

Abstract
Yunis-Varón syndrome (YVS) is an autosomal-recessive disorder with cleidocranial dysplasia, digital anomalies, and severe neurological involvement. Enlarged vacuoles are found in neurons, muscle, and cartilage. By whole-exome sequencing, we identified frameshift and missense mutations of FIG4 in affected individuals from three unrelated families. FIG4 encodes a phosphoinositide phosphatase required for regulation of PI(3,5)P2 levels, and thus endosomal trafficking and autophagy. In a functional assay, both missense substitutions failed to correct the vacuolar phenotype of Fig4-null mouse fibroblasts. Homozygous Fig4-null mice exhibit features of YVS, including neurodegeneration and enlarged vacuoles in neurons. We demonstrate that Fig4-null mice also have small skeletons with reduced trabecular bone volume and cortical thickness and that cultured osteoblasts accumulate large vacuoles. Our findings demonstrate that homozygosity or compound heterozygosity for null mutations of FIG4 is responsible for YVS, the most severe known human phenotype caused by defective phosphoinositide metabolism. In contrast, in Charcot-Marie-Tooth disease type 4J (also caused by FIG4 mutations), one of the FIG4 alleles is hypomorphic and disease is limited to the peripheral nervous system. This genotype-phenotype correlation demonstrates that absence of FIG4 activity leads to central nervous system dysfunction and extensive skeletal anomalies. Our results describe a role for PI(3,5)P2 signaling in skeletal development and maintenance.
Main Text
Yunis and Varón first described the syndrome that bears their name in 1980, based on three Colombian families with a total of five affected children.1 Since then, approximately 25 individuals with Yunis-Varón syndrome (YVS) (MIM 216340) have been described.2–19 Frequent features include structural brain abnormalities, sparse and pale hair, and facial dysmorphisms. Skeletal abnormalities include wide fontanelles with calvarial dysostosis, aplasia or hypoplasia of the clavicles and phalanges in the hands and feet, and absence of thumbs and halluces. Pelvic bone dysplasia, absent sternal ossification centers, and fractures are also frequent.17 Neuropathology shows extensive neuronal loss and diffuse atrophy affecting the cerebellar vermis, corpus callosum, basal ganglia, and frontal lobes. Vacuoles compatible with enlarged lysosomes are seen in neurons, muscle, cartilage, heart, and macrophages.17 In the urine, multiple abnormal oligosaccharide bands appear, suggesting a dysfunction of lysosomal enzymes,8,12 but no consistent storage material could be identified12 and the enzyme activities of oligosaccharidases were normal.8
Six families affected by Yunis-Varón syndrome were included in this study. The clinical features of the eight affected individuals are summarized in Table 1. Pictures and radiographs of most affected individuals are available in previously published case reports.5,7,8,18,20 The study was conducted according to the guidelines of the institutional review board of the Baylor College of Medicine and informed consent was obtained prior to collection of samples. The inclusion criterion was a high index of suspicion of Yunis-Varón syndrome by a clinical geneticist. Frequent features found in the individuals include sparse scalp hair, protruding eyes, low-set ears, a high arched palate, and micrognatia (Table 1). Skeletal features include wide fontanelles and calvarial dysostosis, digital hypoplasia, especially of the thumbs and halluces, pelvic dysplasia with hip dislocations, and absent or hypoplastic clavicles. Affected individuals were significantly hypotonic and presented global developmental delay and often feeding and swallowing difficulties. Central nervous system anomalies in individuals 1 and 2 consisted of frontal lobe atrophy with pachygyria and hypoplasia of the corpus callosum and cerebellar vermis. In individual 3, autopsy revealed an absent olfactory bulb and tract, an atypical ventricular hamartoma, and neuronal loss with vacuolation in layers 3 and 5 of the cerebral cortex, the cerebellar dentate nucleus, and the olivary bodies. Individual 4 had agenesis of the corpus callosum, and individual 5 had neuronal loss with vacuolation in the cerebral cortex, the thalamus, subthalamic nuclei, globus pallidus, and the cerebellar dentate nucleus. Investigations for peripheral neuropathies had not been performed in part because of the absence of suggestive clinical findings, a fact that might be confounded by the severe central neurological disease in these individuals and the frequent early death. At the autopsy of individual 3, muscle histology was compatible with neurogenic atrophy. Electron microscopy of skin fibroblasts from individual 5 revealed the presence of large vacuoles as well as electron-dense inclusions (Figure 1A). Histological examination of muscle revealed variable fiber size and large vacuoles in many fibers (Figures 1B–1D). Electron microscopy of muscle detected membrane-limited vacuoles derived from sarcolemma and myofibrils, filled with amorphous, granular, or membranous material and partially degraded organelles (Figures 1E and 1F). Most vacuoles were membrane limited, with those at the fiber surface also limited by the basal lamina. Mitochondria were normal in size, number, and morphology.
Table 1.
