

Abstract
Cockayne syndrome (CS) is a genetic disorder characterized by developmental abnormalities and photodermatosis resulting from the lack of transcription-coupled nucleotide excision repair, which is responsible for the removal of photodamage from actively transcribed genes. To date, all identified causative mutations for CS have been in the two known CS-associated genes, ERCC8 (CSA) and ERCC6 (CSB). For the rare combined xeroderma pigmentosum (XP) and CS phenotype, all identified mutations are in three of the XP-associated genes, ERCC3 (XPB), ERCC2 (XPD), and ERCC5 (XPG). In a previous report, we identified several CS cases who did not have mutations in any of these genes. In this paper, we describe three CS individuals deficient in ERCC1 or ERCC4 (XPF). Remarkably, one of these individuals with XP complementation group F (XP-F) had clinical features of three different DNA-repair disorders—CS, XP, and Fanconi anemia (FA). Our results, together with those from Bogliolo et al., who describe XPF alterations resulting in FA alone, indicate a multifunctional role for XPF.
Main Text
Cockayne syndrome (CS [MIM 216400 and 133540]) is a rare autosomal-recessive disorder characterized by growth retardation, microcephaly, impairment of nervous system development, pigmentary retinopathy, peculiar facies, and progeria together with abnormal skin photosensitivity.1 On the basis of its clinical severity, CS is classified into three types: (1) CS type I, the classical (moderate) form, which is characterized by normal prenatal growth and the onset of progressive developmental abnormalities in infancy; (2) CS type II, the more severe (early-onset) form, in which prenatal developmental abnormalities are present; and (3) CS type III, a milder form in which progeroid symptoms manifest after middle age (see GeneReview in Web Resources). CS type II is often considered for differential diagnoses of other microcephalic maldevelopmental disorders, such as cerebrooculofacialskeletal syndrome (COFS [MIM 214150], also known as Pena-Shokeir syndrome type II) and Warburg Micro syndrome (MIM 600118).2 In addition to these CS classes, there are rare CS variants that display the combined features of CS and XP (MIM 278700, 610651, 278720, 278730, 278740, 278760, and 278780), termed XPCS. Although CS is commonly associated with sunlight sensitivity, the dermatological phenotypes in CS are milder than those in XP and skin cancers are not found in CS. XPCS individuals, however, present with severe skin phenotypes, including severe photosensitivity, abnormal skin pigmentation, and skin cancer predisposition, all of which are common to XP individuals, who also show developmental abnormalities typical of CS.
XP and CS are associated with deficiencies in nucleotide excision repair (NER), which removes sunlight-induced UV photolesions and bulky DNA base adducts from cellular DNA.3 NER is subdivided into two pathways, global genome NER (GG-NER) and transcription-coupled NER (TC-NER), which are distinct in their initial DNA-damage recognition processes. In contrast to XPCS, which causes defects in both pathways, CS causes specific defects to TC-NER.4 To date, all CS (types I, II, and III)-affected individuals have been associated with mutations in ERCC8 (CSA) or ERCC6 (CSB),5 the products of which function, together with UVSSA (the product of the gene defective in UV-sensitive syndrome 3 [UVSS3 (MIM 614640), also known as UVSS-A])6–8 in the still poorly understood initiation reaction of TC-NER, namely, the processing of the elongating form of RNA polymerase II stalled at the site of UV-induced DNA damage. These products thereby facilitate further recruitment of TFIIH, XPA, and NER incision endonucleases, ERCC1-XPF (ERCC4) and XPG (ERCC5), to carry out the removal of photolesions from actively transcribed genes.4 In contrast to TC-NER, GG-NER is a genome-wide repair mechanism, and it shares the later steps with TC-NER and is deficient in several types of XP and XPCS.
The lack of TC-NER, as found in CS, can be assessed by a deficiency in the RRS test, which measures the recovery of RNA synthesis after UV irradiation. Another marker for DNA repair activity, the unscheduled DNA synthesis (UDS) test measures the GG-NER activity as the level of dNTP incorporation during DNA-repair synthesis after UV damage. Being negative for RRS and positive for UDS is diagnostic for CS. We have developed an efficient system for measuring RRS and UDS activities by using the incorporation of ethynyluracil derivatives and automated fluorescence-based imaging.9,10 In a previous report, we sequenced all coding exons and their neighboring exon-intron boundaries of ERCC8, ERCC6, and UVSSA of 61 CS individuals whose genetic and/or molecular defects had not yet been determined so that we could evaluate whether UVSSA mutations might also result in CS phenotypes.6 Although we did not identify any CS pathogenic mutation in UVSSA, we did identify several CS individuals who did not appear to have any causal mutations in ERCC8, ERCC6, or UVSSA, suggesting that additional gene(s) involved in TC-NER remain to be discovered.
Here, we report on three CS individuals who have normal ERCC8 and ERCC6 but have a defect in either ERCC1 or ERCC4 (XPF). In two cases, the affected individuals displayed typical CS clinical features (CS types I and II). In the third case, the affected individual not only had the combined XPCS phenotype but also had features of a third DNA-repair disorder, Fanconi anemia (FA [MIM 227650]). We propose that ERCC1 and ERCC4 be included as CS- and FA-associated genes.
CS20LO (photographs unavailable) was the daughter of nonconsanguineous parents and had an uneventful antenatal period. At 4–6 months of age, she had microcephaly, micrognathia, and contractures in the knees and elbows and was hypertonic with a dislocated radial head, deep-set eyes, and several moderate skeletal abnormalities, including camptodactyly, adducted thumbs, stiff limbs, steeply sloping acetabulae, wrist contracture, slender long bones with mildly flared metaphyses, and moderate kyphoscoliosis. She displayed brisk reflexes, and there were no feeding concerns. The karyotype was normal. Brain MRI at birth indicated a possible polymicrogyria; a nuclear-magnetic-resonance scan when she was 4 months old revealed large (>2 cm in depth) bilateral subdurals but no major visible malformations. An abnormal electroencephalogram was noticed at 9 months of age. At 16 months of age, she had nystagmus but no additional ophthalmic abnormalities; her corneas were completely clear and showed no evidence of cataracts. The diagnosis was confirmed as CS type II (early onset) because of RRS deficiency (see below). She died at the age of 2.5 years.
CS1USAU (Figure 1A) was born at around 41 weeks with a birth weight of 2.9 kg. He was prenatally diagnosed with microcephaly. His head circumference was at about the 9th percentile in the first few months and was lower than the 2nd percentile by 1 year of age. He developed normally for the first year without obvious abnormalities, except for the microcephaly. At the age of 5 years, he developed multiple unusual plantar warts on his hands and forearms. He had unusual freckling on his hands and the back of his neck and tended to burn easily and quickly, but did not blister badly, when he was exposed to sunlight. Since then, he has been well protected from solar exposure. At 7 years of age, he had deep-set eyes, progressive scoliosis, and multiple contractures in his feet; he required lengthening of the Achilles tendon because of muscle cramps in his hamstrings and calves. His skin was deeply pigmented with rashes and flat freckles. He displayed moderate bilateral hearing impairment, especially in the higher tones (he had normal hearing before the age of 3 years), short stature (height of 110.4 cm [<2nd percentile] and weight of 16.3 kg [<3rd percentile]), microcephaly (head circumference of 45.5 cm [<2nd percentile]), and circulatory problems. He had bilateral astigmatism but no cataracts, attention deficit hyperactivity disorder, and learning disability. There were no obvious abnormalities apart from some delayed myelination on brain MRI at the age of 3 years, although basal ganglia T1 shortening was observed at the age of 7 years. He had severe migraines and headaches. His ambulation lessened over time, and he needed a gastrostomy-jejunostomy tube for feeding. Diagnosis of CS was confirmed at the age of 7 years because of RRS deficiency (see below). He is currently 16 years old.
