

Abstract
Arthrogryposis multiplex congenita (AMC) is caused by heterogeneous pathologies leading to multiple antenatal joint contractures through fetal akinesia. Understanding the pathophysiology of this disorder is important for clinical care of the affected individuals and genetic counseling of the families. We thus aimed to establish the genetic basis of an AMC subtype that is associated with multiple dysmorphic features and intellectual disability (ID). We used haplotype analysis, next-generation sequencing, array comparative genomic hybridization, and chromosome breakpoint mapping to identify the pathogenic mutations in families and simplex cases. Suspected disease variants were verified by cosegregation analysis. We identified disease-causing mutations in the zinc-finger gene ZC4H2 in four families affected by X-linked AMC plus ID and one family affected by cerebral palsy. Several heterozygous females were also affected, but to a lesser degree. Furthermore, we found two ZC4H2 deletions and one rearrangement in two female and one male unrelated simplex cases, respectively. In mouse primary hippocampal neurons, transiently produced ZC4H2 localized to the postsynaptic compartment of excitatory synapses, and the altered protein influenced dendritic spine density. In zebrafish, antisense-morpholino-mediated zc4h2 knockdown caused abnormal swimming and impaired α-motoneuron development. All missense mutations identified herein failed to rescue the swimming defect of zebrafish morphants. We conclude that ZC4H2 point mutations, rearrangements, and small deletions cause a clinically variable broad-spectrum neurodevelopmental disorder of the central and peripheral nervous systems in both familial and simplex cases of both sexes. Our results highlight the importance of ZC4H2 for genetic testing of individuals presenting with ID plus muscle weakness and minor or major forms of AMC.
Introduction
Arthrogryposis multiplex congenita (AMC) is a heterogeneous group of disorders with an incidence of 1 in 12,000 live births, symmetrical gender distribution, and a perinatal mortality of 32%.1 It is characterized by congenital joint contractures and is often associated with decreased fetal movements, so-called fetal akinesia.2–4 AMC is seen in a variety of developmental defects of the musculoskeletal and nervous systems, in connective-tissue disorders, and in chromosomopathies.1 In AMC-affected children, who show abnormal results on neurological examination, fetal akinesia might result from abnormalities of either the central or the peripheral nervous system, the muscle, or the neuromuscular endplate.5 These conditions comprise antenatal spinal muscular atrophy,4 hypomyelinating peripheral neuropathies,6 connatal myasthenia caused by either transplacental antibodies against the acetylcholine receptor2 or mutations in genes encoding components of the neuromuscular endplate,7 connatal myopathies,3 and myotonic dystrophy.8 Inheritance of AMC can be autosomal dominant3 or recessive,1 but rare X-linked cases have been described as well.9–12
Intellectual disability (ID) is a complex disorder that affects 1%–3% of the general population. It is characterized by significant limitations of intellectual functioning, adaptive behavior, or daily living skills with an onset before 18 years of age. Over the past decade, many ID-associated genes have been identified by chromosome breakpoint mapping, candidate-gene approaches, and whole-genome array-based copy-number analysis, the latter of which allows the detection of small deletions and duplications. More recently, massively parallel sequencing has led to the identification of many new genetic defects in familial and simplex cases, further emphasizing the vast genetic heterogeneity of ID.13–15 Similarly, X-linked ID (XLID), which arises from mutations in genes on the X chromosome and accounts for about 10%–12% of the ID seen in males,16 is a genetically heterogeneous disorder for which more than 92 associated genes have been described. Despite the large number of known mutations in genes implicated in XLID, many families assumed to be affected by XLID on the basis of linkage studies or pedigree structure still do not have a molecular diagnosis.
Our aim was to identify pathogenic mutations in families and simplex cases with the combination of AMC plus XLID and to functionally validate the pathogenic nature of the identified DNA variation in model systems. We identified disease-causing mutations in the zinc-finger gene ZC4H2 in five families and three unrelated simplex cases (of both sexes) with this phenotype. In mouse primary neurons, ZC4H2 localized to the postsynaptic compartment of excitatory synapses, and the DNA variation identified in the affected individuals influenced dendritic spine density. In zebrafish, zc4h2 knockdown caused abnormal swimming and impaired α-motoneuron development, which could not be rescued by altered proteins containing the pathogenic substitutions.
Subjects and Methods
Subjects
Individuals were recruited through neuropediatric and clinical genetics outpatient clinics and the EUROMRX consortium and its associated groups. The clinical characteristics of the persons, in whom we later discovered mutations in ZC4H2, are listed in Tables 1 and 2 and Tables S1A and S1B, available online. This study was approved by the local institutional review boards. Written informed consent was obtained from all participants and their legal guardians according to the Declaration of Helsinki. Information about deceased individuals was obtained through relatives and from hospital records.
Table 1.
Overview of Frequent Clinical Symptoms Present in Hemizygous Males
| Characteristics and Symptoms | HPO ID | Family 1 | Family 2 | Family 3 | Family 4 | Family 5 | Simplex Case 1 | Total (per Genotype) |
|---|---|---|---|---|---|---|---|---|
| Mutation | NA | c.187G>C (p.Val63Leu) | c.593G>A (p.Arg198Gln) | c.601C>T (p.Pro201Ser) | c.637C>T (p.Arg213Trp) | c.637C>T (p.Arg213Trp) | de novo X inversion | NA |
| Origin | NA | Germany | Germany | Netherlands | Australia | France | Germany | NA |
| Age at last assessment | NA | 27–74 years (mean = 56 years) | 0.5–8.0 years | 1.4–13.0 weeks | 7.5–13.5 years | 8.0–28.2 years | 3.8 years | NA |
| Age at death | NA | no disease-related deaths | 0.5–8.0 years | 1.4–13.0 weeks | no disease-related deaths | no disease-related deaths | no disease-related deaths | NA |
| Number of affected individuals | NA | 6 | 5 | 5 | 2 | 2 | 1 | 5 genotypes |
| Growth | ||||||||
| Dwarfism | HPO:1516 | – | 2 | 1 | – | 1 | 1 | 4/5 |
| Head and Neck | ||||||||
| Long (flat) philtrum | HPO:299 | 1 | 2 | 5 | – | – | 1 | 4/5 |
| Oral motor dysfunction | HPO:5216 | – | 1 | – | – | 2 | 1 | 3/5 |
| Low-set ears | HPO:369 | – | 2 | – | – | 1 | 1 | 3/5 |
| Ptosis | HPO:1488 | 6 | 3 | 1 | – | 1 | 1 | 5/5 |
| Upturned nares | HPO:463 | – | 2 | 5 | – | – | 1 | 3/5 |
| Broad alveolar ridges | HPO:187 | – | – | 5 | – | 2 | 1 | 3/5 |
| High-arched palate | HPO:218 | – | 2 | 5 | – | 2 | 1 | 4/5 |
| Carp-shaped mouth | HPO:10806 | 1 | 2 | 5 | – | 2 | 1 | 5/5 |
| Drooling | HPO:2307 | – | 3 | – | 2 | 2 | 1 | 3/5 |
| Short neck (with limited rotation) | HPO:470 | – | 2 | 1 | – | 1 | 1 | 4/5 |
| Respiratory | ||||||||
| Neonatal respiratory distress | HPO:2643 | – | 3 | 5 | – | – | 1 | 3/5 |
| Chest | ||||||||
| Humeroscapular mobility restriction | HPO:6467 | – | 2 | 1 | – | – | 1 | 3/5 |
| Narrow shoulders or thorax | HPO:6664 | 1 | 3 | – | – | 1 | 1 | 4/5 |
| Abdomen | ||||||||
| Poor feeding | HPO:2022 | – | 4 | – | – | 1 | 1 | 3/5 |
| Skeletal | ||||||||
| Kyphosis, lordosis, or scoliosis | HPO:2751 | 1 | 3 | 1 | – | – | 1 | 4/5 |
| Congenital hip dislocations or hip flexion contractures | HPO:1374 | – | 4 | 2 | – | – | 1 | 3/5 |
| Short limbs | HPO:9826 | – | – | 2 | – | 1 | 1 | 3/5 |
| Flexion contractures of elbows or knees | HPO:2987, 2978 | – | 3 | 2 | – | 1 | 1 | 4/5 |
| Proximally placed thumbs | HPO:9623 | – | 2 | 1 | – | – | 1 | 3/5 |
| Camptodactyly | HPO:1215 | – | 5 | 2 | – | – | 1 | 3/5 |
| Ulnar deviation of the finger | HPO:9465 | – | 3 | 1 | – | – | 1 | 3/5 |
| Proximally placed toes | HPO:1780 | – | 4 | 1 | – | – | 1 | 3/5 |
| Equinovarus feet or contracture of Achilles tendon | HPO:8110, 1771 | 6 | 5 | 2 | 2 | – | 1 | 5/5 |
| Skin, Nails, and Hair | ||||||||
| High anterior hairline | HPO:294 | – | 2 | 5 | – | – | 1 | 3/5 |
| Muscle and Soft Tissue | ||||||||
| Distal muscle weakness | HPO:2460 | 6 | 2 | – | – | 2 | 1 | 4/5 |
| Edema or fat pads (hands and/or feet) | HPO:7514 | – | 3 | 1 | – | – | 1 | 3/5 |
| Neurologic | ||||||||
| Retardation of motor development | HPO:1263 | 6 | 5 | 1 | 2 | 2 | 1 | 5/5 |
| Intellectual disability | HPO:1249 | 6 | 5 | 1 | 2 | 2 | 1 | 5/5 |
| Spasticity | HPO:1257 | – | 5 | – | 2 | 1 | 1 | 3/5 |
| Seizures | HPO:1250 | – | 4 | 1 | 1 | 2 | 1 | 4/5 |
| Delayed myelination on MRI | HPO:2188 | – | 2 | 1 | – | – | 1 | 3/5 |
| Diffuse cerebral atrophy on MRI | HPO:2283 | – | 3 | – | – | 1 | 1 | 3/5 |
This table shows the clinical symptoms seen in >50% of the five different genotypes of all affected males from families 1–5 and of simplex case 1. The entire set of symptoms is listed in Table S1A. None of the investigated affected males showed abnormalities in motor (n = 4) or sensory (n = 3) peripheral nerve conduction studies or any histological signs of a myopathy (n = 7). All symptoms are listed according to the nomenclature and the systematics of the OMIM “Clinical Synopsis” and are mapped to the Human Phenotype Ontology.17 The phenotypes of families 1 and 3 have been published.9,10 Abbreviations are as follows: HPO, Human Phenotype Ontology; NA, not applicable; and –, symptom absent or unknown.