Clinical Features of Individuals Investigated
|
Individual Number |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | Review of the Literature (n = 17) | |
| Familial relationship and reference of first description | affected siblings18 | affected siblings5 | first affected child8 | first affected child20 | first affected child, unaffected brothers7 | first affected child | |||
| Origin and consanguinity in parents | Mexican, third cousins | English, nonconsanguineous | Italian, nonconsanguineous | German, nonconsanguineous | Anglo-Saxon, nonconsanguineous | French, nonconsanguineous | |||
| FIG4 mutations | c.1260_1261delGT (p.Thr422Glnfs*6) from each parent | maternal mutation: c.311G>A (p.Gly104Asp); paternal mutation: c.831_838delTAAATTTG (p.Lys278Trpfs*6) | c.524T>C (p.Leu175Pro) from each parent | no mutation by Sanger | no mutation by Sanger | no mutation by Sanger | |||
| Gender | F | F | M | M | F | F | M | F | |
| Age at last follow-up | 1 year | death at 2 months | death at 4 months | TOP | death at 4 months | 9 months | death at 7 years after cardiac surgery | 1 month | |
| Enlarged vacuoles demonstrated on pathological material | not on muscle biopsy | NA | + (CNS) | not on bone histology | + (muscle, CNS, fibroblasts) | not on muscle biopsy | NA | NA | |
| Birth weight (g) | 2,200 | 2,470 | 3,030 | 250 at 18 weeks | 2,580 | 3,035 | 2,630 | 3,250 | 2,397 ± 298a |
| BW < 3rd percentile | − | − | − | − | − | − | − | − | 10/17 (59%) |
| Birth length (cm) | 45 | 46 | 52 | 24 at 18 weeks | 47 | 51 | 44 | 49 | 46 ± 2a |
| BL < 3rd percentile | − | − | − | − | − | − | − | − | 10/14 (71%) |
| OFC (cm) | 28 | 30 | 33 | 18 at 18 weeks | 34 | 33 | 31 | 35 | 32 ± 3a |
| Microcephaly | + | + | − | − | − | − | + | − | 9/16 (56%) |
| Head and Neck | |||||||||
| Sparse scalp hair | + | + | + | NA | + | + | + | + | 17/17 (100%) |
| Wide fontanelle/sutures | + | + | + | + | NA | − | − | + | 16/16 (100%) |
| Protruding eyes | + | + | + | + | + | − | − | + | 10/12 (83%) |
| Anteverted nostrils | + | − | + | + | + | − | + | − | 12/13 (92%) |
| Short philtrum | − | − | − | NA | + | − | − | − | 8/8 (100%) |
| Short upper lip | + | + | + | + | + | + | − | + | 13/13 (100%) |
| Labial-gingival retraction | + | + | − | − | + | − | + | − | 8/10 (80%) |
| High arched palate | + | + | + | NA | + | + | − | − | 9/10 (90%) |
| Micrognathia | + | + | + | + | + | − | + | + | 14/14 (100%) |
| Low set/dysplastic ears | + | + | + | + | + | + | + | + | 18/18 (100%) |
| Loose skin in neck | + | + | + | − | NA | + | + | + | 6/7 (86%) |
| Thorax | |||||||||
| Congenital heart defect | + | + | − | − | − | + | + | + | 3/15 (20%) |
| Absent/hypoplastic nipples | − | − | − | − | − | − | + | − | 3/11 (27%) |
| Genital anomalies | − | − | + | − | − | − | + | − | 5/13 (38%) |
| Limbs | |||||||||
| Absent/hypoplastic thumbs | + | + | + | + | + | − | + | + | 16/17 (94%) |
| Short pointed fingers | + | + | + | + | + | − | + | + | 14/15 (93%) |
| Absent/hypoplastic halluces | + | + | + | + | + | − | − | + | 16/16 (100%) |
| Short pointed toes | + | + | + | + | + | + | + | + | 15/16 (94%) |
| Absent/hypoplastic nails | + | + | + | + | + | + | + | + | 12/14 (86%) |
| Single palmar crease | + | + | + | + | NA | − | + | − | 5/7 (71%) |
| Central Nervous System | |||||||||
| Structural brain anomalies (see text) | + | + | + | + | + | + | + | − | 6/12 (50%) |
| Hypotonia | + | + | + | NA | + | + | + | + | 5/5 (100%) |
| Radiological | |||||||||
| Calvarial dysostosis | + | + | + | + | NA | NA | − | − | 15/15 (100%) |
| Craniofacial disproportion | + | + | + | + | NA | − | − | − | 8/8 (100%) |
| Absent/hypoplastic clavicles | + | + | − | − | NA | − | + | − | 13/17 (76%) |
| Absent sternal ossification center | + | + | NA | NA | NA | − | − | − | 7/7 (100%) |
| Pelvic dysplasia | + | + | + | − | NA | − | + | + | 9/10 (90%) |
| Hip dislocation | − | + | + | NA | NA | − | + | − | 5/8 (63%) |
| Thumb aplasia/hypoplasia | + | + | + | + | NA | − | + | + | 13/14 (93%) |
| Agenesia/hypoplasia of distal phalanges of the fingers | + | + | + | + | NA | − | + | + | 13/14 (93%) |
| Agenesia/hypoplasia of middle phalanges of the fingers | + | + | + | + | NA | − | + | + | 10/11 (91%) |
| Number of positive features over features assessed | 28/35 | 28/34 | 26/35 | 18/29 | 17/24 | 10/35 | 23/35 | 18/35 | |
Cases included in the review of the literature are from several researchers.1–5,9,11,12,14–17,21 For individual 4, because a termination of pregnancy was performed, several clinical parameters could not be assessed. For individual 5, skeletal radiographs were not available. Features present in individual 8 not fully captured by this table are asthma, tracheomalacia, dysphagia lusoria, ventricular septal defect, and anemia of unknown etiology. Abbreviations are as follows: TOP, termination of pregnancy; F, female; M, male; BW, birth weight; BL, birth length; OFC, occipitofrontal circumference; NA, not assessed. Plus signs (+) and minus signs (−) indicate presence and absence, respectively, of trait.
Data shown are mean ± SD.
Figure 1.