Figure 1.
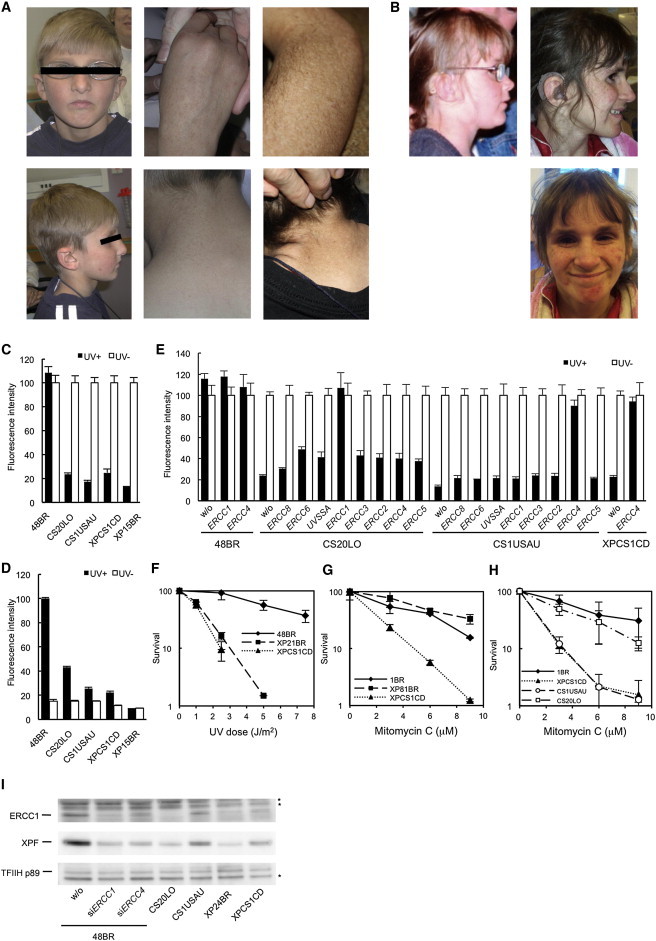
Three CS Individuals Belong to the ERCC1 and XP-F Complementation Groups
(A) CS1USAU, aged 8 years (left and middle panels) and 16 years (right panels). Note the pigmentation and hypopigmented maculas.
(B) XPCS1CD with features of XP, CS, and FA at 7.5 (left) and 11 years old (right).
(C) Reduction of RRS rates after UV irradiation in three CS cell lines (filled bars, 11 J/m2 UVC; empty bars, no UV). RRS was measured by ethynyluridine (EU) incorporation and nuclear fluorescence detection (EU assay). 48BR is a normal control, CS20LO, CS1USAU, and XPCS1CD are CS cells, and XP15BR is affected by XP complementation group A (XP-A).
(D) Reduced level of unscheduled DNA synthesis (UDS) in the CS cells (filled bars, 20 J/m2 UVC; empty bars, no UV).
(E) Complete rescue of the RRS deficiency by the infection of recombinant lentivirus expressing ERCC1 cDNA in CS20LO cells and ERCC4 cDNA in CS1USAU and XPCS1CD cells (filled bars, 10 J/m2 UVC; empty bars, no UV).
(F) The colony-forming ability of XPCS1CD cells after UVC irradiation was compared with that of a normal control (48BR) and that of a cell strain with XP complementation group C (XP21BR).
(G) The colony-forming ability of XPCS1CD cells was compared with that of a normal control (1BR) and that of an XP-A cell strain (XP81BR) after treatment with different doses of mitomycin C (MMC) for 10 min.
(H) MMC sensitivity of different cell strains was determined by MMC treatment for 10 min and measurement of their viability by their ability to incorporate 3H-thymidine (5 μCi/ml; 3 hr incubation) 3 days after treatment.
(I) Immunoblotting of ERCC1 and XPF in cells from CS individuals, ERCC4-deficient XP24BR cells, and normal 48BR cells with the TFIIH-p89 (XPB) subunit as a loading control. 48BR cells were mock transfected or transfected with siRNAs targeting either ERCC1 or ERCC4. Asterisks indicate nonspecific bands. RRS was normalized to activity in nonirradiated cells. UDS activity was normalized to that of normal 48BR cells.
Error bars represent the SD of medians of nuclear fluorescence measurements in quintuplicate samples in (C)–(E) and the SD of means of triplicate experiments in (F)–(H).
XPCS1CD (Figure 1B) was the second child of nonconsanguineous parents. Intrauterine growth failure was noted during pregnancy, and therefore labor was induced at 38 weeks. Her birth weight was between the 0.4th and 2nd percentiles, her developmental milestones were globally delayed, and she required speech therapy. By 18 months of age, she was reported to sunburn with minimal sun exposure in both winter and summer months in the UK. Her parents described that sunburns would develop 24 hr after exposure, peak at 2–3 days, and resolve after 10–14 days. As a result, she was carefully photoprotected and did not have any further sunburn reactions after 4 years of age. When she was 5 years old, she developed progressive bilateral sensorineural deafness, and 2 years later, she presented with worsening ataxia, tremor, and weakness. She had short stature and significant microcephaly. At the age of 7.5 years (Figure 1B, left), her height was 1.4 cm below the 0.4th percentile and her occipitofrontal head circumference (OFC) was 6.5 cm below the 0.4th percentile. By the age of 6 years, she was thrombocytopenic and neutropenic, coinciding with treatment for recurrent infections. Her blood film showed irregularly shaped cells, and she gradually became pancytopenic with transfusion dependence by the age of 9 years. Renal impairment was detected at the age of 10 years, when she presented with hypertension, nephrotic range proteinuria, and rising serum creatinine. The karyotype was normal.
Upon examination at 11 years of age (Figure 1B, right), she was microcephalic with an OFC well below the 0.4th percentile and had extremely short stature. She had deep-set eyes, a prominent nose, small teeth, and freckling over the nose and cheeks. There was marked palmar erythema, and extensive plane warts were seen on the arms. Her skin had an aged appearance, particularly over the dorsal hands. She had five café-au-lait macules. Her neurological examination highlighted a resting tremor, head titubation, and an ataxic gait. She demonstrated saccadic eye movements and an element of vertical nystagmus. There was subtle pyramidal tract weakness distally with extensor plantar reflexes. Sensation was intact. Ophthalmological examination revealed hypermetropia and bilateral blepharitis, but no other ocular abnormalities. MRI of the brain showed increased signal in the peritrigonal white-matter bilaterally. A renal biopsy and bone marrow aspirate were performed under general anesthesia (when she was 11 years old). The bone marrow showed a profoundly hypocellular sample in which all three lineages were present, blast percentage was <1%, and dysplasia was minimal. No clonal cytogenetic abnormalities were detected. A simultaneous full blood count showed a haemoglobin level of 9.4 g/dl (11.5–15.5 g/dl), a red blood cell count of 2.98 × 1012/l (3.40–5.20 × 1012/l), and a mean corpuscular volume of 95.0 fl (80.0–98.0 fl). Studies of liver function were normal. The renal biopsy showed areas of focal segmental glomerular sclerosis and evident thickening of the capillary basement membrane. There was a background of interstitial fibrosis and tubular atrophy with glomerular shrinkage. She died at the age of 12 years after acute deterioration of her renal function. A postmortem examination was not done.
Many of her features were characteristic of CS,11 although pancytopenia and renal failure are not usually associated with this disorder. The exaggerated and prolonged sunburn reactions together with progressive freckling at sun-exposed sites were suggestive of XP, whereas the pancytopenia, bone marrow findings, and recurrent infections are features often seen in FA.
