoform 3 were not detected in immunoprecipitates obtained from culture media of the control or GAPO cases (not shown). In parallel, we studied and were unable to detect any ANTXR1 in paraformaldehyde-fixed cultured control and GAPO fibroblasts by immunofluorescence analysis. This is in accordance with the results of our immunoblot analysis and the available RNaseq data, which both suggest that ANTXR1 levels and corresponding mRNA amounts are very low in these cells. Next, we used phalloidin staining to evaluate the actin cytoskeleton because ANTXR1 has been previously shown to interact with actin. These studies revealed a striking reorganization of the actin cytoskeletal microfilaments specifically in the GAPO fibroblasts, but β-tubulin and vimentin staining of microtubules and intermediate filaments, respectively, were unaltered (Figure 3). This suggests that ANTXR1, a molecule mediating the coupling of extracellular ligands to the actin cytoskeleton, is crucial for actin assembly and that disruption of the actin network might be the major pathogenetic event leading to altered cell-adhesion properties and progressive extracellular-matrix buildup observed in individuals with GAPO syndrome.
Figure 3.

Immunofluorescence Analysis of Cultured Skin Fibroblasts
The cells were grown on 70 mm2 glass chamber slides (Lab-Tek, Nalge Nunc International) for 48 hr. Then the cells were fixed with 4% paraformaldehyde in PBS, permeabilized in 0.1% TRITON, washed, blocked with 5% BSA in PBS, and incubated in a humidified chamber at 4°C overnight with mouse monoclonal β-tubulin antibody (Sigma) or Vimentin (V9) antibody (BioGenex). For fluorescence detection, species-specific secondary antibodies Alexa Fluor 488 or 555 (Molecular Probes, Invitrogen) were used. For actin staining, Alexa Fluor 488 Phalloidin (Molecular Probes, Invitrogen) was used. Slides were mounted in fluorescence mounting medium Immu-Mount (Shandon Lipshaw) and analyzed by confocal microscopy. XYZ images were sampled according to Nyquist criterion with the Nikon TE2000E C1si laser-scanning confocal microscope with a Nikon PlanApo objective (60×, numerical aperture 1.40) and 488 and 543 laser lines. Images were restored with a classic maximum-likelihood restoration algorithm in the Huygens Professional Software (SVI, Hilversum, the Netherlands). Phalloidin staining demonstrated remarkable alterations in the actin cytoskeletal network in cell lines from GAPO cases with p.Arg169* and p.Arg88* substitutions. No abnormalities in microtubules or intermediate filaments were detected with β-tubulin or vimentin staining, respectively.
Unfortunately, we did not have the opportunity to study mRNA processing, protein production, or the actin network in the fourth GAPO case with ANTXR1 mutation c.1435–12A>G. In this case, the mutation theoretically affects splicing of ANTXR1 mRNA and potentially encodes ANTXR1isoforms 1 and 2 with altered C-terminal cytoplasmic tails; if produced, these isoforms could potentially retain some biological functions. In addition to this, the mutation should not theoretically affect proteosynthesis of the secreted ANTXR1 isoform 3. This could help to explain the evidently milder clinical presentation of this case compared to the other three cases with nonsense mutations.
ANTXR1 is most highly produced in tumor endothelial cells and other tumor stromal cells, which might include both pericytes and fibroblasts.25,32,33 Antxr1-mutant mice with targeted deletion of exon 13—encoding the transmembrane domain—are viable and progressively develop misaligned incisor teeth, and female mice are infertile.34 In another mutant mouse model, complete Antxr1 disruption due to removal of exon 1—encoding the start codon and signal peptide—leads to a moderate excess of extracellular matrix in many tissues, including the ovaries, uterus, skin, hair follicles, cranial sutures of the skull, and the periodontal ligament of the incisors, resulting in dental dysplasia.31 These features are consistent with the clinical presentation of individuals afflicted with GAPO syndrome. It is also notable that some of the individuals with GAPO syndrome have infantile hemangiomas,35 which have been associated with germline heterozygosity for missense mutations in ANTXR136 and dysfunction of the complex formed by VEGFR2, β1 integrin, and ANTXR1.37 Generalized extracellular-matrix-homeostasis defects observed in GAPO-syndrome-affected individuals with ANTXR1 mutations are similar to those found in individuals with juvenile hyaline fibromatosis (MIM 228600) and infantile systemic hyalinosis (MIM 236490), which are allelic disorders caused by mutations in anthrax toxin receptor 2 (ANTXR2), also known as capillary morphogenesis gene 2 (CMG2), an ANTXR1 homolog.38
We conclude that our data, together with recapitulation of many of the phenotypic features characteristic of GAPO syndrome in Antxr1-mutant mice and individuals with ANTXR2 mutations, strongly suggest involvement of ANTXR1 mutations in the generalized extracellular-matrix-homeostasis defect characteristic of this disease. From a clinical perspective, our finding provides essential information for DNA testing in other families. In addition, autopsy tissues and cultured skin fibroblasts from these affected individuals represent an interesting cellular model and potential resource for detailed studies on the pathogenesis of individual clinical symptoms present in GAPO syndrome and studies focused on ANTXR1 functions in general.