Abnormal Subcellular Morphology in YVS Tissues
(A) Electron micrograph of cultured skin fibroblasts from individual 5 showing large, empty cytoplasmic vacuoles and electron-dense inclusion bodies. Scale bar represents 1 μm.
(B–D) Muscle sections stained with Gomori trichrome (B), Periodic Acid Schiff (PAS) (C), and acid phosphatase (D) reveal variable fiber size and large vacuoles containing basophilic material stained by acid phosphatase and PAS staining. Scale bars represent 10 μm.
(E and F) Electron micrographs of skeletal muscle showing vacuoles in the subsarcolemmal (E) and intramyofibrillar (F) spaces filled with amorphous, granular, or membranous material and partially degraded organelles. The vacuoles are mostly membrane limited; those located at the fiber surface abutting the extracellular space are also limited by the basal lamina (arrowheads). The mitochondria were normal in number, size, and morphology (inset). Scale bars represent 1 μm.
Candidate gene analysis for RUNX2, mutations in which cause classical cleidocranial dysplasia (MIM 119600), failed to detect mutations.22 Exome sequencing was performed as described previously23,24 on individuals 1 and 5 and the parents of individuals 3 and 4 because high-quality DNA required for exome sequencing was not available from individuals 3 and 4. Sequence analysis of genes with rare (MAF < 1%) or novel mutations yielded FIG4 (RefSeq accession number NM_014845.5) as the best candidate because the spontaneous Fig4-null mouse has multiple related phenotypes including cellular vacuolation, neurodegeneration, and dysplasia of the corpus callosum.25 The FIG4 genotypes of parents and affected offspring in the three families described in Table 1 are shown in Figure 2. In family 1, the parents were third cousins and both were carriers of the same 2 bp frameshift deletion, c.1260_1261delGT, which is predicted to cause protein truncation upstream of the phosphatase catalytic motif CX5RT/S (Figure 3A) and therefore complete loss of function. In family 2, the maternal allele was the missense variant p.Gly104Asp (c.311G>A) and the paternal allele was an 8 bp frameshift deletion (c.831_838delTAAATTTG). After identification of heterozygous mutations in the parents, a pathology specimen demonstrated inheritance of both mutations in individual 3. In family 3, with no reported consanguinity, both parents were heterozygous for the novel missense variant p.Leu175Pro (c.524T>C) and the affected child was homozygous for the variant allele. The missense substitutions in individual 1 (Gly104Asp) and individual 5 (p.Leu175Pro) alter amino acid residues that are highly conserved through evolution (Figure 3B). Glycine 104 is located at the beginning of β sheet 7 within the noncatalytic, protein-binding domain of FIG4 (Figure 3C).27 Leucine 175 is located in α helix 2 of the same domain and appears to interact directly with residue Glu302, the site of a functionally null substitution in a individual with CMT4J26 (Figure 3C). Four protein prediction programs assessed these two missense variants as likely to affect FIG4 function (Table S1 available online).
Figure 2.

Segregation of FIG4 Mutations in Three Unrelated YVS-Affected Families
FIG4 genotypes of family members demonstrating autosomal-recessive inheritance with sequencing chromatograms below. Numbering from RefSeq NM_014845.5.
Figure 3.
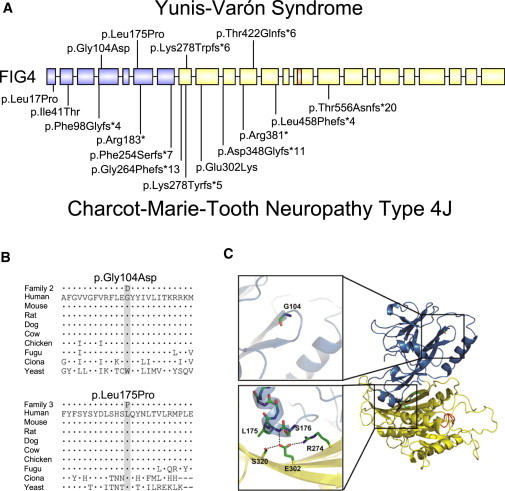
Location of FIG4 Mutations
(A) Location of mutations in FIG4 exons; introns not drawn to scale. Above, YVS; below, CMT4J.26 Protein-interaction domain, blue; catalytic domain, yellow; P loop containing the catalytic CX5R(T/S) motif, orange.
(B) Evolutionary conservation of amino acids around the missense mutations. Alignment was performed with ClustalW2. Dots represent identity; dashes represent gaps.
(C) Positions of substitutions in the 3-dimensional protein structure (courtesy of Yuxin Mao27). Same color scheme as in (A). Abbreviations used to designate amino acids are as follows: G104, p.Gly104; L175, p.Leu175; S176, p.Ser176; S320, p.Ser320; E302, p.Glu302; and R274, p.Arg274. Gly104 is located at the beginning of β sheet 7 within the noncatalytic, protein-binding domain of FIG4.27 p.Leu175 is located in α helix 2 of the same domain and appears to interact directly with residue p.Glu302, the site of a functionally null mutation in an individual with CMT4J.26 GenBank transcript used for FIG4 residue numbering was NM_014845.5. The NCBI protein sequences used for alignment were as follows: human, accession number NP_055660.1; mouse, NP_598760.1; rat, NP_001040561.1; dog, NP_001108097.1; cow, NP_001069482.1; chicken, NP_001108095.1; Fugu, CAG03571.1; Ciona intestinalis, XP_002125633.1; and yeast Saccharomyces cerevisiae, NP_014074.1.