To determine DNA-repair activities in the CS individuals’ cells, we established dermal fibroblast cultures from skin biopsies. Written informed consent was obtained from the individuals’ families, and the experiments were carried out in accordance with the local ethical standards (Nagasaki University Ethical, Legal, and Social Implications committee). We first measured the RRS rate after exposure to 10 J/m2 UVC irradiation.6,9,10 RNA-synthesis activity was significantly reduced in all three CS cell lines compared with normal cells (Figure 1C), indicating a deficiency in TC-NER activity, as expected for CS cells. We also measured UDS, a mark of GG-NER activity,6,9,10 which is not normally affected in CS cells.12 We found a significant reduction in UDS rates in all three CS cell lines (Figure 1D). These results imply that the CS individuals have defective GG-NER and TC-NER pathways, and we therefore presumed that, unlike other CS individuals, they carry defects in genes that commonly function in both the TC-NER and GG-NER pathways. Consequently, we anticipated that these three CS individuals might carry mutations either in ERCC3 (XPB), ERCC2 (XPD), ERCC4, or ERCC5 (XPG) or in ERCC1, and we assayed complementation of the RRS defects by infecting the CS cells lines with recombinant lentiviruses expressing one of these NER-related cDNAs6 (Figure 1E). The RRS defects were dramatically and specifically restored when CS20LO cells were infected with lentiviruses expressing ERCC1 and when CS1USAU and XPCS1CD cells were infected with ERCC4 cDNA, but not when the cells were infected with other viruses (Figure 1E). We conclude that CS20LO is assigned to the ERCC1 complementation group and that, likewise, CS1USAU and XPCS1CD are assigned to XP complementation group F (XP-F).
A hallmark of cells from individuals with FA is a specific defect in the repair of DNA interstrand crosslinks (ICLs). The ERCC1-XPF complex, which is uniquely among the NER proteins, is also involved in ICL repair.13,14 Because several clinical features of XPCS1CD are characteristic of FA, we measured the survival of this individual’s cells after exposure to UV irradiation and to the crosslinking agent mitomycin C (MMC). Figure 1F shows that cells from XPCS1CD are as sensitive to UV irradiation as those from an XP control. In contrast, the XP control cells (in this case, from group A) are, as reported in the literature,15 not sensitive to MMC, whereas XPCS1CD cells are much more sensitive (Figure 1G). Furthermore, chromosome-breakage studies on blood showed increased sensitivity to MMC and an elevated level of chromosome damage upon exposure to the alkylating agent DEB (1,2,3,4-diepoxybutane). However, the levels were lower than expected for a diagnosis of FA (data not shown). In view of these observations, we also examined the MMC sensitivity of CS1USAU and CS20LO cells and found that the former were also very sensitive to MMC, whereas the latter were marginally, if at all, sensitive (Figure 1H).
ERCC1 is needed for stabilizing and enhancing XPF endonuclease activity,16,17 and indeed, using immunoblotting, we observed a significant reduction in the expression level of XPF concurrently with the reduction of ERCC1 expression in CS20LO cells (Figure 1I). In contrast, XPF and ERCC1 levels were modestly reduced in CS1USAU and XPCS1CD cells (Figure 1I).
To determine the genetic changes in the three CS individuals, we carried out (1) PCR-based gDNA amplification of the exons and neighboring exon-intron boundaries of ERCC1 from CS20LO cells and of ERCC4 from CS1USAU cells and subsequent direct sequencing of the amplified gDNA fragments and (2) RT-PCR of mRNA and sequencing of the cDNA from XPCS1CD cells. In CS20LO cells, a homozygous mutation, c.693C>G, in exon 7 of ERCC1 (RefSeq accession number NM_001983.3) resulted in amino acid substitution p.Phe231Leu, located in the C-terminal XPF-interacting helix-hairpin-helix (HhH) domain (Figure 2A). ERCC1 pathogenic changes identified in NER-deficient diseases and in other genetic disorders are extremely rare; only two cases have been reported.18,21 Individual 165TOR,21 who had clinical features overlapping those of CS and was diagnosed with a severe form of COFS, was heterozygous for the ERCC1 c.693C>G (p.Phe231Leu) mutation found in our CS individual. In 165TOR, the other ERCC1 pathogenic allele was c.472C>T (p.Gln158*); however, the mutated transcript is presumed to be subject to nonsense-mediated mRNA decay (NMD). XP202DC18 was a XP individual who developed progressive neurodegeneration during adolescence and died at the age of 37 years. He was compound heterozygous for nonsense substitution p.Lys226* near the C terminus and splice-site mutation c.604-26G>A, which might have affected the expression level of wild-type ERCC1. In this individual, a low amount of the wild-type ERCC1 allele expression might have been sufficient to ameliorate the more severe aspects of the disorder.
Figure 2.
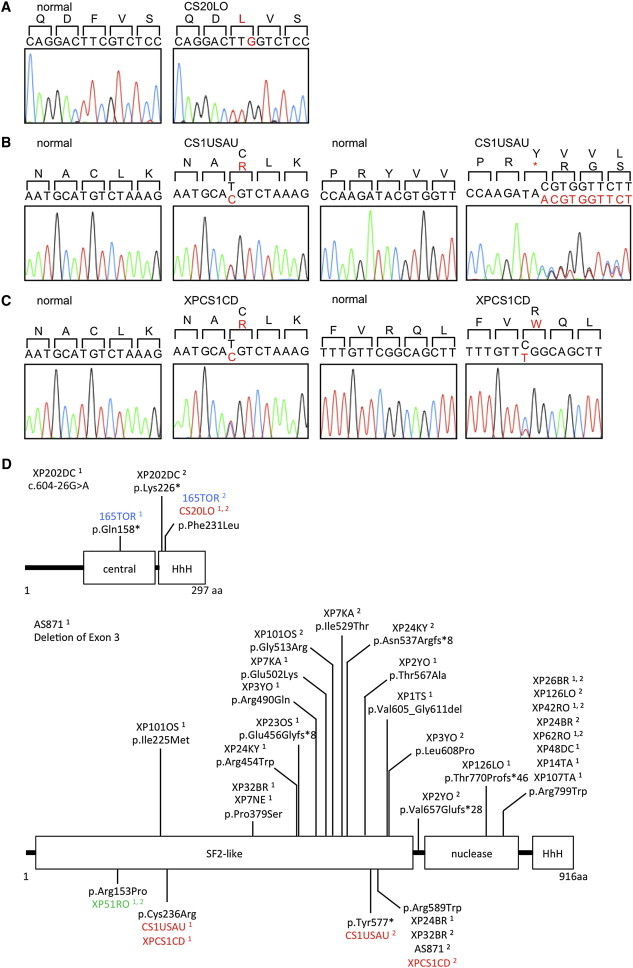
Locations of the ERCC1 and ERCC4 Mutations Identified in the CS Cells and in Known NER Disorders
(A) The homozygous c.693C>G single-nucleotide variant (SNV) in ERCC1 exon 7 in the CS individual, CS20LO; the altered amino acid, Phe231, is shown in red.
(B) The heterozygous c.706T>C SNV in ERCC4 exon 4 and the 1 base insertion, c.1730_1731insA, in ERCC4 exon 8 are identified in the CS individual, CS1USAU. The altered amino acids, Cys236 and Tyr577, are shown in red.
(C) The heterozygous c.706T>C SNV in ERCC4 exon 4 and the heterozygous c.1765C>T SNV in XPCS1CD. The altered amino acids, Cys236 and Arg589, are shown in red.