Table 2.
Overview of Frequent Clinical Symptoms Present in Heterozygous Females
| Characteristics and Symptoms | HPO ID | Family 2 | Family 3 | Family 4 | Family 5 | Simplex Case 2 | Simplex Case 3 | Total (per Genotype) |
|---|---|---|---|---|---|---|---|---|
| Mutation | NA | c.593G>A (p.Arg198Gln) | c.601C>T (p.Pro201Ser) | c.637C>T (p.Arg213Trp) | c.637C>T (p.Arg213Trp) | deletion of chrX: 63,666,909–64,493,512 | deletion of chrX: 64,049,596–64,370,757 | NA |
| Origin | NA | Germany | Netherlands | Australia | France | United States | Spain | NA |
| Age at last assessment | NA | – | – | – | – | 9 years | 13 years | NA |
| Number of affected individuals | NA | 1 | 7 | 1 | 2 | 1 | 1 | 5 genotypes |
| Genitourinary | ||||||||
| Urine incontinence | HPO:20 | – | 1 | – | – | 1 | 1 | 3/5 |
| Skeletal | ||||||||
| Camptodactyly | HPO:1215 | – | 5 | – | – | 1 | 1 | 3/5 |
| Equinovarus foot deformity or contracture of Achilles tendon | HPO:8110, 1771 | 1 | 3 | – | – | 1 | 1 | 4/5 |
| Muscle and Soft Tissue | ||||||||
| Distal muscle weakness | HPO:2460 | – | 3 | – | – | 1 | 1 | 3/5 |
| Neurologic | ||||||||
| Intellectual disability | HPO:1249 | – | 3 | 1 | 2 | 1 | 1 | 4/5 |
| Dysarthria and/or deficit in expressive language | HPO:1260 | – | 1 | – | – | 1 | 1 | 3/5 |
This table shows the clinical symptoms seen in >50% of the five different genotypes of all affected females from families 2–5 and simplex cases 2 and 3. The entire set of symptoms is listed in Table S1B. All symptoms are listed according to the nomenclature and the systematics of the OMIM “Clinical Synopsis” and are mapped to the Human Phenotype Ontology.17 The phenotype of family 3 has been published.10 Abbreviations are as follows: HPO, Human Phenotype Ontology; NA, not applicable; and –, symptom absent or unknown.
Genetic Studies
For next-generation sequencing and genetic analysis, DNA was extracted from peripheral-blood samples or formalin-fixed tissues. For family 1, the mutation was identified in the index male (IV-3 in Figure 1A) by the sequencing of all exons on the X chromosome similarly to the previously described method.18 Family 2 was haplotyped with 21 microsatellite markers (5–10 cM distance) with GeneHunter,19 which revealed a 37 cM interval flanked by DXS6810 and DXS6797 (Figure S1). A BED file with all coding exons plus 100 bp flanking sequences of this region was generated with GeneDistiller software20 and used for custom production of a NimbleGen Sequence Capture Array (4.97 Mb). Fifteen micrograms of genomic DNA from individual V-1 (Figures 1B and S1) and his mother (IV-6) were fragmented, captured, and sequenced on the Genome Sequencer FLX System (Roche). Eighty-five percent of 1.54 million reads (421 ± 52 bp) were aligned (average coverage 65×) and called for variants with GeneiousPro v.5.6.5 (Biomatters). Variants were then evaluated with MutationTaster software.21 For family 3 (Figure 1C), we analyzed all coding exons of ZC4H2 with primer pairs listed in Table S2 by traditional Sanger sequencing.
Figure 1.

Overview of Genetic Results in Individuals with ZC4H2 Mutations Discovered by Massively Parallel Sequencing, Chromosome Breakpoint Mapping, and aCGH
(A–D) Pedigrees of families 1–5, affected by ZC4H2 missense mutations. Asterisks indicate that a mutation is present. Filled-in symbols represent affected individuals, and symbols with slashes represent deceased individuals. The following abbreviation is used: WT, wild-type.
(A) Pedigree of family 1, affected by WWS.9 The c.178G>C (RefSeq NM_018684.3) mutation was verified by Sanger sequencing to segregate with the disease phenotype in the family.
(B) Family 2 is affected by an early lethal AMC subtype that segregates with the c.593G>A mutation.
(C) Family 3, previously published by Hennekam et al.,10 is affected by an early lethal form of AMC due to a ZC4H2 c.601C>T missense mutation.
(D) Families 4 and 5 are affected by a c.637C>T missense mutation.
(E) Multiple-sequence alignment of ZC4H2 domains in five species and conservation of the substituted amino acids (colored and boxed). Amino acid residues of the ZC4H2 zinc-finger domain in the C terminus are underlined.
(F) Schematic view of the X chromosome with the 96 kb breakpoint region in Xq11.2 determined by fluorescence in situ hybridization (FISH) and the position and exon-intron structure of ZC4H2. For simplicity, we show only two out of five alternative transcripts. Arrows above the exons indicate the direction of transcription. Breakpoint-spanning clones in the boy with the X inversion are presented as green horizontal lines.
(G) FISH results for BAC RP13-524D22 from Xq11.2 with split signals in Xq11.2 and Xq28 (arrows) indicate that this clone spans the breakpoint.
(H) RT-PCR expression results of ZC4H2 (upper) and HPRT (lower) in the fibroblasts of the boy with the X inversion (P) and of the control (C). For ZC4H2 RT-PCR amplifications, we used two different primer pairs (indicated by 1 and 2 below the agarose-gel picture) and found with both sets that ZC4H2 transcripts were lacking in the affected individual but that the expected products (three bands in lane 4 and one band in lane 5) were present in a male control. HPRT RT-PCR served as a control for cDNA integrity. Abbreviations are as follows: M, marker; +, addition of reverse transcriptase enzyme (RT); and –, negative control.
(I) Schematic view of the deletions identified by aCGH in two affected girls (simplex cases 2 and 3). Blue horizontal bars indicate the respective deletion in each of the affected individuals. Both deletions include ZC4H2 (indicated by the red bar), but no other known gene.