Acknowledgments
This work was supported by Charles University institutional programs PRVOUK-P24/LF1/3, UNCE 204011, and SVV2012/ 2645; by the Biotechnology and Biomedicine Centre of the Academy of Sciences and Charles University (CZ.1.05/1.1.00/02.0109); by the European Regional Development Fund; by grant NT13116-4/2012 from the Ministry of Education and Ministry of Health of the Czech Republic; and by the intramural research program of the National Cancer Institute, National Institutes of Health, US Department of Health and Social Services. A.H. was supported by the Netherlands Organization for Health Research and Development (ZonMW 916-12-095). We thank clinical colleagues and families who contributed samples used in this study, as well as members of the Genomic Disorders Group Nijmegen for their technical support. A.C. and B.S.C. are coinventors of filed patents related to the development of ANTXR1 antibodies for cancer therapy.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
ESE finder, http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home
GenBank, http://www.ncbi.nlm.nih.gov/genbank/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Tipton R.E., Gorlin R.J. Growth retardation, alopecia, pseudo-anodontia, and optic atrophy—the GAPO syndrome: report of a patient and review of the literature. Am. J. Med. Genet. 1984;19:209–216. doi: 10.1002/ajmg.1320190202. [DOI] [PubMed] [Google Scholar]
- 2.Goloni-Bertollo E.M., Ruiz M.T., Goloni C.B., Muniz M.P., Valério N.I., Pavarino-Bertelli E.C. GAPO syndrome: three new Brazilian cases, additional osseous manifestations, and review of the literature. Am. J. Med. Genet. A. 2008;146A:1523–1529. doi: 10.1002/ajmg.a.32157. [DOI] [PubMed] [Google Scholar]
- 3.Sinha R., Trikha A., Laha A., Raviraj R., Kumar R. Anesthetic management of a patient with GAPO syndrome for glaucoma surgery. Paediatr. Anaesth. 2011;21:910–912. doi: 10.1111/j.1460-9592.2011.03550.x. [DOI] [PubMed] [Google Scholar]
- 4.Nanda A., Al-Ateeqi W.A., Al-Khawari M.A., Alsaleh Q.A., Anim J.T. GAPO syndrome: a report of two siblings and a review of literature. Pediatr. Dermatol. 2010;27:156–161. doi: 10.1111/j.1525-1470.2010.01100.x. [DOI] [PubMed] [Google Scholar]
- 5.Lei S., Iyengar S., Shan L., Cherwek D.H., Murthy S., Wong A.M. GAPO syndrome: a case associated with bilateral interstitial keratitis and hypothyroidism. Clin. Dysmorphol. 2010;19:79–81. doi: 10.1097/MCD.0b013e32833593d1. [DOI] [PubMed] [Google Scholar]
- 6.Castrillon-Oberndorfer G., Seeberger R., Bacon C., Engel M., Ebinger F., Thiele O.C. GAPO syndrome associated with craniofacial vascular malformation. Am. J. Med. Genet. A. 2010;152A:225–227. doi: 10.1002/ajmg.a.33192. [DOI] [PubMed] [Google Scholar]
- 7.Kocabay G., Mert M. GAPO syndrome associated with dilated cardiomyopathy: an unreported association. Am. J. Med. Genet. A. 2009;149A:415–416. doi: 10.1002/ajmg.a.32638. [DOI] [PubMed] [Google Scholar]
- 8.Demirgüneş E.F., Ersoy-Evans S., Karaduman A. GAPO syndrome with the novel features of pulmonary hypertension, ankyloglossia, and prognathism. Am. J. Med. Genet. A. 2009;149A:802–805. doi: 10.1002/ajmg.a.32686. [DOI] [PubMed] [Google Scholar]
- 9.Wajntal A., Koiffmann C.P., Mendonça B.B., Epps-Quaglia D., Sotto M.N., Rati P.B., Opitz J.M. GAPO syndrome (McKusick 23074)—a connective tissue disorder: report on two affected sibs and on the pathologic findings in the older. Am. J. Med. Genet. 1990;37:213–223. doi: 10.1002/ajmg.1320370210. [DOI] [PubMed] [Google Scholar]