(D) The structure of ERCC1 and XPF and the amino acid alterations reported here and published previously.18–20 The cases shown in colors are as follows: XP, black; COFS, blue; XFE, green; and CS or XPCS, red. Superscripted 1 and 2 designate the number of discrete alleles.
CS1USAU cells had two heterozygous mutational changes in ERCC4 (RefSeq NM_005236.2): (1) an exon 4 mutation, c.706T>C, causing an amino acid substitution, p.Cys236Arg, located in the N-terminal large SF2 helicase domain and (2) an exon 8 frameshift insertion, c.1730_1731insA, which generated a premature stop codon, p.Tyr577* (Figure 2B). XPCS1CD had two ERCC4 heterozygous mutations, c.706T>C and c.1765C>T, resulting in changes p.Cys236Arg (the same mutation as we found in CS1USAU) and p.Arg589Trp (previously found in three XP individuals) (Figure 2C), respectively.18 Previously identified alterations in ERCC1 and XPF, together with those identified in this paper, are shown in Figure 2D.
XPF pathogenic alterations are normally associated with mild XP cases without severe developmental abnormalities.18 There has been one exceptional case, XFE progeroid syndrome (MIM 610965). The affected individual, XP51RO, carried a homozygous mutation resulting in a p.Arg153Pro change in the SF2 domain of XPF.22 In all reported cases with mild XP-F, the pathogenic mutations are hypomorphic and there is a substantial amount of residual XPF in the cells; however, in the XFE individual cells, XPF expression was severely reduced.22 In contrast, in CS1USAU and XPCS1CD cells, a substantial amount of XPF remained (Figure 1I), suggesting that the pathogenic allele is fully expressed and the altered protein is stable (also see later purification of the ERCC1-XPF complex). These observations indicate that p.Cys236Arg causes XPF dysfunction and confers a CS phenotype on individuals CS1USAU and XPCS1CD.
XPF and ERCC1 form a heterodimeric structure-specific endonuclease, the ERCC1-XPF complex, which cleaves on the 5′ side of the UV-damaged DNA at the incision step of NER.23–25 The proteins form a complex through their C-terminal HhH domains and stabilize the counterpart when in a complex. To test whether the p.Phe231Leu altered ERCC1 and the p.Cys236Arg altered XPF form proper ERCC1-XPF complexes, we introduced the altered proteins (V5-tagged) into 293FT cells and performed immunoprecipitation to check the binding affinity between ERCC1 and XPF. Although the ERCC1 p.Phe231Leu substitution is located in the XPF-binding HhH domain and the residue interfaces with two alpha helices of the XPF HhH domain, the altered ERCC1 binding capacity to XPF was unaffected by the substitution (Figure 3A). Similarly, we observed no significant decrease in coprecipitation of wild-type ERCC1 in the complex when p.Cys236Arg XPF was expressed (Figure 3B). The ERCC1-XPF complex also interacts with TFIIH, as shown by both in vitro and in vivo experiments.26 We therefore tested the binding affinity of altered ERCC1 and XPF with the XPB-p89 subunit of TFIIH. Although there was no significant reduction in binding affinity of p.Phe231Leu ERCC1 expressed in 293FT cells (Figure 3A), we observed, after UV irradiation, less p89 in immunoprecipitates from cells expressing p.Cys236Arg XPF than from wild-type controls (Figure 3B). Recent studies also demonstrated that the three known structure-specific endonuclease complexes (SSEs)—ERCC1-XPF, SLX4 (FANCP)-SLX1, and MUS81 (SLX3)-EME1 (SLX2)—interact with each other in crosslink-repair pathways. Importantly, mutations in SLX4 have been identified in several FA individuals, and SLX4 acts as a scaffold and plays a key role in the coordination of these interactions.27–29 We therefore tested whether one of these interactions, namely the binding affinity of the altered protein with MUS81 (Figure 3B), is compromised in cells expressing the XPF p.Cys236Arg substitution. There was no impact on the binding affinity, suggesting that this was not the cause of the FA features observed in XPCS1CD.
Figure 3.
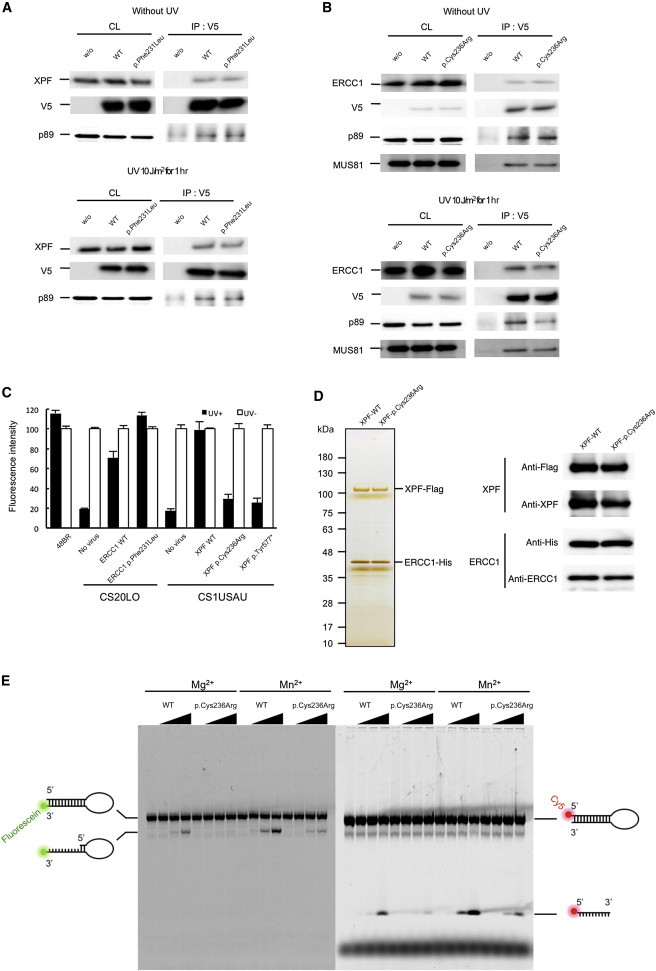
ERCC1 and XPF Interaction in the CS Cells
(A) V5-tagged wild-type and p.Phe231Leu altered ERCC1 interactions with the endogenous wild-type XPF were assayed by immunoprecipitation either without UV irradiation or 1 hr after 10 J/m2 of UV irradiation.
(B) V5-tagged wild-type and p.Cys236Arg altered XPF were expressed in 293T cells, and interactions with the endogenous wild-type ERCC1 were assayed by immunoprecipitation from extracts of cells either without UV irradiation or 1 hr after 10 J/m2 of UV irradiation. Interactions were detected by immunoblotting with antibodies against the V5-tagged ERCC1 and XPF (1H6, MBL), MUS81 (MTA30 2G10/3, Santa Cruz), ERCC1 (8F1, Santa Cruz), and XPF (Ab-1, Thermo scientific). Abbreviations are as follows: CL, crude lysate (10% load); and IP, immunoprecipitate.
(C) Rescue of the RRS deficiency was assayed by the infection of recombinant lentivirus expressing either wild-type or mutant ERCC1 and ERCC4 cDNAs (filled bars, 11 J/m2 UV; empty bars, no UV). RRS was normalized to activity in nonirradiated cells. Error bars represent the SD of medians of nuclear fluorescence measurements in quintuplicate samples.
