

Abstract
The X chromosome lags behind autosomal chromosomes in genome-wide association study (GWAS) findings. Indeed, the X chromosome is commonly excluded from GWAS analyses despite being assayed on all current GWAS microarray platforms. This raises the question: why are so few hits reported on the X chromosome? This commentary aims to examine this question through review of the current X chromosome results in the National Human Genome Research Institute Catalog of Published Genome-Wide Association Studies (NHGRI GWAS Catalog). It will also investigate commonly cited reasons for exclusion of the X chromosome from GWAS and review the tools currently available for X chromosome analysis. It will conclude with recommendations for incorporating X chromosome analyses in future studies.
Main Text
X Chromosome Results in the NHGRI GWAS Catalog
GWAS have identified more than 2,800 associations at p ≤ 5 × 10−8 for nearly 300 traits in more than 1,400 papers, yet only 15 such associations have been reported on the X chromosome (NHGRI GWAS Catalog). Many potential reasons could account for this lag in X chromosome GWAS findings. These include lack of coverage on GWAS chips, differences in the number of genes or variants on the X chromosome compared to the autosomes, differences in the minor-allele frequency of variants on the X chromosome, and current methods' lack of power to detect associations.
Although in the past genotyping chips included few if any X chromosome markers, today genotyping chips include millions of SNPs, tens of thousands of which are on the X chromosome. For example, the Illumina HumanOmni5-Quad BeadChip has over 4.3 million markers, 113,213 of which are on the X chromosome and 511 of which are in the pseudo-autosomal region. Popular content-specific chips, such as the Affymetrix Axiom Exome Genotyping Array, also include X chromosome content: of more than 300,000 markers on this array, 6,900 are specific to the X chromosome. The X chromosome continues to lag behind, though, in GWAS results.
Table 1 compares the chromosome size in base pairs, number of genes per chromosome, number of associations in the NHGRI GWAS Catalog, and number of loci in the NHGRI GWAS Catalog for all 22 autosomal chromosomes along with the X and Y chromosomes. The X chromosome lags far behind all of the autosomal chromosomes both in associations and in distinct loci found in published GWAS. Even chromosome 22, at one-third the size of the X chromosome, has four times as many associations, and only the Y chromosome has fewer (0 associations and 0 loci). The 15 associations at nine distinct loci (Table 2) on the X chromosome (total number of genes = 1,669) also contrast sharply with the 120 reported associations at 33 distinct loci on chromosome 7 (total = 1,880 genes) and the autosomal averages of 128 associations and 26 loci. Although the X chromosome and chromosome 7 are of a similar size (155 Mb and 159 Mb, respectively), the X chromosome has even fewer associations than the much smaller chromosomes 21 (48 Mb) and 22 (51 Mb), which have 28 and 62 associations respectively at 7 and 10 loci. Thus, the number of genes on the X chromosome does not appear to account for the lack of GWAS findings.
Table 1.
A Comparison of Autosomal Chromosomes to the X and Y Chromosomes
| Chr | Size Chr (bp)a | Genes per Chrb | Associations in GWAS Catalogc | Loci in GWAS Catalog |
|---|---|---|---|---|
| 1 | 249,250,621 | 3536 | 260 | 49 |
| 2 | 243,199,373 | 2346 | 248 | 47 |
| 3 | 198,022,430 | 1924 | 136 | 41 |
| 4 | 191,154,276 | 1470 | 118 | 28 |
| 5 | 180,915,260 | 1615 | 141 | 32 |
| 6 | 171,115,067 | 2054 | 309 | 37 |
| 7 | 159,138,663 | 1880 | 120 | 33 |
| 8 | 146,364,022 | 1317 | 123 | 24 |
| 9 | 141,213,431 | 1522 | 110 | 29 |
| 10 | 135,534,747 | 1457 | 147 | 32 |
| 11 | 135,006,516 | 2149 | 197 | 29 |
| 12 | 133,851,895 | 1706 | 176 | 34 |
| 13 | 115,169,878 | 703 | 50 | 18 |
| 14 | 107,349,540 | 1526 | 60 | 20 |
| 15 | 102,531,392 | 1272 | 99 | 23 |
| 16 | 90,354,753 | 1344 | 119 | 21 |
| 17 | 81,195,210 | 1770 | 94 | 19 |
| 18 | 78,077,248 | 546 | 52 | 12 |
| 19 | 59,128,983 | 2085 | 109 | 13 |
| 20 | 63,025,520 | 884 | 67 | 15 |
| 21 | 48,129,895 | 449 | 28 | 7 |
| 22 | 51,304,566 | 855 | 62 | 10 |
| X | 155,270,560 | 1669 | 15 | 9 |
| Y | 59,373,566 | 426 | 0 | 0 |
| Avg bp | Avg Genes | Avg Associations | Avg Loci | |
| 128,986,559 | 1521 | 118 | 24 | |
The comparison considered chromosome size in base pairs, number of genes per chromosome, number of associations in the NHGRI GWAS Catalog (p ≤ 5 × 10−8), and number of loci in the NHGRI GWAS Catalog.