For five individuals of family 4 (I-1, I-2, II-1,II-2, and II-5 in Figure 1D), whole-exome sequencing (WES) was performed with Illumina TrueSeq WES protocols. The enriched DNA (350–400 bp products) was sequenced on the Illumina HiSeq2000 platform (Axeq Technologies), which returned on average 48.3 Mb of 100 bp paired-end reads per individual. These reads were quality trimmed with the FASTX toolkit and aligned to UCSC Genome Browser hg19 with the Burrows-Wheeler Aligner with default parameters, except for -d 5 and -l 35. SAMtools was used for generating BAM files. Sequence variants were realigned, recalibrated, and reported with the Genome Analysis Toolkit and categorized with Annovar. We applied different inheritance models for this family: autosomal recessive (homozygous or compound heterozygous), X-linked, and de novo (postulated germline mosaicism was applied for one of the parents).
For family 5, all X-chromosome-specific exons were enriched from genomic DNA of the index male (IV-1 in Figure 1D) with droplet-based multiplex PCR similarly to the previously described method,22 and DNAs were sequenced on the Illumina HiSeq2000 platform (ATLAS).
For segregation analysis of the ZC4H2 mutations identified in this study, we used gene-specific primers flanking the mutations and determined genotypes for all available family members by Sanger sequencing of PCR products. The c.593G>A (RefSeq accession number NM_018684) (p.Arg198Gln) mutation was further verified by restriction-fragment-length-polymorphism analysis and excluded in 100 alleles of healthy control subjects from the same ethnic background: the 290 bp PCR product generated with the oligonucleotide primers 5′-CTGAACTGGGTTATGTCCTTCC-3′ (forward) and 5′-CTTCCGTTTCGGCTTTTTG-3′ (reverse) was cleaved by HpaII into 263 + 27 bp in the presence of the mutation and 218 + 45 + 27 bp in the absence of the mutation (data not shown).
We performed X-inversion breakpoint mapping by fluorescence in situ hybridization (FISH) on metaphase chromosomes of the affected boy according to standard protocols by using bacterial artificial clones (BACs) selected from the regions of interest (Table S3). For expression analysis in fibroblasts, we isolated total RNA and performed RT-PCR with two different sets of ZC4H2 primer pairs and one HPRT primer pair (Table S2), which served as a control for cDNA integrity. ZC4H2-specific bands were excised from agarose gels, extracted (QIAGEN), and Sanger sequenced with the same primers as for the amplification.
For simplex case 1, array comparative genomic hybridization (aCGH) was performed with the NimbleGen CGX-6 format (which includes 135K oligonucleotide probes) according to the manufacturer’s protocol, and the scanned data were processed with Genoglyphix software (Signature Genomics). For simplex cases 2 and 3, aCGH was performed with a 105K feature (case 2) or a 135K feature (case 3) whole-genome microarray (SignatureChip Oligo Solution version 1.0 or 2.0, custom-designed by Signature Genomic Laboratories and manufactured by Agilent Technologies or Roche NimbleGen) as previously described.23,24
Generation of Human Wild-Type and Altered ZC4H2 Constructs for Overexpression Studies
For transient expression, full-length open reading frames of human wild-type and altered ZC4H2 constructs from family 1 were amplified with primer sets (1) ZC4H2-EGFP-N2-EcoRI-forw and ZC4H2-EGFP-N2-SalI-rev and (2) ZC4H2-EGFP-C2-EcoRI-forw and ZC4H2-EGFP-C2-SalI-rev (Table S2). PCR products were double digested with EcoRI and SalI (New England Biolabs), and the fragments were ligated into pEGFP-C2 and pEGFP-N2 (Clontech). The clones with c.637C>T (p.Arg213Trp), c.593G>A (p.Arg198Gln), and c.601C>T (p.Pro201Ser) mutations were generated from the wild-type constructs by site-directed mutagenesis with the Quick Change II Site-Directed Mutagenesis Kit (Stratagene). Myc-tagged ZC4H2 constructs were generated from the pEGFP-C2 clones after double digestion with EcoRI and SalI and ligation into the pCMVTag3B vector (Stratagene). Correctness of all sequences was determined by Sanger sequencing.
Immunofluorescence Studies of Wild-Type and Altered ZC4H2 in Cell Lines and Primary Neurons
Neuro2A cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) in the presence of antibiotics. Hippocampi or cortices were dissected from embryonic day (E) 17.5 C57BL/6J mouse embryos, dispersed in cold PBS, and triturated with papain (10 U/ml) (Worthington) for 22 min at 37°C. Cells were resuspended in DMEM/Ham’s F-12 with 10% FBS and centrifuged at 400 × g (for 3 min), and the pellet was resuspended in primary neuron basal medium, neuronal survival factor, and 0.5 mM glutamine (Lonza). Dissociated cells were plated onto poly-D-lysine (SIGMA)- and laminin (Invitrogen)-coated substrates at a density of 400 cells/mm2. For immunofluorescence studies, human embryonic kidney (HEK) 293T and Neuro2A cells were fixed with 4% PFA 24 hr after transfection, permeabilized in 0.2% Triton X-100, and washed at room temperature (RT). Cells were incubated with 0.5 μg DAPI/ml and mounted with Fluoromount (SouthernBiotech). Transfected mouse hippocampal neurons were fixed with 4% PFA for 20 min and permeabilized with PBS, 3% BSA, and 0.2% Triton X-100 for 1 hr at RT.
Per well, 5 × 104 HEK293T cells or 8 × 104 Neuro2A cells were transfected with 1 μg plasmid DNA and 1 μl Lipofectamine 2000 (Invitrogen). Images were acquired with a confocal laser-scanning microscope (Zeiss LSM 700) with a 63× objective and were analyzed with the ZEISS LSM Image Browser. Mouse hippocampal neurons were transfected at 14 days in vitro (DIV) with the use of the Magnetofectamine transfection kit (OZ Biosciences). Microscopy was carried out 24–48 hr after transfection. Cells were incubated with primary antibodies mouse anti-PSD95-mAb (1:100; ThermoFisher), goat anti-GAD65-67 (1:50; SCBT), goat anti-VGLUT1 (1:50; SCBT), and rabbit anti-Gephyrin (1:1,000; Abcam) diluted in blocking solution (PBS, 3% BSA, and 0.2% Triton X-100) and with a secondary antibody (FluoProbes FP-594 donkey anti-mouse, anti rabbit, or anti-goat antibody [1:300; Interchim]) for 45 min. Sequential acquisitions were made, and high-resolution z stack images of neurons were taken with the 63× objective of a confocal FluoView 500 microscope (Olympus) with optical section separation (z interval) of 0.5 μm and maximal projections. The extent of colocalization was measured with the “colocalization” module of the 64-bit version of Imaris 5.0.2 (Bitplane AG).25
Colocalization of ZC4H2-GFP with presynaptic and postsynaptic markers was statistically analyzed with Prism Software and the Mann-Whitney U test. The number of dendritic spines was statistically analyzed with a one-way ANOVA (posthoc Newman-Kewls).
Immunoblot Analysis
A total of 5 × 105 HEK293T cells were grown in 75 cm2 flasks, and 8 μg of plasmid DNA was transfected with the use of Lipofectamine2000 (Invitrogen) according to the manufacturer’s recommendations. Twenty-four hours after transfection, cells were lysed in lysis buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 2 mM MgCl2, 0.1% NP40, and protease inhibitors [Roche]), sonicated, and centrifuged for 15 min at 13,000 rpm at 4°C. Immunoblots were incubated with the following primary and secondary antibodies: goat polyclonal anti-GFP-HRP (1:10,000; Abcam), rabbit polyclonal anti-actin (1:10,000; Sigma), peroxidase-conjugated anti-mouse-HRP (1:3,000; Dianova), and anti-rabbit-HRP (1:5,000; Amersham).
Zc4h2 Expression Analysis in Mouse Brain Tissues and Primary Cortical Cultures
All mouse experiments were performed according to protocols approved by the Committee on Use and Care of Animals at the University François-Rabelais (Tours, France) and by the Institut National de la Santé et de la Recherche Médicale (Paris, France). TissueScan qPCR Arrays (OriGene) were used for screening for Zc4h2 expression in 48 samples covering five stages of embryonic, postnatal, and adult brain tissues with the use of Soadvance Syber (BioRad) gene expression assays. The prestandardized cDNAs were amplified by PCR using a primer set specific to Zc4h2 (Table S2).