(D) Purification of the recombinant ERCC1-XPF complexes. FLAG-tagged (C terminus) wild-type and p.Cys236Arg altered XPF were coexpressed with the 6 × His-tagged ERCC1 (C terminus) in 293T cells. The cells were harvested and lysed in lysis buffer (50 mM Tris-HCl, 150 mM KCl, 1 mM EDTA, 1% NP-40, 10% glycerol, 0.25 mM PMSF, protease-inhibitor cocktail [PIC, Roche], pH 7.5). Tandem affinity purification was performed with mouse anti-FLAG IgG-conjugated beads (M2 agarose, SIGMA) followed by TALON metal affinity resin (Clontech). Cell lysates were incubated with anti-FLAG agarose beads and were subsequently washing with salt buffer (20 mM Tris-HCl, 1 M KCl, 0.1 mM EDTA, 10% glycerol, PIC, pH 7.5) and starting buffer (20 mM Tris-HCl, 800 mM KCl, 10% glycerol, 10 mM imidazole, PIC, pH 7.5). Binding proteins were eluted with starting buffer containing 1 mg/ml FLAG peptide. The eluted fractions were incubated with TALON metal affinity resin (Clontech) and were then sequentially washed with buffer A (40 mM HEPES, 1,000 mM NaCl, 10% glycerol, 10 mM Imidazole, PIC, pH 7.5) and buffer B (40 mM HEPES, 100 mM NaCl, 10% glycerol, 10 mM Imidazole, PIC, pH 7.5). The purified complex was sequentially eluted with elution buffer A (40 mM HEPES, 100 mM NaCl, 10% glycerol, 100 mM Imidazole, PIC, pH 7.5) and elution buffer B (40 mM HEPES, 100 mM NaCl, 10% glycerol, 300 mM Imidazole, pH 7.5). Purified complex was dialyzed against GF buffer (25 mM HEPES, 150 mM NaCl, 10% glycerol, 5 mM beta-mercaptoethanol, PIC, pH 8.0) and concentrated. The purified complexes were resolved by SDS-PAGE and detected by silver staining (100 ng of total protein). Purity of the complexes was assessed by immunoblotting with antibodies against the FLAG (MBL) and 6 × His (MBL) tags, as well as ERCC1 and XPF. No endogenous XPF was detected.
(E) Endonuclease activity of the wild-type and altered ERCC1-XPF complexes was determined with fluorescently labeled 12 bp stem-dT22-loop DNA templates. Either 3′-fluorescein- or 5′-Cy5-labeled oligonucleotides (5′-GCCAGCGCTCGG-dT22-CCGAGCGCTGGC-3′) were purchased from SIGMA and self-annealed in TE buffer. Nuclease incision reactions were performed on 200 fmol of the DNA templates and 0, 25, 50, 100 ng of the purified ERCC1-XPF complexes in a total volume of 15 μl nuclease reaction buffer (25 mM HEPES, 40 mM NaCl, 10% glycerol, 0.5 mM beta-mercaptoethanol, and 0.1 mg/ml BSA and either 2 mM MgCl2 or 0.4 mM MnSO4, pH 8.0). The reaction mixtures were incubated at 30°C for 1 hr. Samples were separated on urea-denatured 15% PAGE, and the cleaved products were analyzed by a Typhoon imager (GE).
To further test whether the altered ERCC1 and XPF proteins retain any DNA-repair functions, we performed complementation analyses of the RRS defects by expressing mutant ERCC1 and ERCC4 cDNAs in the ERCC1- and XPF-deficient CS cells, respectively (Figure 3C). Consistent with the previous report describing a correction of UV sensitivity after transfection with the ERCC1 c.693C>G (p.Phe231Leu) pathogenic mutant cDNA into ERCC1-deficient 165TOR cells,21 we observed that RRS in the ERCC1-deficient CS20LO cells was fully restored upon infection with the lentivirus expressing c.693C>G (p.Phe231Leu) mutant ERCC1 cDNA, whereas expression of the p.Cys236Arg altered XPF failed to restore RRS levels of the XPF-deficient CS1USAU cells.
We then purified recombinant ERCC1-XPF complexes and determined their endonuclease activity. As observed in Figure 1I, the altered p.Cys236Arg XPF complex was stable (Figure 3D). Because stem-loop structures are good substrates for the ERCC1-XPF complex,30 we performed in vitro incision assays with fluorescent-labeled DNA templates containing a 12 bp stem-dT22-loop.19,30 We observed significant reduction of the endonuclease activity of the p.Cys236Arg altered complex (Figure 3E).
These data indicate that XPF malfunction, caused by the lack of proper interaction between TFIIH and ERCC1-XPF, and a substantial reduction of the stem-loop incision activity of the endonuclease complex cause the CS and FA phenotypes in CS1USAU and XPCS1CD, whereas the reason for ERCC1 deficiency in CS20LO cells remains unclear.
Unlike NER-associated genes that are responsible exclusively for CS and UVSS phenotypes (i.e., ERCC8, ERCC6, and UVSSA), ERCC4 and ERCC1 are presumably indispensable for fetal development because there is no known individual carrying homozygous or compound-heterozygous null mutations in either of the genes. Given that we found that the p.Phe231Leu altered ERCC1 retains normal NER function, we decided to determine the precise expression levels of the mutant ERCC1 and ERCC4 alleles identified in two of the CS individuals. We undertook allele-specific quantitative RT-PCR (qRT-PCR) with sets of primers that selectively amplify the wild-type or the mutant ERCC1 and ERCC4 cDNAs (Table 1). In CS20LO cells, primer set ERCC1-WT and ERCC1-Rv1 allowed specific amplification of the wild-type ERCC1 allele (Figure 4A, left), whereas another set of primers, ERCC1-p.Phe231Leu and ERCC1-Rv1, amplified the c.693C>G (p.Phe231Leu) allele in exon 7 (Figure 4A, middle); primers ERCC1-com1 and ERCC1-Rv1 amplified both the wild-type and the mutant alleles (Figure 4A, right). We observed a very low level (50-fold < wild-type) of expression of the ERCC1 c.693C>G (p.Phe231Leu) allele in CS20LO cells (Figure 4A, middle and right), suggesting that the ERCC1-null phenotype can be ascribed to this drastic attenuation of ERCC1 mRNA expression. Subsequently, expression of the wild-type and the two mutated ERCC4 alleles in CS1USAU cells was examined. Similarly to how we quantified ERCC1 mRNA, we designed sets of primers to specifically amplify the c.706T>C (p.Cys236Arg) allele located in exon 4 (Figure 4B, middle) and the c.1730_1731insA (p.Tyr577*) allele in exon 8 (Figure 4C, middle), as well as their corresponding wild-type alleles (Figure 4B and 4C, left). We confirmed the expression of the c.706T>C (p.Cys236Arg) allele specifically in CS1USAU cells (Figure 4B, middle and right) with primers XPF-p.Cys236Arg and XPF-Rv1, whereas the c.1730_1731insA (p.Tyr577*) allele, measured by primers XPF-p.Tyr577* and XPF-Rv2, was subjected to NMD, because its expression level was very low in the CS cells (Figure 4C, middle). Given these qRT-PCR data and the protein expression levels in the CS cells (Figure 1I), we conclude that the ERCC1-deficiency phenotype in CS20LO results from a very low level of expression of the mutant allele and that the XPF deficiency in CS1USAU and XPCS1CD is mainly caused by protein malfunction. A summary of our findings is presented in Table 2.
Table 1.