The UCSC Genome Browser (Human Feb. 2009 [GRCh37/hg19] assembly) was used for assessing chromosome size in bp.
The NCBI Map Viewer build 37.2 was used for genes assessing per chromosome.
GWAS Catalog results are for the period through 12/2011. p ≤ 5 × 10−8.
Table 2.
Traits Associated with the Nine Loci and 15 Associations Identified on the X Chromosome in the NHGRI GWAS Catalog
| Locus | Trait |
|---|---|
| Xp22.33 | height10,11 |
| Xp22.2 | colorectal cancer12 (MIM 114500) |
| Xp22.13 | Wilms tumor13 (MIM 194070) |
| Xp22.11 | immune response to smallpox14 |
| Xp11.22 | prostate cancer15–17 (MIM 176807) |
| hypospadias18 (MIM 300856) | |
| Xq12 | prostate cancer19 |
| male-pattern baldness20 (MIM 109200) | |
| Xq13.1 | primary tooth development (number of teeth)21 |
| primary tooth development (time to first tooth eruption)21 | |
| Xq25 | immune response to smallpox14 |
| Xq28 | bilirubin levels22 (MIM 601816) |
| type 1 diabetes23 (MIM 222100) | |
| type 2 diabetes24 (MIM 125853) | |
| immune response to smallpox14 |
p ≤ 5 × 10−8 for associations identified from the NHGRI GWAS Catalog.
Over the last several months, we have reviewed every GWAS paper published from January 2010 to December 2011 and included in the NHGRI GWAS Catalog1, and found that only 33% (242 of 743 papers) reported including the X chromosome in analyses (Figure 1). Examining X chromosome GWAS associations and autosomal associations in the same papers, we were able to compare the minor-allele frequency (MAF), variant functional classification, and p values between X chromosome variants and autosomal variants. By comparing variants only within papers that identified X chromosome hits at p < 1 × 10−5 in the NHGRI GWAS Catalog, we were able to account in part for variations in power, sample size, and genotyping chip across GWA studies; such variations might have prevented detection of X chromosome variants. For the 42 GWASs that were reported from January 2005 to December 2011 and had at least one X chromosome association at p < 1 × 10−5 in the NHGRI GWAS Catalog, minor-allele frequencies for X chromosome variants were similar to those of autosomal variants (0.36 versus 0.38, p = 0.6), but median p values for X chromosome associations were higher than those for autosomal associations by roughly an order of magnitude (2 × 10−6 versus 8 × 10−7, p = 0.2), although not significantly different. Comparing the functional classes assigned to these variants, we found that none of 59 X chromosome variants (p < 1 × 10−5 in the NHGRI GWAS Catalog) was exonic, whereas 4% (31/715) of autosomal variants were. Genic variants were similarly found in a higher proportion on autosomes than on the X chromosome (50% and 32%, respectively), whereas intergenic variants were more common among the X chromosome hits (68% versus 49%, p = 0.01). Given the similarity between the X chromosome and other autosomes in variant MAFs and gene number, these factors cannot explain the under-representation of X chromosome SNPs in GWAS results. However, the higher p values of X chromosome associations, increased chromosome anomalies along with missing call rates, and lack of missense or synonymous variants all point toward potential problems with genotyping accuracy and power.
Figure 1.
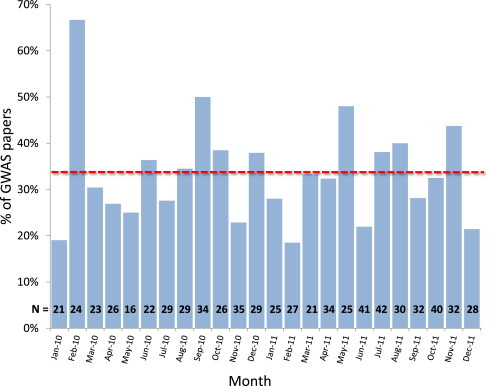
Percent of GWAS Papers with X Chromosome Analyses by Month
Only 33% of papers on average analyzed the X chromosome (dashed line), and there is no discernible trend toward increased analysis. N is the number of papers published each month in the NHGRI GWAS Catalog.