We developed a functional assay for FIG4 activity based on rescue of the cytoplasmic vacuolization seen in cultured fibroblasts from the Fig4-null mouse.25,28 Null fibroblasts are transfected with Fig4 cDNA plus GFP cDNA to permit identification of successfully transfected cells by their fluorescence. Representative transfected cells are shown in Figure 4A. The wild-type Fig4 cDNA rescued vacuolization in 73% of transfected cells, whereas the p.Gly104Asp and p.Leu175Pro mutant cDNAs failed to correct vacuolization (Figure 4B). Comparable levels of expression were obtained with the different transgenes (see Figure S1). The complete lack of rescue in this assay indicates that p.Gly104Asp and p.Leu175Pro are loss-of-function, null substitutions.
Figure 4.

Functional Effects of FIG4 Mutations
Transfection assay for FIG4 function. Low-passage (<7) embryonic fibroblasts from E13 homozygous Fig4−/− mice (MEFs) were cotransfected with 6 μg GFP cDNA and 6 μg of the indicated Fig4 cDNA via Fugene-6 (Promega). The presence of vacuoles in fluorescent cells was assessed with an inverted Leica DMIRB microscope equipped with epifluorescence and an Olympus DP30 BW digital camera. Fluorescent cells were classified as vacuolated if they contained ≥6 vacuoles. Representative transfected cells are shown in (A) (GFP on the left). The percent of transfected (fluorescent) cells lacking vacuoles resulting from correction by the transfected Fig4 cDNA is shown in (B), with the number of nonvacuolated cells and the total number of cells assayed.
Sanger sequencing in affected individuals from three additional families did not detect FIG4 mutations. Some of these individuals did not present with all the classical clinical features of Yunis-Varón syndrome (see Table 1 for details). Apart from the presence of calvarial dysostosis, no other feature distinguished the individuals with FIG4 mutations from those without. There was a trend toward a higher number of features in individuals with FIG4 mutations (Table 1, bottom row). The individuals without identified FIG4 mutations might carry undetected mutations within introns or regulatory elements of FIG4. Alternatively, Yunis-Varón syndrome may be genetically heterogeneous with some cases caused by mutations in other gene(s).
FIG4 encodes a lipid phosphatase that is part of a multiprotein complex regulating the abundance of the signaling phosphoinositide PI(3,5)P2.29,30 The protein complex, which is localized on the membranes of late endosomes and lysosomes, also contains the kinase PIKfyve/FAB1 that converts PI(3)P to PI(3,5)P2 and the scaffold protein VAC14. The transient production of PI(3,5)P2 at the cytoplasmic surface of intracellular vesicles is thought to mediate vesicle fusion and trafficking via interaction with effector proteins such as WIPI1.31 Deficiency of FIG4 results in instability of the biosynthetic complex, reduced PI(3,5)P2 concentration, and accumulation of enlarged intracellular vacuoles containing the lysosomal membrane proteins LAMP1 and LAMP2.25,32,33
The pale tremor mouse carries a spontaneous null mutation of Fig4 caused by a retroposon insertion that results in loss of Fig4 mRNA25 and protein.28 Fig4-null mice have small body size, diluted hair color, spongiform degeneration of the central and peripheral nervous systems, and juvenile lethality.25 This phenotype is recapitulated by global deletion of the floxed Fig4 allele.34 Fig4 is expressed ubiquitously and in the brain the highest expression is in hippocampus, striatum, and hindbrain.35 Neurodegeneration in Fig4-null mice is most pronounced in layers 4 and 5 of the cortex, brain stem, and deep cerebellar nucleus and progresses from accumulation of large intracellular vacuoles to extensive spongiform degeneration. Inclusion bodies containing p62 and LAMP1 accumulate in astrocytes of Fig4-null mice, apparently as a result of impaired resolution of autophagolysosomes.25,32,36 Deficiency of FIG4 also results in defective myelination of the central and peripheral nervous systems and hypoplasia of the corpus callosum.25,32,37 Neuron-specific expression of a Fig4 transgene in the null mice substantially rescues neurodegeneration, myelination, and survival, and the neuron-specific knockout recapitulates the null phenotype.34
Mutations of human FIG4 were previously identified in individuals with Charcot-Marie-Tooth disease type 4J (MIM 611228), an autosomal-recessive peripheral neuropathy that progresses to wheelchair dependence and death.25,26 Individuals with CMT4J are compound heterozygotes carrying one null mutation and one missense substitution. All but one CMT4J individual carry the missense substitution Ile41Thr,26 which reduces binding affinity for the scaffold protein VAC1428 and destabilizes the protein.28,38 Overexpression of FIG4-Ile41Thr in transgenic mice can rescue the null phenotype,28 and the mutant protein retains activity in a yeast vacuolization assay,39 demonstrating that this variant retains partial function. In contrast, the more severe Yunis-Varón phenotype is the result of complete loss of FIG4 activity. The striking differences between Yunis-Varón and CMT4J provide a clear example of diverse clinical consequences resulting from different allelic mutations of the same gene.
The clinical effects of heterozygous FIG4 mutations remain unclear. We previously described heterozygous null mutations in amyotrophic lateral sclerosis (ALS).39 However, two smaller studies of ALS did not detect mutations.40,41 Further, heterozygous null parents in CMT4J pedigrees do not report symptoms of motor neuron disease.25,26 Heterozygosity for FIG4-null mutations may constitute a risk factor or modifier of neurological disease rather than an independent cause.