Primers Used for Allele-Specific Amplifications
| Primer Designation | Primer Sequence |
|---|---|
| ERCC1-WT1 | 5′- CTGATGGAGAAGCTAGAGCAGGACTTC-3′ |
| ERCC1-p.Phe231Leu | 5′- CTGATGGAGAAGCTAGAGCAGGACTTG-3′ |
| ERCC1-com1 | 5′- CCTGATGGAGAAGCTAGAGCAGGACTT-3′ |
| ERCC1-Rv1 | 5′- GGTCAGACATTCAGTCACCCGC-3′ |
| XPF-WT1 | 5′- GACTGCTATACTGGACATTTTAAATGCAT-3′ |
| XPF-p.Cys236Arg | 5′- GACTGCTATACTGGACATTTTAAATGCAC-3′ |
| XPF-com1 | 5′- AGACTGCTATACTGGACATTTTAAATGCA-3′ |
| XPF-Rv1 | 5′- TCCAAGCTGGTGCCACAAA-3′ |
| XPF-WT2 | 5′- AGGGTACTACATGAAGTGGAGCCAAGATAC-3′ |
| XPF-p.Tyr577* | 5′- AGGGTACTACATGAAGTGGAGCCAAGATAA-3′ |
| XPF-com2 | 5′- AAGGGTACTACATGAAGTGGAGCCAAGATA-3′ |
| XPF-Rv2 | 5′- CCCTCAGAGGTTTCCCAGGC-3′ |
Figure 4.
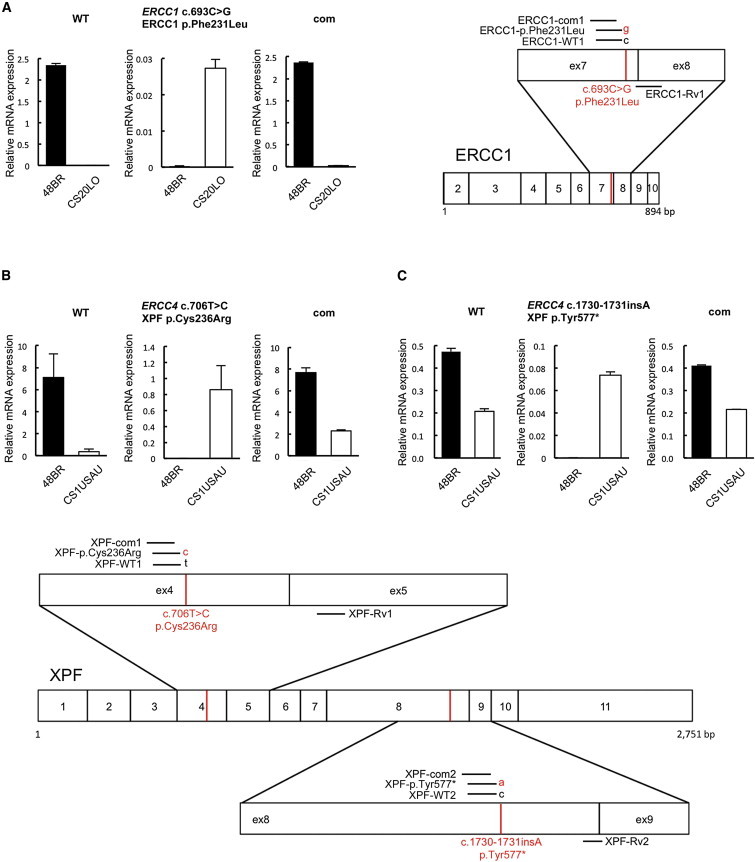
Expression of the Pathogenic Alleles in the CS Cells
Total RNA was extracted with ISOGEN reagent (NIPPON GENE) according to the manufacturer’s instruction. One microgram of total RNA was reverse transcribed with a high-capacity RNA-to-cDNA kit (Applied Biosystems, Life Technologies). Quantitative PCR amplification and real-time detection were carried out in a Thermal Cycler Dice Real-Time System (TaKaRa Bio) with SYBR Premix Ex TaqII (TaKaRa Bio) and a QuantiTect SYBR Green PCR kit (QIAGEN). For each sample, relative mRNA levels were normalized against HPRT1 mRNA expression. Error bars represent the SD of means of triplicate experiments.
(A) Selective quantitative amplification of the wild-type and the mutated c.693C>G (p.Phe231Leu) ERCC1 alleles in CS20LO cells and normal 48BR cells. Allele-specific primers selectively amplified the wild-type (c.693C) allele (ERCC1-WT1 and ERCC1-Rv1, left panel), the CS pathogenic mutant (c.693C>G) allele (ERCC1-p.Phe231Leu and ERCC1-Rv1, middle panel), and both alleles at once (ERCC1-com1 and ERCC1-Rv1, right panel).
(B) Selective quantitative amplification of the wild-type and the mutated c.706T>C (p.Cys236Arg) ERCC4 alleles in CS1USAU cells and normal 48BR cells. Allele-specific primers selectively amplified the wild-type (c.706T) allele (XPF-WT1 and XPF-Rv1, left panel), the CS pathogenic mutant (c.706T>C) allele (XPF-p.Cys236Arg and XPF-Rv1, middle panel), and both alleles at once (XPF-com1 and XPF-Rv1, right panel).
(C) Selective quantitative amplification of the wild-type and the c.1730_1731insA (p.Tyr577*) ERCC4 alleles. Allele-specific primers selectively amplified the wild-type (c.1731C) allele (XPF-WT2 and XPF-Rv2, left panel), the CS pathogenic mutant (c.1730_1731insA) allele (XPF-p.Tyr577* and XPF-Rv2, middle panel), and both alleles at once (XPF-com2 and XPF-Rv2). Locations of the primer sets used for the qRT-PCR experiments are depicted on the right-hand side. Transcripts from HPRT1 were used as a quantification control. Primers used for the qPCR are listed in Table 1.
Table 2.
Summary of Phenotypes of CS Cells
|
Individual |
|||
|---|---|---|---|
| CS20LO | CS1USAU | XPCS1CD | |
| RRS | – | – | – |
| UDS | – | – | – |
| Virus complementation with ERCC1 wild-type cDNA (RRS)a | + | – | – |
| Virus complementation with ERCC4 wild-type cDNA (RRS)a | – | + | + |
| XPF expression | – | +/– | +/– |
| ERCC1 expression | – | +/– | +/– |
| Protein alteration | ERCC1 p.Phe231Leu | XPF p.Cys236Arg and p.Tyr577* | XPF p.Cys236Arg and p.Arg589Trp |
| ERCC1-XPF IP | + | + | + (p.Cys236Arg) |
| p89 IP | + | +/– | +/– (p.Cys236Arg) |
| Virus complementation with ERCC1 c.693C>G (p.Phe231Leu) cDNA (RRS)a | + | ND | ND |
| Virus complementation with ERCC4 c.706T>C (p.Cys236Arg) cDNA (RRS)a | ND | – | ND |
| ERCC1-XPF nuclease activity | ND | – (p.Cys236Arg) | – (p.Cys236Arg) |
| Mutant mRNA level | V low | +/– | ND |
Abbreviations are as follows: +, normal; –, defective or absent; +/–, intermediate; IP, immunoprecipitation; ND, not done.
Effect of the indicated cDNA on the RRS response.
We have identified three CS individuals who carry pathogenic mutations in either ERCC1 or ERCC4. Approximately 25 XP-F individuals have been described in the literature.18 The majority of them are XP individuals, who, compared with individuals affected by XP complementation group C, have relatively mild skin problems and skin cancers that appear relatively later in life. Only XFE individual XP51RO had a much more severe phenotype.22 Like our CS individuals, he had short stature, cachexia, and microcephaly. He had mild learning difficulties, hearing loss, and visual impairment. He was sensitive to the sun from birth and had dry, atrophic skin and irregular pigmentation on sun-exposed areas. Laboratory studies indicated renal insufficiency, and he died at the age of 16 years from severe pneumonia and multisystem organ failure. This individual had many features in common with XPCS1CD, but there was no report of pancytopenia. He was homozygous for p.Arg153Pro in XPF.