- 10.Meguid N.A., Afifi H.H., Ramzy M.I., Hindawy A., Temtamy S.A. GAPO syndrome: first Egyptian case with ultrastructural changes in the gingiva. Clin. Genet. 1997;52:110–115. doi: 10.1111/j.1399-0004.1997.tb02527.x. [DOI] [PubMed] [Google Scholar]
- 11.Baxova A., Kozlowski K., Obersztyn E., Zeman J. GAPO syndrome (Radiographic clues to early diagnosis) Radiol. Med. (Torino) 1997;93:289–291. [PubMed] [Google Scholar]
- 12.van de Steeg E., Stránecký V., Hartmannová H., Nosková L., Hřebíček M., Wagenaar E., van Esch A., de Waart D.R., Oude Elferink R.P., Kenworthy K.E. Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J. Clin. Invest. 2012;122:519–528. doi: 10.1172/JCI59526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nosková L., Stránecký V., Hartmannová H., Přistoupilová A., Barešová V., Ivánek R., Hůlková H., Jahnová H., van der Zee J., Staropoli J.F. Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am. J. Hum. Genet. 2011;89:241–252. doi: 10.1016/j.ajhg.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Becker J., Semler O., Gilissen C., Li Y., Bolz H.J., Giunta C., Bergmann C., Rohrbach M., Koerber F., Zimmermann K. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2011;88:362–371. doi: 10.1016/j.ajhg.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilissen C., Arts H.H., Hoischen A., Spruijt L., Mans D.A., Arts P., van Lier B., Steehouwer M., van Reeuwijk J., Kant S.G. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am. J. Hum. Genet. 2010;87:418–423. doi: 10.1016/j.ajhg.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoischen A., van Bon B.W., Gilissen C., Arts P., van Lier B., Steehouwer M., de Vries P., de Reuver R., Wieskamp N., Mortier G. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat. Genet. 2010;42:483–485. doi: 10.1038/ng.581. [DOI] [PubMed] [Google Scholar]
- 17.Hoischen A., van Bon B.W., Rodríguez-Santiago B., Gilissen C., Vissers L.E., de Vries P., Janssen I., van Lier B., Hastings R., Smithson S.F. De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat. Genet. 2011;43:729–731. doi: 10.1038/ng.868. [DOI] [PubMed] [Google Scholar]
- 18.Cartegni L., Wang J., Zhu Z., Zhang M.Q., Krainer A.R. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith P.J., Zhang C., Wang J., Chew S.L., Zhang M.Q., Krainer A.R. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum. Mol. Genet. 2006;15:2490–2508. doi: 10.1093/hmg/ddl171. [DOI] [PubMed] [Google Scholar]
- 20.St Croix B., Rago C., Velculescu V., Traverso G., Romans K.E., Montgomery E., Lal A., Riggins G.J., Lengauer C., Vogelstein B., Kinzler K.W. Genes expressed in human tumor endothelium. Science. 2000;289:1197–1202. doi: 10.1126/science.289.5482.1197. [DOI] [PubMed] [Google Scholar]
- 21.Carson-Walter E.B., Watkins D.N., Nanda A., Vogelstein B., Kinzler K.W., St Croix B. Cell surface tumor endothelial markers are conserved in mice and humans. Cancer Res. 2001;61:6649–6655. [PubMed] [Google Scholar]
- 22.Bradley K.A., Mogridge J., Mourez M., Collier R.J., Young J.A. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414:225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 23.Liu S., Leppla S.H. Cell surface tumor endothelium marker 8 cytoplasmic tail-independent anthrax toxin binding, proteolytic processing, oligomer formation, and internalization. J. Biol. Chem. 2003;278:5227–5234. doi: 10.1074/jbc.M210321200. [DOI] [PubMed] [Google Scholar]
- 24.Vargas M., Karamsetty R., Leppla S.H., Chaudry G.J. Broad expression analysis of human ANTXR1/TEM8 transcripts reveals differential expression and novel splice variants. PLoS ONE. 2012;7:e43174. doi: 10.1371/journal.pone.0043174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nanda A., Carson-Walter E.B., Seaman S., Barber T.D., Stampfl J., Singh S., Vogelstein B., Kinzler K.W., St Croix B. TEM8 interacts with the cleaved C5 domain of collagen alpha 3(VI) Cancer Res. 2004;64:817–820. doi: 10.1158/0008-5472.can-03-2408. [DOI] [PubMed] [Google Scholar]