Skeletal dysplasia is a consistent feature of YVS not previously investigated in the Fig4-null mouse. Animal experimentation was performed under approval from the Institutional Animal Care and Use Committee of the University of Michigan. We assessed Fig4 expression in wild-type mouse tissues and found that expression in calvaria, osteoblasts, and bone marrow cells is comparable to expression in brain (Figure S2). We previously described in detail the neuropathology found in Fig4-null mice, but we did not investigate skeletal phenotypes. Unlike individuals with YVS, Fig4-null mice do not exhibit aplasia or hypoplasia of digits on the front or rear limb (Figure S3A). Homozygous null animals have reduced body weight and impaired growth (Figure S3B). The skeleton is normal at birth but is noticeably smaller at postnatal day 21 (P21), when long bones and clavicles are 20%–25% smaller than those of wild-type littermates (see Figure S3C for femoral length). Normal shape of the clavicles and pelvic bones were demonstrated in newborn mice and in dissections and 3D reconstructions from microcomputed tomography (microCT) of mice at P21 (Figure 5). Although the bones were normally shaped and had a normal external appearance, they had an obvious lower trabecular and cortical density, as can be observed by the porous appearance in 3D reconstructions with a density thresholding adequate for wild-type mouse skeletons (Figure 5). To investigate trabecular and cortical bone density in a quantitative manner, we carried out microCT of vertebrae and femurs at P21 (Figure 6) as described previously.42 The reduced size and density of the trabeculae is evident in the vertebral cross-section (Figures 6A and 6B). Bone volume fraction, bone surface, trabecular number, and connectivity density were reduced to less than 50% of wild-type values, whereas trabecular separation was increased more than 3-fold (Figures 6C–6H). Moreover, femoral cortical thickness was reduced to less than 50% of wild-type values (Figures 6J and 6K). Extensive vacuolization was observed in cultures of isolated osteoblasts from calvarial tissue (Figure 6L), isolated as described previously,43 and in cultured bone marrow mesenchymal stromal (not shown). The analysis of skeletal development in Fig4-null mice thus demonstrates abnormal ossification and vacuolized osteoblasts. A comparison of the phenotypes in humans and mice is given in Table 2.
Figure 5.

3D Reconstruction after MicroCT Scanning of Mouse Skeletons
Developmental defects of the skeletons are not present in Fig4-null mice (age P21). Multiple views are provided for the cranium, clavicles, and pelvis.
Figure 6.

MicroCT Analysis of Fig4-Null Mice
(A) 3D reconstruction of L4 vertebrae of P21 littermates.
(B) Trabecular bone of L4 vertebrae.
(C–K) MicroCT analysis of L4 vertebral bone. Bone volume fraction, BV/TV (C); bone surface, BS (D); trabecular number, Tb.N (E); trabecular thickness, Tb.Th (F); trabecular separation, Tb.Sp (G); connectivity density, Conn.D (H); density of trabecular bone, Density of BV (I).
(J) 3D reconstruction of femurs of P21 littermates, with a cut plane through the third trochanter.
(K) Femoral cortical thickness, from below the third trochanter to the middiaphysis. n = 4 per group for the vertebrae and femurs; *p < 0.05.
(L) Vacuolization is clearly seen in cultured primary osteoblasts from Fig4-null mice.
All errors bars represent standard deviation.
Table 2.
Comparison of Phenotypes of Individuals with Yunis-Varón Syndrome and Fig4-Null Mice
| Feature | Individuals with Yunis-Varón Syndrome | Fig4-Null Mice |
|---|---|---|
| Severe CNS disease with extensive vacuolation | + | + |
| Postnatal growth deficiency | + | + |
| Hair/fur | sparse, light-colored | pale |
| Digital hypoplasia | + | − |
| Calvarial dysostosis | + | − |
| Clavicular defects | + | − |
| Pelvic dysplasia | + | − |
| Abnormal bone ossification or maintenance | + (narrow diaphyses and fracture susceptibility) | + (low trabecular volume and thin cortices) |
| Dental anomalies | + (hypodontia, impaction, premature loss) | relative overgrowth of incisors or altered craniofacial morphology |
Plus signs (+) and minus signs (−) indicate presence and absence, respectively, of trait.
In addition to Charcot-Marie-Tooth disease (FIG4 and MTMR2 genes), defects of phosphoinositide metabolism can result in a wide variety of conditions as listed in Table S2.44–46 Opsismodysplasia (MIM 258480) was recently identified to be caused by defects in the inositol-1,4,5-trisphosphate 5-phosphatase INPPL1.47,48 Although clearly different from YVS notably because of the rhizomelia, platyspondyly, and frontal bossing in opsismodysplasia, there are overlapping clinical features such as a large anterior fontanelle, anteverted nostrils, pelvic bone anomalies, metaphyseal anomalies (cupping in opsismodysplasia, flaring in YVS), delayed ossification, shortened digits, hypotonia, and early death.
Calvarial and clavicular development is most dependent on intramembranous bone formation, yet endochondral ossification is also involved in clavicular development.49 Abnormal formation of the clavicles and cranial vault in humans is observed in cleidocranial dysplasia resulting from mutations of RUNX2, a transcription factor critical for both intramembranous and endochondroal ossification of the skeleton, and also in several other syndromes (Table S3). In view of the abnormal development of cranium, clavicles, pelvic bones, digits, and sternal ossification in humans and the low bone mass in mice, it is likely that FIG4 deficiency affects membranous ossification and endochondral ossification as well as skeletal maintenance. The report of abnormal ossification and increased markers of bone turnover in YVS12 suggest an underlying imbalance of bone formation and resorption that may result from intrinsic defects of both osteoblasts and osteoclasts. Future investigation of vesicular trafficking and autophagy in mice with conditional knockout in specific lineages will permit identification of the stages of skeletal development and maintenance that require FIG4.