Somewhat surprisingly, the previously reported ERCC1-deficient severe COFS individual was compound heterozygous for the same pathogenic p.Phe231Leu change together with a termination substitution, p.Gln158*,21 whereas our CS type II individual was homozygous for this substitution. Because the p.Phe231Leu altered ERCC1 retained normal function, differences in severity of the clinical features might be explained by the dosage of ERCC1 expression. We hypothesize that the relatively milder clinical features observed in our CS individual might be due to the fact that the biallelic expression of the mutated ERCC1 allele produced twice the level of functional ERCC1 in the CS individual as in the COFS individual. This idea is also supported by the observation that the other ERCC1-deficient XP individual, who displayed milder clinical features, harbored a splice-site mutation that most likely allowed readthrough of a significant amount of normal protein.
We identified a heterozygous XPF alteration, p.Cys236Arg, in CS individuals CS1USAU and XPCS1CD. CS1USAU displayed mild skin problems in the range defined by normal CS features, but he has been very well protected from sun exposure. He displayed hypopigmented macules (Figure 1A), which are typical of XP and often occur on the limbs and other sun-exposed sites before “freckling.” In view of this and his early freckling, it seems appropriate to classify this affected individual as also having the combined XPCS phenotype. However, XPCS1CD, but not CS1USAU, also exhibited hematological abnormalities characteristic of FA. Surprisingly, the second allele in the milder individual, CS1USAU, was a functionally null termination mutation, which was subjected to NMD, whereas in the more severely affected XPCS1CD, the second allele harbored c.1765C>T (p.Arg589Trp), which is found in several XP individuals and results in mislocalization of the ERCC1-XPF complex to the cytoplasm and thus causes a severe XP phenotype when combined with certain other alleles.19 XP32BR (with p.Arg589Trp and p.Pro379Ser) was a mildly affected XP individual in his teens and had no neurological problems; XP24BR (with p.Arg589Trp and p.Arg799Trp) was also mildly affected but is now showing some abnormal neurological and CS-like facial features in her 40s (our unpublished data); AS871 (with p.Arg589Trp resulting from the deletion of exon 3) had severe skin symptoms and neurodegeneration.19 The reason why a partially functional allele (c.1765C>T [p.Arg589Trp]) in XPCS1CD results in a more severe phenotype than the null allele in CS1USAU is currently unclear.
Mice homozygous for a frameshift mutation in Ercc4 (Xpf) die by 3 weeks of age.31 Likewise, mice in which Ercc1 has been deleted die soon after birth.32,33 They develop progressive neurodegeneration, leucopenia and thrombocytopenia,22 and impaired liver function.32 Many of these characteristics resemble those of XPCS1CD. Pancytopenia is a hallmark of FA, a disorder associated with cellular hypersensitivity to DNA-ICL-inducing agents.34 Recent data have indicated that endogenous aldehydes generate DNA damage that requires the FA pathway for resolution.35 We therefore favor the explanation that pancytopenia arises from a defect in the response to such crosslinkers; this defect is common to the FA individuals and XPCS1CD (Figure 1G). Support for this idea comes from Bogliolo et al.,36 who report on two FA individuals with mutations in ERCC4 but without features of CS or XP.
Finally, our results demonstrate that ERCC4 and ERCC1 must be added to the list of genes in which defects can result in either CS or the combined XP-CS-FA phenotype.
Acknowledgments
This work was supported by KAKENHI Grants-in-Aid for Young Scientists A (24681008), Exploratory Research (24659533) from the Japan Society for the Promotion of Science, a science research grant from Inamori Foundation, a medical research grant from Mochida Memorial Founds for Medical and Pharmaceutical Research, a medical research grant from the Daiichi-Sankyo Foundation of Life Science, a grant for basic science research from The Sumitomo Foundation, and a medical research grant from Takeda Science Foundation to T.O.; Special Coordination Funds for Promoting Science and Technology from the Japan Science and Technology Agency to Y.N.; and a grant from the Associazione Italiana per la Ricerca sul Cancro (AIRC) to M.S. We are grateful to the National Commissioning Group of the UK National Health Service (NHS) for funding the xeroderma pigmentosum service. In addition, the authors acknowledge financial support from the UK Department of Health via the National Institute for Health Research comprehensive Biomedical Research Centre award to Guy’s & St. Thomas’ NHS Foundation Trust in partnership with King’s College London and King’s College Hospital NHS Foundation Trust. Some early experiments were carried out by Di J. Sun. We are grateful to the families of the affected individuals for their helpful cooperation with these studies.
Contributor Information
Alan R. Lehmann, Email: a.r.lehmann@sussex.ac.uk.
Tomoo Ogi, Email: togi@nagasaki-u.ac.jp.
Web Resources
The URLs for data presented herein are as follows:
GeneReviews, Laugel, V. (1993). Cockayne Syndrome, http://www.ncbi.nlm.nih.gov/books/NBK1342/
References
- 1.Kleijer W.J., Laugel V., Berneburg M., Nardo T., Fawcett H., Gratchev A., Jaspers N.G., Sarasin A., Stefanini M., Lehmann A.R. Incidence of DNA repair deficiency disorders in western Europe: Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair (Amst.) 2008;7:744–750. doi: 10.1016/j.dnarep.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 2.Laugel V., Dalloz C., Tobias E.S., Tolmie J.L., Martin-Coignard D., Drouin-Garraud V., Valayannopoulos V., Sarasin A., Dollfus H. Cerebro-oculo-facio-skeletal syndrome: three additional cases with CSB mutations, new diagnostic criteria and an approach to investigation. J. Med. Genet. 2008;45:564–571. doi: 10.1136/jmg.2007.057141. [DOI] [PubMed] [Google Scholar]
- 3.Friedberg E.C., Walker G.C., Siede W., Wood R.D., Schultz R.A., Ellenberger T. ASM Press; Washington, DC: 2006. DNA Repair and Mutagenesis, Second Edition. [Google Scholar]
- 4.Hanawalt P.C., Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 5.Laugel V., Dalloz C., Durand M., Sauvanaud F., Kristensen U., Vincent M.C., Pasquier L., Odent S., Cormier-Daire V., Gener B. Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. Hum. Mutat. 2010;31:113–126. doi: 10.1002/humu.21154. [DOI] [PubMed] [Google Scholar]
- 6.Nakazawa Y., Sasaki K., Mitsutake N., Matsuse M., Shimada M., Nardo T., Takahashi Y., Ohyama K., Ito K., Mishima H. Mutations in UVSSA cause UV-sensitive syndrome and impair RNA polymerase IIo processing in transcription-coupled nucleotide-excision repair. Nat. Genet. 2012;44:586–592. doi: 10.1038/ng.2229. [DOI] [PubMed] [Google Scholar]
- 7.Zhang X., Horibata K., Saijo M., Ishigami C., Ukai A., Kanno S., Tahara H., Neilan E.G., Honma M., Nohmi T. Mutations in UVSSA cause UV-sensitive syndrome and destabilize ERCC6 in transcription-coupled DNA repair. Nat. Genet. 2012;44:593–597. doi: 10.1038/ng.2228. [DOI] [PubMed] [Google Scholar]
- 8.Schwertman P., Lagarou A., Dekkers D.H., Raams A., van der Hoek A.C., Laffeber C., Hoeijmakers J.H., Demmers J.A., Fousteri M., Vermeulen W., Marteijn J.A. UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat. Genet. 2012;44:598–602. doi: 10.1038/ng.2230. [DOI] [PubMed] [Google Scholar]