RNA was extracted from primary cortical neurons after 5, 10, 14, and 21 days in culture. cDNA was synthesized with the QuantiTect RT kit (QIAGEN). Quantitative PCR (qPCR) was done with the use of 50 ng cDNA as a template with SsoFast Eva Green (BioRad) reagent. The same specific primer set was used for expression assays in mouse tissues. The assays were performed in duplicate on LightCycler480 (Roche). Quantification was carried out by the 2-ΔΔCp method. The normalized ratio was obtained for each target gene with the use of LC480 software, qPCR efficiencies were considered, and reference genes Cyc1, Sdha, and Gapdh were used for normalization.
Zebrafish Whole-Mount In Situ Hybridization
Zebrafish were bred and maintained according to approved guidelines prescribed by the Committee on Use and Care of Animals at the National Institute of Genetics (Japan). For in situ hybridization, embryos at 24 and 48 hr postfertilization (hpf) were fixed overnight with 4% PFA in PBS. Embryos were then dehydrated with ascending concentrations of methanol and stored at −20°C overnight before rehydration with descending concentrations of methanol and treatment with proteinase K to increase permeability. Sense and antisense cRNA probes were generated with the DIG RNA labeling kit (Roche) according to the manufacturer’s instructions. Probe hybridization was performed overnight at 65°C and was detected with a DIG antibody conjugated to alkaline phosphatase (Roche), resulting in color development when the substrates nitro blue tetrazolium chloride and 5-bromo-4-chloro-indolyl phosphate were added. In situ hybridization using a sense probe did not detect any significant signal.
Knockdown and Rescue Experiments in Zebrafish
Zebrafish zc4h2 (RefSeq NM_199642) was cloned into a pCR4-TOPO vector (Invitrogen) and used for cRNA synthesis. Antisense morpholino oligonucleotides (MOs) were designed against the translation initiation codon (MO1) and the splice donor site between exon 2 and intron 2 (MO2) of zebrafish zc4h2. Zebrafish embryos were injected with 5 ng of MOs at 1- to 2-cell stages and studied as previously published.26 At these dosages, control MOs produced no discernible phenotype change. For rescue experiments, zebrafish zc4h2 and mouse Zc4h2 (RefSeq NM_001003916) were cloned into the pCS2+ expression vector and missense mutations were introduced into the mouse clone by site-directed mutagenesis. All primer sequences are presented in Table S2. Capped RNA was synthesized with the mMESSAGE mMACHINE SP6 kit (Life Technologies) according to the manufacturer’s protocol. Capped RNA (100 pg) was coinjected with MO2 (4 ng) into zebrafish embryos at 1- to 2-cell stages. At 2 days postfertilization (dpf), normal zebrafish embryos swam away rapidly (>2 cm/s) upon tactile stimulation. The number of embryos that exhibited slow swimming (<2 cm/s) after touch was counted. For labeling of motoneurons and neuromuscular junctions (NMJs) in zebrafish, embryos were fixed in 4% PFA, washed in PBS, and labeled. Motor axon projections were labeled with a znp-1 antibody (antisynaptotagmin 2, 1:100, DSHB) and an AlexaFluor488-conjugated anti-mouse IgG secondary antibody (Life Technologies). NMJs were labeled with α-bungarotoxin conjugated with AlexaFluor594 (Life Technologies).
Results
Clinical Delineation of a Syndrome with AMC plus ID
We initially studied two large families (families 1 and 2) who are compatible with X-linked recessive inheritance and in whom multiple malformations are associated with ID. Although contractures of the feet were one of the main clinical symptoms in family 1, affected by Wieacker-Wolff syndrome (WWS [MIM 314580]),9 the children of family 2 presented with neonatal respiratory distress, AMC, muscle weakness, and ptosis. These features suggested dysfunction of neuromuscular transmission in utero. As development progressed, it became clear that the affected boys of family 2 were also severely intellectually disabled. Correspondingly, the affected males from family 1 also had signs of a developmental defect of neuromuscular transmission, albeit in a much more attenuated form, as seen by congenital equinovarus foot deformity and ptosis. Central symptoms—besides ID—that were seen in the majority of individuals were spasticity due to involvement of upper motor neurons and seizures as a manifestation of disturbed formation of the cerebral network. Histological investigation did not provide evidence of a demyelinating or axonal neuropathy or a myopathy (see below). Phenotypic analysis of heterozygous females revealed mild ID, and in several individuals, additional minor dysmorphic signs, such as camptodactyly and equinovarus feet, were present. The phenotype of family 3 had been published previously.10 As a result of our findings and the presence of multiple contractures, respiratory distress, and severe ID in the family, we contacted the authors and enrolled this family into our study. Family 4 had been included in a larger study aimed at identifying novel genes for cerebral palsy (CP). Because the inheritance pattern of CP in the family was not unequivocally X linked, five individuals (affected and unaffected) were analyzed by WES. The heterozygous girl from this family (II-7 in Figure 1D) was assessed at the age of 3.75 years. She had a significant deficit of language development (especially of perceptive language) and of personal and social skills. Her fine and gross motor abilities, however, were appropriate for her age. She had neither CP nor joint contractures. The family 5 index individual, who presented with ID and additional clinical symptoms similar to those of the affected males of families 2 and 3, had been included in the EUROMRX cohort and thus has been sequenced for all X-chromosome-specific exons. For more clinical details on all affected individuals from families 1–5 and simplex cases 1–3, see Tables 1, 2, S1A, and S1B. We can single out retardation of motor development, ID, and equinovarus feet as the minimum common phenotypic denominators of WWS for all different mutations described in this article. The syndrome thus represents a clinically distinct and phenotypically broad neurodevelopmental disorder of the central and peripheral nervous systems.
Next-Generation Sequencing Identifies Missense Mutations in ZC4H2
In an effort to find pathogenic mutations in families affected by syndromic and nonsyndromic forms of XLID, we identified the disease-causing mutation in a family afflicted with WWS27,28 by sequencing all X chromosome exons. After all identified variants were filtered against publicly available data, the only one that remained was a missense mutation in exon 3b (c.187G>C [p.Val63Leu]) of the zinc-finger gene ZC4H2 (RefSeq NM_018684.3). This mutation cosegregated with the disease in the WWS-affected family (family 1 in Figure 1A). In family 2, linkage analysis localized the responsible locus between DXS6810 and DXS6797, and deep sequencing of the linkage interval and subsequent segregation analysis indicated a cosegregating mutation in exon 6 of ZC4H2 (c.593G>A [p.Arg198Gln]) (Figure 1B). Clinical photographs and MRIs of two affected boys are presented in Figures 2A–2C. For family 3,10 we used gene-specific primers to search all ZC4H2 exons and exon-intron boundaries by PCR and Sanger sequencing for a mutation in the mother (III-14 in Figure 1C) of one affected male. This analysis revealed in ZC4H2 exon 6 another transition (c.601C>T [p.Pro201Ser]), which was also present in the heterozygous form in her two sisters (III-6 and III-7 in Figure 1C). For family 4, we performed WES on five family members (I-1, I-2, II-1, II-2, and II-5 in Figure 1D) because the inheritance pattern was inconclusive. In seven genes, we identified unique sequence variants, which were subsequently validated. The three X chromosome variants—c.637C>T (p.Arg213Trp) in ZC4H2 (Figure 1C), c.20T>A (RefSeq NM_152423.4) (p.Leu7Gln) in MUM1L1, and c.3140G>A (RefSeq NM_032968.3) (p.Gly1047Glu) in PCDH11X (MIM 300246) (data not shown)—were predicted to result in amino acid changes. All three variants segregated in the extended family (whole-exome-sequenced individuals, as well as the remaining sibling) according to postulated X chromosome inheritance (data not shown), and as such, we could not distinguish which one was causative. However, during data analysis, the latter two variants (in MUM1L1 and PCDH11X) appeared as SNPs in the latest release of dbSNP135 and were thus considered to be rare, noncausative variants. Of the identified autosomal variants, the potentially compound-heterozygous change in SLC11A2 (MIM 600523) was not confirmed by Sanger sequencing and the two MYH14 (MIM 608568) changes were both on the same allele (i.e., not compound heterozygous). These MYH14 variants did not segregate, given that the healthy mother and her healthy daughter had them as well. The de novo heterozygous variants in FLNB (MIM 603381) and PLAC4 (MIM 613770) were not confirmed by Sanger sequencing (data not shown).
Figure 2.