Acknowledgments
We thank Yuqing Chen for technical assistance, Alyssa Tran for clinical research support, and Shalini N. Jhangiani for exome sequencing coordination. We thank Brian Dawson and Megan Bagos for expert technical assistance with microCT analysis. Funding was provided by National Institutes of Health grants PO1 HD22657 and PO1 HD070394 (B.H.L.), R01 GM24872 (M.H.M.), UL1 TR000433 (G.M.L.), and U54 HG003273-09 (R.A.G.). Funding was also provided by The Rolanette and Berdon Lawrence Bone Disease Program of Texas (B.H.L.). P.M.C. is supported by a CIHR clinician-scientist training award and the O’Malley Foundation. G.M.L. is a fellow of the Postdoctoral Translational Scholars Program of the Michigan CTSA. J.T.L. is supported by Ruth L. Kirschstein National Research Service Award F30 MH098571-01. A.T.V.v.S. is supported by EU 7th FP under GA nr. 241995, project GENCODYS. The EuroBioBank and Telethon Network of Genetic Biobanks (GTB12001F to M.M.) are gratefully acknowledged for providing biological samples.
Contributor Information
Miriam H. Meisler, Email: meislerm@umich.edu.
Brendan H. Lee, Email: blee@bcm.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Allen Mouse Brain Atlas, http://mouse.brain-map.org
ClustalW2 - Multiple Sequence Alignment, http://www.ebi.ac.uk/Tools/msa/clustalw2/
NCBI Gene, http://www.ncbi.nlm.nih.gov/gene
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Yunis E., Varón H. Cleidocranial dysostosis, severe micrognathism, bilateral absence of thumbs and first metatarsal bone, and distal aphalangia: a new genetic syndrome. Am. J. Dis. Child. 1980;134:649–653. doi: 10.1001/archpedi.1980.02130190017005. [DOI] [PubMed] [Google Scholar]
- 2.Hughes H.E., Partington M.W. Brief clinical report: the syndrome of Yunis and Varón—report of a further case. Am. J. Med. Genet. 1983;14:539–544. doi: 10.1002/ajmg.1320140318. [DOI] [PubMed] [Google Scholar]
- 3.Pfeiffer R.A., Diekmann L., Stock H.J. Aplasia of the thumbs and great toes as the outstanding feature of Yunis and Varon syndrome. A new entity. A new observation. Ann. Genet. 1988;31:241–243. [PubMed] [Google Scholar]
- 4.Hennekam R.C., Vermeulen-Meiners C. Further delineation of the Yunis-Varon syndrome. J. Med. Genet. 1989;26:55–58. doi: 10.1136/jmg.26.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garrett C., Berry A.C., Simpson R.H., Hall C.M. Yunis-Varon syndrome with severe osteodysplasty. J. Med. Genet. 1990;27:114–121. doi: 10.1136/jmg.27.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lapeer G.L., Fransman S.L. Hypodontia, impacted permanent teeth, spinal defects, and cardiomegaly in a previously diagnosed case of the Yunis-Varon syndrome. Oral Surg. Oral Med. Oral Pathol. 1992;73:456–460. doi: 10.1016/0030-4220(92)90324-j. [DOI] [PubMed] [Google Scholar]
- 7.Adès L.C., Morris L.L., Richardson M., Pearson C., Haan E.A. Congenital heart malformation in Yunis-Varón syndrome. J. Med. Genet. 1993;30:788–792. doi: 10.1136/jmg.30.9.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dworzak F., Mora M., Borroni C., Cornelio F., Blasevich F., Cappellini A., Tagliavini F., Bertagnolio B. Generalized lysosomal storage in Yunis Varón syndrome. Neuromuscul. Disord. 1995;5:423–428. doi: 10.1016/0960-8966(94)00089-r. [DOI] [PubMed] [Google Scholar]
- 9.Rabe H., Brune T., Rossi R., Steinhorst V., Jorch G., Horst J., Wittwer B. Yunis-Varon syndrome: the first case of German origin. Clin. Dysmorphol. 1996;5:217–222. [PubMed] [Google Scholar]
- 10.Oyer C.E., Tatevosyants N.G., Cortez S.C., Hornstein A., Wallach M. Cleidocranial dysplasia with neonatal death due to central nervous system injury in utero: case report and literature review. Pediatr. Dev. Pathol. 1998;1:314–318. doi: 10.1007/s100249900045. [DOI] [PubMed] [Google Scholar]
- 11.Christie J., Sacks S., Decorato D., Bergasa N.V. Atrophy of the left lobe of the liver and anomalous hepatic vessel in a patient with Yunis-Varon syndrome. J. Clin. Gastroenterol. 1999;29:210–211. doi: 10.1097/00004836-199909000-00025. [DOI] [PubMed] [Google Scholar]
- 12.Walch E., Schmidt M., Brenner R.E., Emons D., Dame C., Pontz B., Wiestler O.D., Bartmann P. Yunis-Varon syndrome: evidence for a lysosomal storage disease. Am. J. Med. Genet. 2000;95:157–160. doi: 10.1002/1096-8628(20001113)95:2<157::aid-ajmg12>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 13.Nagai T. [Yunis-Varon syndrome] Ryoikibetsu Shokogun Shirizu. 2001;34 Part 2:839–840. [PubMed] [Google Scholar]
- 14.Sumi M., Kusumoto T., Kondoh T., Moriuchi H., Miyamoto M., Masuzaki H., Ishimaru T. A case of Yunis-Varon syndrome complicated with complete cleft lip and palate. Am. J. Med. Genet. A. 2004;125A:92–93. doi: 10.1002/ajmg.a.20414. [DOI] [PubMed] [Google Scholar]