Quality and Power Concerns
The human X chromosome is 155 Mb and contains 1,669 known genes, almost 5% of the genes in the human genome (UCSC Genome Browser).2 In the Online Mendelian Inheritance in Man (OMIM) catalog of human genes and genetic disorders, approximately 7% of phenotypes with a known molecular basis, including autoimmune, cognitive, and behavioral conditions, are X linked, providing ample evidence of the importance of the X chromosome in human disease. Of the 53 studies that have analysis data posted in dbGaP, only 31 (58%) posted results on the X chromosome. Furthermore, less than half of the initial GWAS papers published on these 53 studies (24/49 papers) included analysis of the X chromosome in their publication. This raises the question: why is the X chromosome so often excluded from analysis?
Removal of nonautosomal data is common in GWAS quality-control procedures, but few reasons are given for it. An informal poll of leading GWAS geneticists has reinforced the perception that X chromosome data are under-utilized but shows little consensus as to why. Although some are optimistic that improved imputation methods including the X chromosome will lead to a natural uptake of X chromosome analysis in meta-analyses over time, many available data sets might not lend themselves to meta-analyses or reanalysis without improved methods for X chromosome analysis. Despite the availability of X chromosome imputation methods since 2008, and improved algorithms in 2010, little increase in uptake has been noted over the last 2 years. Concerns were also expressed that current GWAS arrays are still poorly designed for these regions; this problem is seen as one that is unlikely to be overcome by technologic development because sequence data will be required for definitive analyses. The Illumina HumanOmni1-Quad assays only 27,000 X chromosome SNPs, for example, as compared to more than 60,000 on comparably sized chromosome 7. Others felt that array quality was not the problem but rather that the special handling needed for sex-specific analyses and their reduced power represented a significant barrier to their inclusion in analyses and to the ability to detect associations. For example, there might be problems with genotype calling for hemizygous males as a result of the lower intensity of some X chromosome variants, and so such males might cluster differently than females. Additionally, HWE checks and MAF checks might need to be conducted separately for the X chromosome because the expected frequencies are sex dependent. Moreover, most initial GWAS reports produced many useful autosomal findings while excluding the X chromosome from analysis, perhaps setting expectations that autosomal data alone were sufficient for high-profile publications. Challenges in analyzing and interpreting X chromosome data, combined with the plethora of findings obtainable from the autosomes alone, might therefore lead many investigators to under-utilize X chromosome data given that important associations can often be found without it. Overall, though, most geneticists polled agreed that the X chromosome deserved more attention despite its being more difficult to analyze.
A review of X chromosome literature and more than 700 GWASs suggests that the genotyping accuracy on the X chromosome is also often lower than that on other chromosomes because of difficulties involving clustering algorithms, higher frequencies of chromosome anomalies, and more missing data on X chromosome variants. Data from the Gene Environment Association Studies (GENEVA) consortium, for example, showed 4-fold more genotype calls removed as a result of missing call rate filters (p = 0.005) and 14-fold more frequent calls filtered out as a result of chromosome anomalies (p = 0.002) on the X chromosome than on autosomes. Genotyping of the pseudo-autosomal region shared with the Y chromosome and hemizygous males can also be problematic. In addition, random X inactivation in women could potentially obscure important association signals, although up to 15% of X chromosome genes might also escape X inactivation.3 These analytic complexities could further reduce power for X chromosome analyses and make detecting associations even more difficult.
Available Tools
Although tools have been available for X-chromosome-specific analysis since 20074–6 and for X chromosome imputation since the availability of IMPUTEv0.5 in 2008,7 the availability of these tools has not coincided with increases in the overall percentage of GWASs conducting X chromosome analyses; this percentage has remained steadily ∼33% from Jan 2010 to Jan 2012. Methods by Clayton, Zheng, and Thornton4–6 have explored improvements for X chromosome analyses in case-control studies with and without related individuals. These methods take into account the effects of Hardy-Weinberg equilibrium, X-linked SNPs, and X inactivation on X chromosome association analyses and provide multiple valid methods that can be more powerful than commonly used association methods, developed primarily for the autosomes, in detecting associations on the X chromosome.4–6,8 However, of the 42 papers with at least one X chromosome association at p < 1 × 10−5 in the NHGRI GWAS Catalog, only three used X-chromosome-specific methods for their analysis. Of the other 39 papers, 18 conducted their analysis in PLINK,9 which currently codes X chromosome alleles for association tests as A = 0 and B = 1 for males and AA = 0, AB = 1, and BB = 2 for females, which does not account for X inactivation in females. Thus, although improved genotyping and methods developed specifically for the X chromosome might improve the power to detect important associations on the X chromosome in future GWASs, it is also important to make these methods widely accessible to the GWAS community to promote uptake.