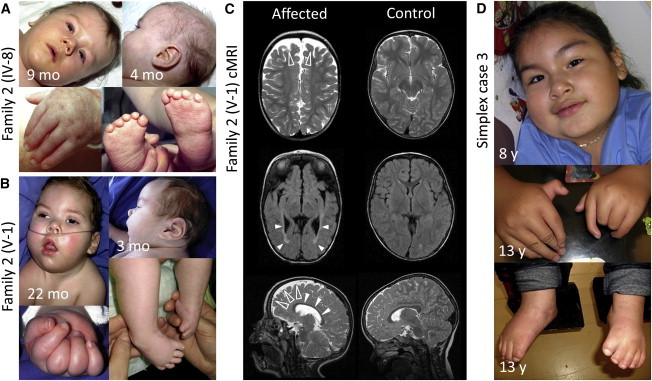
Clinical Photographs and Cranial MRIs of Two Affected Boys from Family 2 and a Simplex Case
(A) Male child IV-8 (family 2) shows facial weakness, low-set and posteriorly rotated ears, and strabismus. He has bilateral camptodactyly and equinovarus feet with small, proximally placed toes.
(B) Male child V-1 (family 2) shows prominent facial weakness, a flat philtrum, strabismus, upturned nares, a carp-shaped mouth, narrow shoulders, and muscular hypotonia. The contracted fingers cannot be opened passively and show characteristic fat pads over the extensor surfaces.
(C) Axial and parasagittal cranial MRIs (cMRIs) of individual V-1 (family 2) at the age of 22 months (left column) and an age-matched control (right column). In the affected child, frontal brain atrophy (open arrowheads) and hypoplasia of the corpus callosum (closed arrowheads) are depicted in the sagittal T2-weighted image. The fluid-attenuated-inversion-recovery image (middle panel) shows abnormality of the periventricular white matter mainly in the occipital region (closed arrowheads).
(D) Female simplex case 3 shows narrow shoulders, proximally placed thumbs, camptodactyly, radial and ulnar deviation of the fingers, and mild equinovarus feet.
The same ZC4H2 mutation as was found in family 4 (c.637C>T [p.Arg213Trp]) was identified in the affected male from family 5 (IV-1 in Figure 1D) by massively parallel sequencing of all X-chromosome-specific exons with the use of a PCR-based enrichment strategy. The mutation was also present in his mother (III-2), his affected uncle (III-6), and two other female relatives (II-2 and III-4) (Figure 1D). Representative sequence chromatograms of all mutations from families 1–5 are shown in Figure S2.
All ZC4H2 amino acid positions affected by identified mutations are highly conserved (Figures 1E and S3). The new amino acids are predicted to be damaging to ZC4H2 function by PolyPhen-2 and MutationTaster.17 Such changes were not detected in publicly available databases, including the 1000 Genomes Project database, dbSNP135, and the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server. Moreover, when tested in 100 ethnic controls (males), the c.637C>T (p.Arg213Trp) mutation was not detected. None of the changes were present among >450 X chromosome exomes from males with XLID (data not shown). Importantly, no other variants (X linked or autosomal) identified and segregating in these families were predicted to be deleterious.
An X Chromosome Inversion Abolishes ZC4H2 Expression in a Severely Affected Boy
We also examined a severely affected boy (simplex case 1 in Tables 1 and S1A) with a phenotype similar to that of the affected boys of families 2 and 3; he was found to have a paracentric de novo inversion on the X chromosome with breakpoints at Xq11.2 (location of ZC4H2) and Xq28. Using serial FISH, we mapped both breakpoints to intervals of <150 kb (Figures 1F and 1G and Table S3). Whereas the breakpoint region at Xq28 did not contain any known gene (Figure S4), the mapped region at Xq11.2 contained ZC4H2, which led us to speculate that the rearrangement might interfere with the normal expression of this gene. We thus performed RT-PCR experiments with two different primer sets on RNA isolated from cultured skin fibroblasts of the affected male and a control. The results showed that unlike in the normal control, no ZC4H2 transcripts were detectable in mRNA from the affected male (Figure 1H), indicating that the rearrangement abolished ZC4H2 expression.
aCGH Identifies Mildly Affected Females with Single Allelic Deletions of the ZC4H2 Locus, and ZC4H2 Is Subject to X Inactivation
Using whole-genome microarray analysis, we also identified small heterozygous deletions at Xq11.2 in two mildly affected girls (simplex cases 2 and 3 in Tables 2 and S1B and Figures 1I and S5). In simplex case 2, the deletion removed about 826 Kb of the X chromosome genomic DNA (chrX: 63,666,909–64,493,512 [UCSC Genome Browser hg19]) and was absent in her mother. In simplex case 3, the deletion encompassed about 321 Kb (chrX: 64,049,596–64,370,757 [UCSC Genome Browser hg19]). Clinical photographs are presented in Figure 2D. Additional parental DNA was unavailable for further testing. Both deletions included ZC4H2, but not any other adjacent gene, and allowed us to conclude that heterozygous deletions of ZC4H2 can result in a clinical phenotype.
To investigate whether ZC4H2 might be subject to X inactivation, we determined its expression level in blood lymphocytes from healthy males and females by microarray analysis. Its relative expression level compared to those of other genes known to be expressed from only one or from both X chromosomes suggested that ZC4H2 is subject to X inactivation in females (Figure S6). We next determined the X inactivation status of healthy and affected females who carried a heterozygous missense mutation or deletion of ZC4H2. We observed preferential inactivation of the mutated X chromosome in all females tested, except for the affected girl of family 4 and her unaffected mother (data not shown). An overview of all genetic results is provided in Table S4.
ZC4H2 Is Expressed in the Central and Peripheral Nervous Systems of Humans, Mice, and Zebrafish
The functional role of ZC4H2 is not well characterized. We therefore first investigated its expression pattern and found it to be transcribed in human fetal brain and other tissues (Figure S7). In mouse and zebrafish embryos, Zc4h2 and zc4h2, respectively, are strongly expressed throughout the brain and in the spinal cord (Figure 5A and Mouse Genome Informatics [see Web Resources]). These results prompted us to investigate the in vivo expression pattern of Zc4h2 during neurodevelopment in various mouse brain regions and spinal cord of different developmental stages. Interestingly, in all brain areas, Zc4h2 expression was highest during embryonic development and declined postnatally, suggesting an important role of Zc4h2 during brain development (Figure 3). A similar dynamic gene expression was evident in cultures of mouse primary cortical neurons: there was a significant decline of Zc4h2 expression in more mature neurons than in immature neurons (p < 0.05) (Figure S8).
Figure 5.
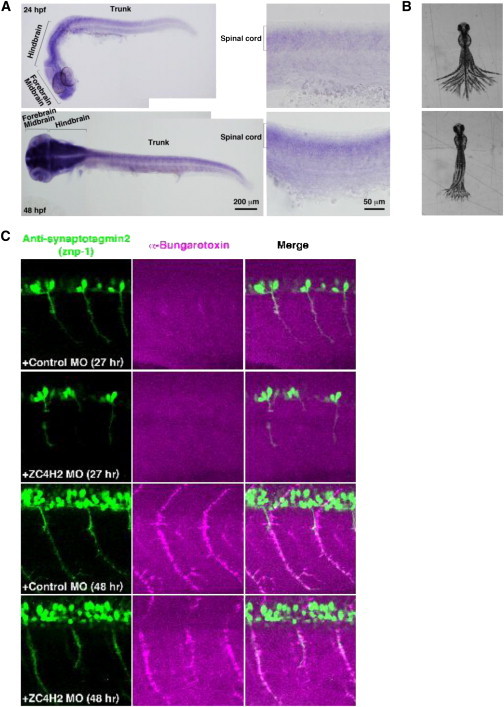
Zebrafish zc4h2 Is Expressed in the Brain and Spinal Cord, and its Knockdown Causes Compromised Swimming Contractions and Abnormal α-Motoneuron Development
(A) Whole-mount in situ hybridization with a zc4h2 probe at 24 hpf (upper row) and 48 hpf (lower row). Embryos show zc4h2 expression in the forebrain, midbrain, hindbrain, and spinal cord.
(B) Superimposed images of tail movement at 48 hpf. Compared to animals injected with control MOs, zc4h2-knockdown zebrafish (lower) show weak swimming contraction.
(C) At 27 and 48 hpf, outgrowth of spinal cord motoneurons and formation of NMJs are morphologically affected in zc4h2 morphants, but not in controls.
Figure 3.