- 15.Bhatia S., Holla R.G. Yunis-Varon syndrome. Indian Pediatr. 2005;42:373–375. [PubMed] [Google Scholar]
- 16.Kulkarni M.L., Vani H.N., Nagendra K., Mahesh T.K., Kumar A., Haneef S., Mohammed Z., Kulkarni P.M. Yunis Varon syndrome. Indian J. Pediatr. 2006;73:353–355. doi: 10.1007/BF02825832. [DOI] [PubMed] [Google Scholar]
- 17.Basel-Vanagaite L., Kornreich L., Schiller O., Yacobovich J., Merlob P. Yunis-Varon syndrome: further delineation of the phenotype. Am. J. Med. Genet. A. 2008;146A:532–537. doi: 10.1002/ajmg.a.32135. [DOI] [PubMed] [Google Scholar]
- 18.Corona-Rivera J.R., Romo-Huerta C.O., López-Marure E., Ramos F.J., Estrada-Padilla S.A., Zepeda-Romero L.C. New ocular findings in two sisters with Yunis-Varón syndrome and literature review. Eur. J. Med. Genet. 2011;54:76–81. doi: 10.1016/j.ejmg.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 19.Elizondo-Dueñaz R., Rivera-Silva G., Marcos Abdala H., López-Altamirano M., Martínez-Menchaca H.R. [Yunis-Varon syndrome: a case report] Gac. Med. Mex. 2012;148:81–82. [PubMed] [Google Scholar]
- 20.Reutter H., Bagci S., Müller A., Gembruch U., Geipel A., Berg C., Eggermann T., Spengler S., Bartmann P., Rudnik-Schöneborn S. Primary pulmonary hypertension, congenital heart defect, central nervous system malformations, hypo- and aplastic toes: another case of Yunis-Varón syndrome or report of a new entity. Eur. J. Med. Genet. 2012;55:27–31. doi: 10.1016/j.ejmg.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 21.Partington M.W. Cardiomyopathy added to the Yunis-Varon syndrome. Proc. Greenwood Genetic Center. 1988;7:224–225. [Google Scholar]
- 22.Lee B., Thirunavukkarasu K., Zhou L., Pastore L., Baldini A., Hecht J., Geoffroy V., Ducy P., Karsenty G. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat. Genet. 1997;16:307–310. doi: 10.1038/ng0797-307. [DOI] [PubMed] [Google Scholar]
- 23.Campeau P.M., Kim J.C., Lu J.T., Schwartzentruber J.A., Abdul-Rahman O.A., Schlaubitz S., Murdock D.M., Jiang M.M., Lammer E.J., Enns G.M. Mutations in KAT6B, encoding a histone acetyltransferase, cause Genitopatellar syndrome. Am. J. Hum. Genet. 2012;90:282–289. doi: 10.1016/j.ajhg.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campeau P.M., Lu J.T., Sule G., Jiang M.M., Bae Y., Madan S., Högler W., Shaw N.J., Mumm S., Gibbs R.A. Whole-exome sequencing identifies mutations in the nucleoside transporter gene SLC29A3 in dysosteosclerosis, a form of osteopetrosis. Hum. Mol. Genet. 2012;21:4904–4909. doi: 10.1093/hmg/dds326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chow C.Y., Zhang Y., Dowling J.J., Jin N., Adamska M., Shiga K., Szigeti K., Shy M.E., Li J., Zhang X. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448:68–72. doi: 10.1038/nature05876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicholson G., Lenk G.M., Reddel S.W., Grant A.E., Towne C.F., Ferguson C.J., Simpson E., Scheuerle A., Yasick M., Hoffman S. Distinctive genetic and clinical features of CMT4J: a severe neuropathy caused by mutations in the PI(3,5)P2 phosphatase FIG4. Brain. 2011;134:1959–1971. doi: 10.1093/brain/awr148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manford A., Xia T., Saxena A.K., Stefan C., Hu F., Emr S.D., Mao Y. Crystal structure of the yeast Sac1: implications for its phosphoinositide phosphatase function. EMBO J. 2010;29:1489–1498. doi: 10.1038/emboj.2010.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lenk G.M., Ferguson C.J., Chow C.Y., Jin N., Jones J.M., Grant A.E., Zolov S.N., Winters J.J., Giger R.J., Dowling J.J. Pathogenic mechanism of the FIG4 mutation responsible for Charcot-Marie-Tooth disease CMT4J. PLoS Genet. 2011;7:e1002104. doi: 10.1371/journal.pgen.1002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin N., Chow C.Y., Liu L., Zolov S.N., Bronson R., Davisson M., Petersen J.L., Zhang Y., Park S., Duex J.E. VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P(2) in yeast and mouse. EMBO J. 2008;27:3221–3234. doi: 10.1038/emboj.2008.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Botelho R.J., Efe J.A., Teis D., Emr S.D. Assembly of a Fab1 phosphoinositide kinase signaling complex requires the Fig4 phosphoinositide phosphatase. Mol. Biol. Cell. 2008;19:4273–4286. doi: 10.1091/mbc.E08-04-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Proikas-Cezanne T., Ruckerbauer S., Stierhof Y.D., Berg C., Nordheim A. Human WIPI-1 puncta-formation: a novel assay to assess mammalian autophagy. FEBS Lett. 2007;581:3396–3404. doi: 10.1016/j.febslet.2007.06.040. [DOI] [PubMed] [Google Scholar]
- 32.Ferguson C.J., Lenk G.M., Meisler M.H. Defective autophagy in neurons and astrocytes from mice deficient in PI(3,5)P2. Hum. Mol. Genet. 2009;18:4868–4878. doi: 10.1093/hmg/ddp460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weisman L.S. Yeast vacuole inheritance and dynamics. Annu. Rev. Genet. 2003;37:435–460. doi: 10.1146/annurev.genet.37.050203.103207. [DOI] [PubMed] [Google Scholar]