Conclusions and Recommendations
With such diversity of opinions and such a wide range of analytical issues, improvements in genotype calling accuracy and methods developed specifically for the X chromosome could facilitate improvements in the power to enhance the detection of important associations. Moreover, SNP data from the X chromosome already exist on many of today’s GWAS arrays. Although such data might not be perfect, the analysis of such existing underutilized data could enhance discovery and further understanding of the genetics of human disease at a modest additional cost. Comparison to targeted sequencing data could also reveal important information about improvements necessary for capturing these underutilized regions of the genome better in future analyses.
Along with improved methods, it is also crucial to recognize the importance of disseminating knowledge of both new and currently available methods broadly to the greater GWAS community and ensuring that these methods are easy to adopt. Current X chromosome analysis methods often require greater bioinformatics expertise to run and therefore might discourage some investigators from looking beyond the easier-to-analyze autosomal regions. However, given that the X chromosome contains ∼5% of the genes in the human genome, many interesting biological insights could be revealed if we end the exclusion of the X chromosome in future GWASs.
Acknowledgments
The authors would like to thank Ian Marpuri for valuable discussions and assistance.
Web Resources
The URLs for data presented herein are as follows:
NHGRI GWAS Catalog, accessed July 25th, 2012, www.genome.gov/gwastudies
UCSC Genome Browser, GRCh37/hg19 Assembly, http://genome.ucsc.edu/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Gene Environment Association Studies: Data Cleaning Reports, https://www.genevastudy.org/Data_Cleaning_Reports
IMPUTE v0.5, https://mathgen.stats.ox.ac.uk/impute/impute_v0.5.html#chrX
References
- 1.Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carrel L., Willard H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- 4.Zheng G., Joo J., Zhang C., Geller N.L. Testing association for markers on the X chromosome. Genet. Epidemiol. 2007;31:834–843. doi: 10.1002/gepi.20244. [DOI] [PubMed] [Google Scholar]
- 5.Clayton D. Testing for association on the X chromosome. Biostatistics. 2008;9:593–600. doi: 10.1093/biostatistics/kxn007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thornton T., Zhang Q., Cai X., Ober C., McPeek M.S.X.M. XM: association testing on the X-chromosome in case-control samples with related individuals. Genet. Epidemiol. 2012;36:438–450. doi: 10.1002/gepi.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marchini J., Howie B., Myers S., McVean G., Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 8.Hickey P.F., Bahlo M. X chromosome association testing in genome wide association studies. Genet. Epidemiol. 2011;35:664–670. doi: 10.1002/gepi.20616. [DOI] [PubMed] [Google Scholar]
- 9.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.N’Diaye A., Chen G.K., Palmer C.D., Ge B., Tayo B., Mathias R.A., Ding J., Nalls M.A., Adeyemo A., Adoue V. Identification, replication, and fine-mapping of Loci associated with adult height in individuals of african ancestry. PLoS Genet. 2011;7:e1002298. doi: 10.1371/journal.pgen.1002298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carty C.L., Johnson N.A., Hutter C.M., Reiner A.P., Peters U., Tang H., Kooperberg C. Genome-wide association study of body height in African Americans: the Women’s Health Initiative SNP Health Association Resource (SHARe) Hum. Mol. Genet. 2012;21:711–720. doi: 10.1093/hmg/ddr489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dunlop M.G., Dobbins S.E., Farrington S.M., Jones A.M., Palles C., Whiffin N., Tenesa A., Spain S., Broderick P., Ooi L.Y., Colorectal Tumour Gene Identification (CORGI) Consortium. Swedish Low-Risk Colorectal Cancer Study Group. COIN Collaborative Group Common variation near CDKN1A, POLD3 and SHROOM2 influences colorectal cancer risk. Nat. Genet. 2012;44:770–776. doi: 10.1038/ng.