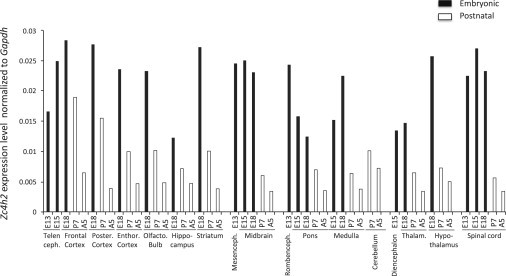
Mouse Zc4h2 Is Expressed in Various Brain Tissues and the Spinal Cord, and its Expression Is Reduced Postnatally
Zc4h2 mRNA expression was assessed by quantitative RT-PCR using SYBR Green I protocol and arrays containing cDNA from 48 mouse samples. The tissues represented are telencephalon, frontal cortex, posterior cortex, entorhinal cortex, olfactory bulb, hippocampus, striatum, mesencephalon, midbrain, rhombencephalon, pons, medulla, cerebellum, diencephalon, thalamus, hypothalamus, and spinal cord. Five developmental stages were investigated: E13, E15, E18, postnatal day 7 (P7), and adult week 5 (A5). Expression of Zc4h2 mRNA is shown relative to the Gapdh reference gene.
High Levels of Altered ZC4H2 Reduce the Number of Dendritic Spines
To gain further insight into the functional role of ZC4H2, we studied the subcellular localization of wild-type and altered proteins in cell lines and mouse primary neurons. Transient transfection of tagged ZC4H2 constructs led to the stable production of the corresponding protein (Figure S9). Immunofluorescence analysis of altered and wild-type ZC4H2 in Neuro2A and HEK293T cells showed its presence in both the nucleus and the cytosol. Compared to that of wild-type constructs, overexpression of constructs containing the missense mutations identified in the families did not reveal any obvious changes (Figure S10 and data not shown). Subsequent analysis showed that human ZC4H2 was distributed in mouse primary hippocampal neurons similarly to in Neuro2A and HEK293T cells (Figure 4A). A more detailed analysis showed that both wild-type and altered ZC4H2 localized at excitatory postsynaptic sites, as verified by a predominant colocalization with PSD95, but not in inhibitory synapses (Figures 4B, 4C, and S11). In subsequent follow-up studies, using altered constructs containing the missense mutations, we found that compared to wild-type ZC4H2 and GFP alone, the p.Val63Leu, p.Arg198Gln, and p.Pro201Ser substitutions caused a significant decrease in synapse number and density. Remarkably, the p.Arg198Gln and p.Pro201Ser substitutions, which caused a more severe clinical phenotype in the affected individuals, were most deleterious (Figures 4D and 4E). We obtained similar results with Myc-tagged constructs, which we used to exclude any potential effect of GFP on ZC4H2 (data not shown).
Figure 4.

Recombinant ZC4H2 Localizes to Excitatory Postsynaptic Sites in Primary Hippocampal Neurons, and Altered Proteins Cause a Significant Decrease in Dendritic Spine Number and Density
(A) Confocal image of a living mature mouse primary hippocampal neuron (DIV 16) with transiently produced ZC4H2-GFP (36 hr after transfection) shows that the protein is present throughout the cell (three independent transfections; 15 cells; scale bar represents 50 μm). The right-hand panel shows a higher magnification of a single neurite (scale bar represents 2 μm). The comparative analysis of neurons transfected with the pEGFP vector used for cloning did not reveal any change in neuronal morphology, nor did we see any differences between GFP-tagged and Myc-tagged constructs (data not shown).
(B) Confocal images of fixed primary hippocampal neurons (DIV 16) with ZC4H2-GFP (36 hr after transfection). Immunostaining with PSD95 (excitatory postsynaptic marker) or VGLUT (excitatory presynaptic marker) antibodies revealed a postsynaptic localization of ZC4H2-GFP in excitatory synapses (n = 3 transfections; 12 cells; scale bar represents 5 μm). Arrows indicate domains of colocalization.
(C) Quantification of the mean percentage of colocalization of ZC4H2 with endogenous PSD95 and VGLUT (excitatory synapse) and with endogenous gephyrin and GAD (inhibitory synapse) (Mann-Whitney U test; **p < 0.01). The following abbreviation is used: ns, not significant. Error bars represent the SEM.
(D) Confocal images of dendrites and dendritic spines of mature neurons (DIV 16) transfected with pEGFP (negative control), ZC4H2-GFP, ZC4H2 p.Arg213Trp-GFP, ZC4H2 p.Val63Leu-GFP, ZC4H2 p.Arg198Gln-GFP, or ZC4H2 p.Pro201Ser-GFP plasmids. For each neuron that was further analyzed, we counted the number of spines in 10 μm sections: pEGFP (1 transfection; 8 cells; 47 spines), ZC4H2 (3 transfections; 18 cells; 111 spines), ZC4H2 p.Arg213Trp (3 transfections; 23 cells; 151 spines), ZC4H2 p.Val63Leu (3 transfections; 7 cells; 47 spines), ZC4H2 p.Arg198Gln (3 transfections; 11 cells; 94 spines), and ZC4H2 p.Pro201Ser (3 transfections; 18 cells; 156 spines).
(E) Quantification of the mean number of dendritic spines for the transiently expressed GFP, wild-type ZC4H2, and altered ZC4H2. Error bars represent the SEM.
The statistical comparative analysis was performed with PRISM software and a one-way ANOVA test (posthoc Newman-Kewls). *p < 0.05; **p < 0.01; ***p < 0.001. The following abbreviation is used: ns, not significant.
Knockdown of zc4h2 in Zebrafish Results in Impaired Swimming and a Reduced Number and Disorganized Pattern of Neuromuscular Endplates
Finally, we investigated the loss of zc4h2 function in zebrafish by knockdown with two different antisense MOs, one designed against the translation initiation codon and the other against the splice donor site between exon 2 and intron 2 of zc4h2. This knockdown did not result in any gross morphological abnormalities, but the morphants showed impaired swimming capability at 2–3 dpf as a result of compromised swimming contractions (Figure 5B and Movies S1 and S2). This defect could be rescued with wild-type zebrafish zc4h2 and with mouse Zc4h2 cDNA constructs, but not with constructs carrying the missense mutations of families 1–5 (Table S5). Because slow swimming might be due to muscle weakness, we next investigated the effect of zc4h2 ablation on the development of the motoneurons, the myotome, and the NMJ. In contrast to control embryos, zc4h2 morphants had shorter and less branched motoneurons projecting from the spinal cord by 27 and 48 hpf, demonstrating a slower pace of axon growth and branching. α-bungarotoxin staining revealed a reduced number and disorganized pattern of neuromuscular endplates in zc4h2 morphants compared to controls (Figure 5C).
Because muscle-fiber growth depends on the timely and correct establishment of the NMJ,29 we measured fiber diameters in muscle-biopsy specimens taken from five affected boys of family 2 at different ages. Until 6 weeks of age, the fibers were smaller and later normalized in size, suggesting delayed innervation (Figure S12A). A disorder of myelination was excluded by electron microscopy (Figure S12C). The formation of the neuromuscular endplate is a carefully orchestrated process of axon trajectories through growth-cone choices and targeting of the myofiber, and an imbalance of muscle agonists and antagonists results in permanent contractures and arthrogryposis.30
Discussion
We report the association between mutations in the X-linked zinc-finger gene ZC4H2 and a disease phenotype. Mutations in ZC4H2 were identified in intellectually impaired individuals in four AMC-affected families and in one CP-affected family. On the basis of in silico analysis, ZC4H2 had previously been suggested as a candidate for XLID.31 In several families, females with heterozygous ZC4H2 mutations were also affected, but to a lesser degree. Furthermore, we found two ZC4H2 deletions and one rearrangement in two female and one male unrelated simplex cases, respectively. These findings, along with the clinical findings in heterozygous females from families 2–5, indicate that females with ZC4H2 mutations might also be affected. This is corroborated by a recently published female who has joint contractures32 and other symptoms and who carries a heterozygous deletion that removes ZC4H2 in addition to other genes.