- 34.Ferguson C.J., Lenk G.M., Jones J.M., Grant A.E., Winters J.J., Dowling J.J., Giger R.J., Meisler M.H. Neuronal expression of Fig4 is both necessary and sufficient to prevent spongiform neurodegeneration. Hum. Mol. Genet. 2012;21:3525–3534. doi: 10.1093/hmg/dds179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sunkin S.M., Ng L., Lau C., Dolbeare T., Gilbert T.L., Thompson C.L., Hawrylycz M., Dang C. Allen Brain Atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res. 2013;41(Database issue):D996–D1008. doi: 10.1093/nar/gks1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferguson C.J., Lenk G.M., Meisler M.H. PtdIns(3,5)P2 and autophagy in mouse models of neurodegeneration. Autophagy. 2010;6:170–171. doi: 10.4161/auto.6.1.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winters J.J., Ferguson C.J., Lenk G.M., Giger-Mateeva V.I., Shrager P., Meisler M.H., Giger R.J. Congenital CNS hypomyelination in the Fig4 null mouse is rescued by neuronal expression of the PI(3,5)P(2) phosphatase Fig4. J. Neurosci. 2011;31:17736–17751. doi: 10.1523/JNEUROSCI.1482-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ikonomov O.C., Sbrissa D., Fligger J., Delvecchio K., Shisheva A. ArPIKfyve regulates Sac3 protein abundance and turnover: disruption of the mechanism by Sac3I41T mutation causing Charcot-Marie-Tooth 4J disorder. J. Biol. Chem. 2010;285:26760–26764. doi: 10.1074/jbc.C110.154658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chow C.Y., Landers J.E., Bergren S.K., Sapp P.C., Grant A.E., Jones J.M., Everett L., Lenk G.M., McKenna-Yasek D.M., Weisman L.S. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 2009;84:85–88. doi: 10.1016/j.ajhg.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsai C.P., Soong B.W., Lin K.P., Tu P.H., Lin J.L., Lee Y.C. FUS, TARDBP, and SOD1 mutations in a Taiwanese cohort with familial ALS. Neurobiol. Aging. 2011;32:553. doi: 10.1016/j.neurobiolaging.2010.04.009. e13–e21. [DOI] [PubMed] [Google Scholar]
- 41.Verdiani S., Origone P., Geroldi A., Bandettini Di Poggio M., Mantero V., Bellone E., Mancardi G., Caponnetto C., Mandich P. The FIG4 gene does not play a major role in causing ALS in Italian patients. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:228–229. doi: 10.3109/21678421.2012.760605. [DOI] [PubMed] [Google Scholar]
- 42.Tao J., Chen S., Yang T., Dawson B., Munivez E., Bertin T., Lee B. Osteosclerosis owing to Notch gain of function is solely Rbpj-dependent. J. Bone Miner. Res. 2010;25:2175–2183. doi: 10.1002/jbmr.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abo-Dalo B., Kim H.-G., Roes M., Stefanova M., Higgins A., Shen Y., Mundlos S., Quade B.J., Gusella J.F., Kutsche K. Extensive molecular genetic analysis of the 3p14.3 region in patients with Zimmermann-Laband syndrome. Am. J. Med. Genet. A. 2007;143A:2668–2674. doi: 10.1002/ajmg.a.32034. [DOI] [PubMed] [Google Scholar]
- 44.Nicot A.S., Laporte J. Endosomal phosphoinositides and human diseases. Traffic. 2008;9:1240–1249. doi: 10.1111/j.1600-0854.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wen P.J., Osborne S.L., Meunier F.A. Dynamic control of neuroexocytosis by phosphoinositides in health and disease. Prog. Lipid Res. 2011;50:52–61. doi: 10.1016/j.plipres.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 46.Hnia K., Vaccari I., Bolino A., Laporte J. Myotubularin phosphoinositide phosphatases: cellular functions and disease pathophysiology. Trends Mol. Med. 2012;18:317–327. doi: 10.1016/j.molmed.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 47.Below J.E., Earl D.L., Shively K.M., McMillin M.J., Smith J.D., Turner E.H., Stephan M.J., Al-Gazali L.I., Hertecant J.L., Chitayat D., University of Washington Center for Mendelian Genomics Whole-genome analysis reveals that mutations in inositol polyphosphate phosphatase-like 1 cause opsismodysplasia. Am. J. Hum. Genet. 2013;92:137–143. doi: 10.1016/j.ajhg.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huber C., Faqeih E.A., Bartholdi D., Bole-Feysot C., Borochowitz Z., Cavalcanti D.P., Frigo A., Nitschke P., Roume J., Santos H.G. Exome sequencing identifies INPPL1 mutations as a cause of opsismodysplasia. Am. J. Hum. Genet. 2013;92:144–149. doi: 10.1016/j.ajhg.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang L.F., Fukai N., Selby P.B., Olsen B.R., Mundlos S. Mouse clavicular development: analysis of wild-type and cleidocranial dysplasia mutant mice. Dev. Dyn. 1997;210:33–40. doi: 10.1002/(SICI)1097-0177(199709)210:1<33::AID-AJA4>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.