- 26.Hotchkiss K.A., Basile C.M., Spring S.C., Bonuccelli G., Lisanti M.P., Terman B.I. TEM8 expression stimulates endothelial cell adhesion and migration by regulating cell-matrix interactions on collagen. Exp. Cell Res. 2005;305:133–144. doi: 10.1016/j.yexcr.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 27.Yang M.Y., Chaudhary A., Seaman S., Dunty J., Stevens J., Elzarrad M.K., Frankel A.E., St Croix B. The cell surface structure of tumor endothelial marker 8 (TEM8) is regulated by the actin cytoskeleton. Biochim. Biophys. Acta. 2011;1813:39–49. doi: 10.1016/j.bbamcr.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Werner E., Kowalczyk A.P., Faundez V. Anthrax toxin receptor 1/tumor endothelium marker 8 mediates cell spreading by coupling extracellular ligands to the actin cytoskeleton. J. Biol. Chem. 2006;281:23227–23236. doi: 10.1074/jbc.M603676200. [DOI] [PubMed] [Google Scholar]
- 29.Gu J., Faundez V., Werner E. Endosomal recycling regulates Anthrax Toxin Receptor 1/Tumor Endothelial Marker 8-dependent cell spreading. Exp. Cell Res. 2010;316:1946–1957. doi: 10.1016/j.yexcr.2010.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verma K., Gu J., Werner E. Tumor endothelial marker 8 amplifies canonical Wnt signaling in blood vessels. PLoS ONE. 2011;6:e22334. doi: 10.1371/journal.pone.0022334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cullen M., Seaman S., Chaudhary A., Yang M.Y., Hilton M.B., Logsdon D., Haines D.C., Tessarollo L., St Croix B. Host-derived tumor endothelial marker 8 promotes the growth of melanoma. Cancer Res. 2009;69:6021–6026. doi: 10.1158/0008-5472.CAN-09-1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davies G., Rmali K.A., Watkins G., Mansel R.E., Mason M.D., Jiang W.G. Elevated levels of tumour endothelial marker-8 in human breast cancer and its clinical significance. Int. J. Oncol. 2006;29:1311–1317. [PubMed] [Google Scholar]
- 33.Chaudhary A., St Croix B. Selective blockade of tumor angiogenesis. Cell Cycle. 2012;11:2253–2259. doi: 10.4161/cc.20374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu S., Crown D., Miller-Randolph S., Moayeri M., Wang H., Hu H., Morley T., Leppla S.H. Capillary morphogenesis protein-2 is the major receptor mediating lethality of anthrax toxin in vivo. Proc. Natl. Acad. Sci. USA. 2009;106:12424–12429. doi: 10.1073/pnas.0905409106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goucha S., Fazaa B., Ezzine N., Jaber K., Elandaloussi H., Abid R., Kamoun M.R. [GAPO syndrome] Ann. Dermatol. Venereol. 2000;127:501–504. [PubMed] [Google Scholar]
- 36.Jinnin M., Medici D., Park L., Limaye N., Liu Y., Boscolo E., Bischoff J., Vikkula M., Boye E., Olsen B.R. Suppressed NFAT-dependent VEGFR1 expression and constitutive VEGFR2 signaling in infantile hemangioma. Nat. Med. 2008;14:1236–1246. doi: 10.1038/nm.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nicolae C., Olsen B.R. Unexpected matrix diseases and novel therapeutic strategies. Cell Tissue Res. 2010;339:155–165. doi: 10.1007/s00441-009-0874-y. [DOI] [PubMed] [Google Scholar]
- 38.Hanks S., Adams S., Douglas J., Arbour L., Atherton D.J., Balci S., Bode H., Campbell M.E., Feingold M., Keser G. Mutations in the gene encoding capillary morphogenesis protein 2 cause juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am. J. Hum. Genet. 2003;73:791–800. doi: 10.1086/378418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.