ZC4H2 amino acids, which are affected by damaging mutations, are highly conserved and predicted to be functionally relevant. In families 1–5, the disease severity correlated with the severity of the underlying mutation. The location of the p.Val63Leu substitution (family 1) in a potential coiled-coil domain is predicted to alter secondary structure and slightly destabilize the local protein conformation.33 This substitution caused a comparatively mild phenotype in females with this mutation. The p.Leu198Gln and p.Pro201Ser substitutions (families 2 and 3, respectively), associated with a severe lethal phenotype, lie within the highly conserved zinc-finger domain (Figure S3) and are predicted to compromise the domain’s function.33 The p.Arg213Trp substitution (families 4 and 5) is located in the protein’s highly conserved C terminus, which cannot be assigned a known domain function or structure. The disease severity of these individuals is intermediate. Importantly, some females with the p.Arg213Trp substitution have developmental delay but are normal otherwise. We hypothesize that the different affection statuses of females with a heterozygous ZC4H2 mutation might be related to varying X inactivation. The results obtained in peripheral-blood lymphocytes were consistent with the presence of symptoms in some heterozygous females, but not in others. However, because the X inactivation pattern in the nervous system can differ from that seen in the blood, as reported for Rett syndrome,34 investigation of blood cells might not be suitable for predicting the phenotype.
Given the clinical phenotypes, we conclude that ZC4H2 should have a neuronal function during fetal growth. Our expression studies in mouse brain tissues of different developmental stages indicate that the gene has its highest level of expression during embryogenesis and that mRNA levels decline after birth. In primary hippocampal mouse neurons, transiently produced ZC4H2 controls the number of dendritic spines, and the magnitude of spine reduction reflects the severity of the clinical phenotype. In zebrafish, zc4h2 knockdown caused abnormal swimming and impaired α-motoneuron development. The deleterious nature of the ZC4H2 variants found in this study is supported experimentally by failure of all missense mutations to rescue the swimming defect of zebrafish morphants.
Apart from in our studies, the role of ZC4H2 has only been investigated in Caenorhabditis elegans, where a homozygous deletion of zc4h2 (called vab-23) was found to be lethal at early larval stages because of failed ventral closure. Neuron-specific rescue of vab-23 in ventral neuroblasts entirely rescued the phenotype. The authors hypothesized that vab-23 might be a nuclear transcription factor with an influence on gene expression for secreted cues that regulate different steps of morphogenesis.35 It was subsequently demonstrated that vab-23 acts under the control of the hox-gene lin-39 and binds directly to the smp-1 promoter to induce smp-1 (semaphorin 1a) transcription.36 Semaphorins are a group of key signal proteins that act through their receptor proteins (neuropilins and plexins) and are essential for neuronal pathfinding, axon sorting and branching, endplate formation, dendrite specification, and synaptic specificity.37 The general importance of axonal-guidance ligands in the pathophysiology of arthrogryposis has recently been supported by the discovery of mutations in endothelin-converting enzyme-like 1 (ECEL1) (MIM 605896 and 615065). Corresponding alterations of endopeptidase ECEL1 prevent the intramuscular branching of α-motoneurons and the formation of sufficient NMJs, resulting in distal arthrogryposis without central nervous symptoms.38,39
Dysregulation of axonal-guidance molecules (e.g., semaphorins or ephrins) might thus be the common denominator for the complex phenotype seen in the individuals with symptoms that affect central neuronal circuits (ID, seizures, gyral and white-matter abnormalities, and, occasionally, CP) and the peripheral nervous system (arthrogryposis and muscle weakness).
Taken together, the X-linked gene ZC4H2 plays an important role during embryonic development of the central and peripheral nervous systems. Although the variable clinical manifestation of ZC4H2 mutations, ranging from lethality in some affected males to very mild or absent symptoms in some females who have the mutation, might be explained in part by skewed X inactivation, other genetic, environmental, or stochastic factors might also be involved. Our results suggest the importance of including ZC4H2 in genetic testing of individuals presenting with ID plus muscle weakness and AMC (or at least equinovarus feet).
Acknowledgments
We thank all family members and affected individuals for participating in this study, Susanne Freier and Susanne Lützkendorf for excellent technical assistance, Cordula Nuener for help with fluorescence in situ hybridization, Karoline Gemeinhardt for initial RT-PCR experiments, and the Department of Microscopy (P.P.F.) of the University of Tours. J.A.R. is an employee of Signature Genomic Laboratories, a subsidiary of Perkin Elmer, Inc. This work was financed by a grant from the German Ministry of Education and Research through the German Mental Retardation Network; by the Deutsche Forschungsgemeinschaft (SFB665 TP C4); by the Einsteinstiftung Berlin (A-2011-63); by Project GENCODYS (241995), which is funded by the European Union Framework Program 7; by Australian National Health and Medical Research Council (NHMRC) grants APP1019928 and APP1008077; by Australian NHMRC Research Fellowship APP508043 (to J.G.); by the Australian Cerebral Palsy Foundation; and by the Adelaide Women’s & Children’s Hospital Research Foundation.
Contributor Information
Markus Schuelke, Email: markus.schuelke@charite.de.
Vera M. Kalscheuer, Email: kalscheu@molgen.mpg.de.
Supplemental Data
Zebrafish embryo injected with control MOs at 2 dpf swims away rapidly (>2 cm/s) upon tactile stimulation.
Zebrafish embryo injected with zc4h2 MOs at 2 dpf exhibits slow swimming (<2 cm/s) after touch.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org/index.html
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Mouse Genome Informatics, http://www.informatics.jax.org/image/MGI:4505977
MutationTaster, http://www.mutationtaster.org
PolyPhen-2, www.genetics.bwh.harvard.edu/pph2/
Project HOPE, http://www.cmbi.ru.nl/hope/home
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Hoff J.M., Loane M., Gilhus N.E., Rasmussen S., Daltveit A.K. Arthrogryposis multiplexa congenita: an epidemiologic study of nearly 9 million births in 24 EUROCAT registers. Eur. J. Obstet. Gynecol. Reprod. Biol. 2011;159:347–350. doi: 10.1016/j.ejogrb.2011.09.027. [DOI] [PubMed] [Google Scholar]
- 2.Reimann J., Jacobson L., Vincent A., Kornblum C. Endplate destruction due to maternal antibodies in arthrogryposis multiplex congenita. Neurology. 2009;73:1806–1808. doi: 10.1212/WNL.0b013e3181c34a65. [DOI] [PubMed] [Google Scholar]
- 3.Tajsharghi H., Oldfors A. Myosinopathies: pathology and mechanisms. Acta Neuropathol. 2013;125:3–18. doi: 10.1007/s00401-012-1024-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bürglen L., Amiel J., Viollet L., Lefebvre S., Burlet P., Clermont O., Raclin V., Landrieu P., Verloes A., Munnich A., Melki J. Survival motor neuron gene deletion in the arthrogryposis multiplex congenita-spinal muscular atrophy association. J. Clin. Invest. 1996;98:1130–1132. doi: 10.1172/JCI118895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bamshad M., Van Heest A.E., Pleasure D. Arthrogryposis: a review and update. J. Bone Joint Surg. Am. 2009;91(Suppl 4):40–46. doi: 10.2106/JBJS.I.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boylan K.B., Ferriero D.M., Greco C.M., Sheldon R.A., Dew M. Congenital hypomyelination neuropathy with arthrogryposis multiplex congenita. Ann. Neurol. 1992;31:337–340. doi: 10.1002/ana.410310318. [DOI] [PubMed] [Google Scholar]
- 7.Steinlein O.K. Genetic disorders caused by mutated acetylcholine receptors. Life Sci. 2007;80:2186–2190. doi: 10.1016/j.lfs.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Nazir M.A., Dillon W.P., McPherson E.W. Myotonic dystrophy in pregnancy. Prenatal, neonatal and maternal considerations. J. Reprod. Med. 1984;29:168–172. [PubMed] [Google Scholar]