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turnbull C., Perdeaux E.R., Pernet D., Naranjo A., Renwick A., Seal S., Munoz-Xicola R.M., Hanks S., Slade I., Zachariou A. A genome-wide association study identifies susceptibility loci for Wilms tumor. Nat. Genet. 2012;44:681–684. doi: 10.1038/ng.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kennedy R.B., Ovsyannikova I.G., Pankratz V.S., Haralambieva I.H., Vierkant R.A., Poland G.A. Genome-wide analysis of polymorphisms associated with cytokine responses in smallpox vaccine recipients. Hum. Genet. 2012;131:1403–1421. doi: 10.1007/s00439-012-1174-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eeles R.A., Kote-Jarai Z., Giles G.G., Olama A.A., Guy M., Jugurnauth S.K., Mulholland S., Leongamornlert D.A., Edwards S.M., Morrison J., UK Genetic Prostate Cancer Study Collaborators. British Association of Urological Surgeons’ Section of Oncology. UK ProtecT Study Collaborators Multiple newly identified loci associated with prostate cancer susceptibility. Nat. Genet. 2008;40:316–321. doi: 10.1038/ng.90. [DOI] [PubMed] [Google Scholar]
- 16.Gudmundsson J., Sulem P., Rafnar T., Bergthorsson J.T., Manolescu A., Gudbjartsson D., Agnarsson B.A., Sigurdsson A., Benediktsdottir K.R., Blondal T. Common sequence variants on 2p15 and Xp11.22 confer susceptibility to prostate cancer. Nat. Genet. 2008;40:281–283. doi: 10.1038/ng.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eeles R.A., Kote-Jarai Z., Al Olama A.A., Giles G.G., Guy M., Severi G., Muir K., Hopper J.L., Henderson B.E., Haiman C.A., UK Genetic Prostate Cancer Study Collaborators/British Association of Urological Surgeons’ Section of Oncology. UK ProtecT Study Collaborators. PRACTICAL Consortium Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat. Genet. 2009;41:1116–1121. doi: 10.1038/ng.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Zanden L.F., van Rooij I.A., Feitz W.F., Knight J., Donders A.R., Renkema K.Y., Bongers E.M., Vermeulen S.H., Kiemeney L.A., Veltman J.A. Common variants in DGKK are strongly associated with risk of hypospadias. Nat. Genet. 2011;43:48–50. doi: 10.1038/ng.721. [DOI] [PubMed] [Google Scholar]
- 19.Kote-Jarai Z., Olama A.A., Giles G.G., Severi G., Schleutker J., Weischer M., Campa D., Riboli E., Key T., Gronberg H., UK Genetic Prostate Cancer Study Collaborators/British Association of Urological Surgeons’ Section of Oncology. UK ProtecT Study Collaborators, The Australian Prostate Cancer BioResource. PRACTICAL Consortium Seven prostate cancer susceptibility loci identified by a multi-stage genome-wide association study. Nat. Genet. 2011;43:785–791. doi: 10.1038/ng.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards J.B., Yuan X., Geller F., Waterworth D., Bataille V., Glass D., Song K., Waeber G., Vollenweider P., Aben K.K. Male-pattern baldness susceptibility locus at 20p11. Nat. Genet. 2008;40:1282–1284. doi: 10.1038/ng.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pillas D., Hoggart C.J., Evans D.M., O’Reilly P.F., Sipilä K., Lähdesmäki R., Millwood I.Y., Kaakinen M., Netuveli G., Blane D. Genome-wide association study reveals multiple loci associated with primary tooth development during infancy. PLoS Genet. 2010;6:e1000856. doi: 10.1371/journal.pgen.1000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanna S., Busonero F., Maschio A., McArdle P.F., Usala G., Dei M., Lai S., Mulas A., Piras M.G., Perseu L. Common variants in the SLCO1B3 locus are associated with bilirubin levels and unconjugated hyperbilirubinemia. Hum. Mol. Genet. 2009;18:2711–2718. doi: 10.1093/hmg/ddp203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barrett J.C., Clayton D.G., Concannon P., Akolkar B., Cooper J.D., Erlich H.A., Julier C., Morahan G., Nerup J., Nierras C., Type 1 Diabetes Genetics Consortium Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voight B.F., Scott L.J., Steinthorsdottir V., Morris A.P., Dina C., Welch R.P., Zeggini E., Huth C., Aulchenko Y.S., Thorleifsson G., MAGIC investigators. GIANT Consortium Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat. Genet. 2010;42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]