- 9.Limsirichaikul S., Niimi A., Fawcett H., Lehmann A., Yamashita S., Ogi T. A rapid non-radioactive technique for measurement of repair synthesis in primary human fibroblasts by incorporation of ethynyl deoxyuridine (EdU) Nucleic Acids Res. 2009;37:e31. doi: 10.1093/nar/gkp023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakazawa Y., Yamashita S., Lehmann A.R., Ogi T. A semi-automated non-radioactive system for measuring recovery of RNA synthesis and unscheduled DNA synthesis using ethynyluracil derivatives. DNA Repair (Amst.) 2010;9:506–516. doi: 10.1016/j.dnarep.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 11.Nance M.A., Berry S.A. Cockayne syndrome: review of 140 cases. Am. J. Med. Genet. 1992;42:68–84. doi: 10.1002/ajmg.1320420115. [DOI] [PubMed] [Google Scholar]
- 12.Lehmann A.R. DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie. 2003;85:1101–1111. doi: 10.1016/j.biochi.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 13.Hoy C.A., Thompson L.H., Mooney C.L., Salazar E.P. Defective DNA cross-link removal in Chinese hamster cell mutants hypersensitive to bifunctional alkylating agents. Cancer Res. 1985;45:1737–1743. [PubMed] [Google Scholar]
- 14.Wood R.D. Mammalian nucleotide excision repair proteins and interstrand crosslink repair. Environ. Mol. Mutagen. 2010;51:520–526. doi: 10.1002/em.20569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujiwara Y. Defective repair of mitomycin C crosslinks in Fanconi’s anemia and loss in confluent normal human and xeroderma pigmentosum cells. Biochim. Biophys. Acta. 1982;699:217–225. doi: 10.1016/0167-4781(82)90110-5. [DOI] [PubMed] [Google Scholar]
- 16.van Vuuren A.J., Appeldoorn E., Odijk H., Yasui A., Jaspers N.G., Bootsma D., Hoeijmakers J.H. Evidence for a repair enzyme complex involving ERCC1 and complementing activities of ERCC4, ERCC11 and xeroderma pigmentosum group F. EMBO J. 1993;12:3693–3701. doi: 10.1002/j.1460-2075.1993.tb06044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biggerstaff M., Szymkowski D.E., Wood R.D. Co-correction of the ERCC1, ERCC4 and xeroderma pigmentosum group F DNA repair defects in vitro. EMBO J. 1993;12:3685–3692. doi: 10.1002/j.1460-2075.1993.tb06043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gregg S.Q., Robinson A.R., Niedernhofer L.J. Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease. DNA Repair (Amst.) 2011;10:781–791. doi: 10.1016/j.dnarep.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahmad A., Enzlin J.H., Bhagwat N.R., Wijgers N., Raams A., Appledoorn E., Theil A.F., J Hoeijmakers J.H., Vermeulen W., J Jaspers N.G. Mislocalization of XPF-ERCC1 nuclease contributes to reduced DNA repair in XP-F patients. PLoS Genet. 2010;6:e1000871. doi: 10.1371/journal.pgen.1000871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsumura Y., Nishigori C., Yagi T., Imamura S., Takebe H. Characterization of molecular defects in xeroderma pigmentosum group F in relation to its clinically mild symptoms. Hum. Mol. Genet. 1998;7:969–974. doi: 10.1093/hmg/7.6.969. [DOI] [PubMed] [Google Scholar]
- 21.Jaspers N.G., Raams A., Silengo M.C., Wijgers N., Niedernhofer L.J., Robinson A.R., Giglia-Mari G., Hoogstraten D., Kleijer W.J., Hoeijmakers J.H., Vermeulen W. First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Am. J. Hum. Genet. 2007;80:457–466. doi: 10.1086/512486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niedernhofer L.J., Garinis G.A., Raams A., Lalai A.S., Robinson A.R., Appeldoorn E., Odijk H., Oostendorp R., Ahmad A., van Leeuwen W. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–1043. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- 23.Bardwell A.J., Bardwell L., Tomkinson A.E., Friedberg E.C. Specific cleavage of model recombination and repair intermediates by the yeast Rad1-Rad10 DNA endonuclease. Science. 1994;265:2082–2085. doi: 10.1126/science.8091230. [DOI] [PubMed] [Google Scholar]
- 24.O’Donovan A., Davies A.A., Moggs J.G., West S.C., Wood R.D. XPG endonuclease makes the 3′ incision in human DNA nucleotide excision repair. Nature. 1994;371:432–435. doi: 10.1038/371432a0. [DOI] [PubMed] [Google Scholar]
- 25.Aboussekhra A., Biggerstaff M., Shivji M.K., Vilpo J.A., Moncollin V., Podust V.N., Protić M., Hübscher U., Egly J.M., Wood R.D. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell. 1995;80:859–868. doi: 10.1016/0092-8674(95)90289-9. [DOI] [PubMed] [Google Scholar]
- 26.Araújo S.J., Nigg E.A., Wood R.D. Strong functional interactions of TFIIH with XPC and XPG in human DNA nucleotide excision repair, without a preassembled repairosome. Mol. Cell. Biol. 2001;21:2281–2291. doi: 10.1128/MCB.21.7.2281-2291.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim Y., Lach F.P., Desetty R., Hanenberg H., Auerbach A.D., Smogorzewska A. Mutations of the SLX4 gene in Fanconi anemia. Nat. Genet. 2011;43:142–146. doi: 10.1038/ng.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crossan G.P., van der Weyden L., Rosado I.V., Langevin F., Gaillard P.H., McIntyre R.E., Gallagher F., Kettunen M.I., Lewis D.Y., Brindle K., Sanger Mouse Genetics Project Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat. Genet. 2011;43:147–152. doi: 10.1038/ng.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stoepker C., Hain K., Schuster B., Hilhorst-Hofstee Y., Rooimans M.A., Steltenpool J., Oostra A.B., Eirich K., Korthof E.T., Nieuwint A.W. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat. Genet. 2011;43:138–141. doi: 10.1038/ng.751. [DOI] [PubMed] [Google Scholar]
- 30.de Laat W.L., Appeldoorn E., Jaspers N.G., Hoeijmakers J.H. DNA structural elements required for ERCC1-XPF endonuclease activity. J. Biol. Chem. 1998;273:7835–7842. doi: 10.1074/jbc.273.14.7835. [DOI] [PubMed] [Google Scholar]
- 31.Tian M., Shinkura R., Shinkura N., Alt F.W. Growth retardation, early death, and DNA repair defects in mice deficient for the nucleotide excision repair enzyme XPF. Mol. Cell. Biol. 2004;24:1200–1205. doi: 10.1128/MCB.24.3.1200-1205.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McWhir J., Selfridge J., Harrison D.J., Squires S., Melton D.W. Mice with DNA repair gene (ERCC-1) deficiency have elevated levels of p53, liver nuclear abnormalities and die before weaning. Nat. Genet. 1993;5:217–224. doi: 10.1038/ng1193-217. [DOI] [PubMed] [Google Scholar]
- 33.Weeda G., Donker I., de Wit J., Morreau H., Janssens R., Vissers C.J., Nigg A., van Steeg H., Bootsma D., Hoeijmakers J.H.J. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr. Biol. 1997;7:427–439. doi: 10.1016/s0960-9822(06)00190-4. [DOI] [PubMed] [Google Scholar]
- 34.Kim H., D’Andrea A.D. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26:1393–1408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garaycoechea J.I., Crossan G.P., Langevin F., Daly M., Arends M.J., Patel K.J. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature. 2012;489:571–575. doi: 10.1038/nature11368. [DOI] [PubMed] [Google Scholar]
- 36.Bogliolo M., Schuster B., Stoepker C., Derkunt B., Su Y., Raams A., Trujillo J.P., Minguillón J., Ramírez M.J., Pujol R. Mutations in ERCC4, Encoding the DNA-Repair Endonuclease XPF, Cause Fanconi Anemia. Am. J. Hum. Genet. 2013;92 doi: 10.1016/j.ajhg.2013.04.002. Published online April 25, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]