- 9.Wieacker P., Wolff G., Wienker T.F., Sauer M. A new X-linked syndrome with muscle atrophy, congenital contractures, and oculomotor apraxia. Am. J. Med. Genet. 1985;20:597–606. doi: 10.1002/ajmg.1320200405. [DOI] [PubMed] [Google Scholar]
- 10.Hennekam R.C., Barth P.G., Van Lookeren Campagne W., De Visser M., Dingemans K.P. A family with severe X-linked arthrogryposis. Eur. J. Pediatr. 1991;150:656–660. doi: 10.1007/BF02072628. [DOI] [PubMed] [Google Scholar]
- 11.Hall J.G., Reed S.D., Scott C.I., Rogers J.G., Jones K.L., Camarano A. Three distinct types of X-linked arthrogryposis seen in 6 families. Clin. Genet. 1982;21:81–97. doi: 10.1111/j.1399-0004.1982.tb00742.x. [DOI] [PubMed] [Google Scholar]
- 12.Ramser J., Ahearn M.E., Lenski C., Yariz K.O., Hellebrand H., von Rhein M., Clark R.D., Schmutzler R.K., Lichtner P., Hoffman E.P. Rare missense and synonymous variants in UBE1 are associated with X-linked infantile spinal muscular atrophy. Am. J. Hum. Genet. 2008;82:188–193. doi: 10.1016/j.ajhg.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Najmabadi H., Hu H., Garshasbi M., Zemojtel T., Abedini S.S., Chen W., Hosseini M., Behjati F., Haas S., Jamali P. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
- 14.Rauch A., Wieczorek D., Graf E., Wieland T., Endele S., Schwarzmayr T., Albrecht B., Bartholdi D., Beygo J., Di Donato N. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 15.de Ligt J., Willemsen M.H., van Bon B.W., Kleefstra T., Yntema H.G., Kroes T., Vulto-van Silfhout A.T., Koolen D.A., de Vries P., Gilissen C. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 16.Ropers H.H. Genetics of early onset cognitive impairment. Annu. Rev. Genomics Hum. Genet. 2010;11:161–187. doi: 10.1146/annurev-genom-082509-141640. [DOI] [PubMed] [Google Scholar]
- 17.Robinson P.N., Köhler S., Bauer S., Seelow D., Horn D., Mundlos S. The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet. 2008;83:610–615. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zanni G., Calì T., Kalscheuer V.M., Ottolini D., Barresi S., Lebrun N., Montecchi-Palazzi L., Hu H., Chelly J., Bertini E. Mutation of plasma membrane Ca2+ ATPase isoform 3 in a family with X-linked congenital cerebellar ataxia impairs Ca2+ homeostasis. Proc. Natl. Acad. Sci. USA. 2012;109:14514–14519. doi: 10.1073/pnas.1207488109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kruglyak L., Daly M.J., Reeve-Daly M.P., Lander E.S. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am. J. Hum. Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 20.Seelow D., Schwarz J.M., Schuelke M. GeneDistiller—distilling candidate genes from linkage intervals. PLoS ONE. 2008;3:e3874. doi: 10.1371/journal.pone.0003874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 22.Hu H., Wrogemann K., Kalscheuer V., Tzschach A., Richard H., Haas S.A., Menzel C., Bienek M., Froyen G., Raynaud M. Mutation screening in 86 known X-linked mental retardation genes by droplet-based multiplex PCR and massive parallel sequencing. Hugo J. 2009;3:41–49. doi: 10.1007/s11568-010-9137-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ballif B.C., Theisen A., McDonald-McGinn D.M., Zackai E.H., Hersh J.H., Bejjani B.A., Shaffer L.G. Identification of a previously unrecognized microdeletion syndrome of 16q11.2q12.2. Clin. Genet. 2008;74:469–475. doi: 10.1111/j.1399-0004.2008.01094.x. [DOI] [PubMed] [Google Scholar]
- 24.Duker A.L., Ballif B.C., Bawle E.V., Person R.E., Mahadevan S., Alliman S., Thompson R., Traylor R., Bejjani B.A., Shaffer L.G. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur. J. Hum. Genet. 2010;18:1196–1201. doi: 10.1038/ejhg.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marvizón J.C., Pérez O.A., Song B., Chen W., Bunnett N.W., Grady E.F., Todd A.J. Calcitonin receptor-like receptor and receptor activity modifying protein 1 in the rat dorsal horn: localization in glutamatergic presynaptic terminals containing opioids and adrenergic alpha2C receptors. Neuroscience. 2007;148:250–265. doi: 10.1016/j.neuroscience.2007.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogino K., Ramsden S.L., Keib N., Schwarz G., Harvey R.J., Hirata H. Duplicated gephyrin genes showing distinct tissue distribution and alternative splicing patterns mediate molybdenum cofactor biosynthesis, glycine receptor clustering, and escape behavior in zebrafish. J. Biol. Chem. 2011;286:806–817. doi: 10.1074/jbc.M110.125500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wieacker P., Wolff G., Wienker T.F. Close linkage of the Wieacker-Wolff syndrome to the DNA segment DXYS1 in proximal Xq. Am. J. Med. Genet. 1987;28:245–253. doi: 10.1002/ajmg.1320280137. [DOI] [PubMed] [Google Scholar]
- 28.Kloos D.U., Jakubiczka S., Wienker T., Wolff G., Wieacker P. Localization of the gene for Wieacker-Wolff syndrome in the pericentromeric region of the X chromosome. Hum. Genet. 1997;100:426–430. doi: 10.1007/s004390050528. [DOI] [PubMed] [Google Scholar]
- 29.Sanes J.R., Lichtman J.W. Development of the vertebrate neuromuscular junction. Annu. Rev. Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- 30.Germiller J.A., Lerner A.L., Pacifico R.J., Loder R.T., Hensinger R.N. Muscle and tendon size relationships in a paralyzed chick embryo model of clubfoot. J. Pediatr. Orthop. 1998;18:314–318. [PubMed] [Google Scholar]
- 31.Lombard Z., Park C., Makova K.D., Ramsay M. A computational approach to candidate gene prioritization for X-linked mental retardation using annotation-based binary filtering and motif-based linear discriminatory analysis. Biol. Direct. 2011;6:30. doi: 10.1186/1745-6150-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holman S.K., Morgan T., Baujat G., Cormier-Daire V., Cho T.J., Lees M., Samanich J., Tapon D., Hove H.D., Hing A. Osteopathia striata congenita with cranial sclerosis and intellectual disability due to contiguous gene deletions involving the WTX locus. Clin. Genet. 2013;83:251–256. doi: 10.1111/j.1399-0004.2012.01905.x. [DOI] [PubMed] [Google Scholar]
- 33.Venselaar H., Te Beek T.A., Kuipers R.K., Hekkelman M.L., Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics. 2010;11:548. doi: 10.1186/1471-2105-11-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gibson J.H., Williamson S.L., Arbuckle S., Christodoulou J. X chromosome inactivation patterns in brain in Rett syndrome: implications for the disease phenotype. Brain Dev. 2005;27:266–270. doi: 10.1016/j.braindev.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 35.Pellegrino M.W., Gasser R.B., Sprenger F., Stetak A., Hajnal A. The conserved zinc finger protein VAB-23 is an essential regulator of epidermal morphogenesis in Caenorhabditis elegans. Dev. Biol. 2009;336:84–93. doi: 10.1016/j.ydbio.2009.09.036. [DOI] [PubMed] [Google Scholar]
- 36.Pellegrino M.W., Farooqui S., Fröhli E., Rehrauer H., Kaeser-Pebernard S., Müller F., Gasser R.B., Hajnal A. LIN-39 and the EGFR/RAS/MAPK pathway regulate C. elegans vulval morphogenesis via the VAB-23 zinc finger protein. Development. 2011;138:4649–4660. doi: 10.1242/dev.071951. [DOI] [PubMed] [Google Scholar]
- 37.Pasterkamp R.J. Getting neural circuits into shape with semaphorins. Nat. Rev. Neurosci. 2012;13:605–618. doi: 10.1038/nrn3302. [DOI] [PubMed] [Google Scholar]
- 38.Dieterich K., Quijano-Roy S., Monnier N., Zhou J., Fauré J., Smirnow D.A., Carlier R., Laroche C., Marcorelles P., Mercier S. The neuronal endopeptidase ECEL1 is associated with a distinct form of recessive distal arthrogryposis. Hum. Mol. Genet. 2013;22:1483–1492. doi: 10.1093/hmg/dds514. [DOI] [PubMed] [Google Scholar]
- 39.McMillin M.J., Below J.E., Shively K.M., Beck A.E., Gildersleeve H.I., Pinner J., Gogola G.R., Hecht J.T., Grange D.K., Harris D.J., University of Washington Center for Mendelian Genomics Mutations in ECEL1 cause distal arthrogryposis type 5D. Am. J. Hum. Genet. 2013;92:150–156. doi: 10.1016/j.ajhg.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Zebrafish embryo injected with control MOs at 2 dpf swims away rapidly (>2 cm/s) upon tactile stimulation.
Zebrafish embryo injected with zc4h2 MOs at 2 dpf exhibits slow swimming (<2 cm/s) after touch.