

Abstract
Idiopathic pulmonary fibrosis (IPF) is a progressive fibrotic lung disease whose underlying molecular mechanisms are largely unknown. Herein, we show that focal adhesion kinase–related nonkinase (FRNK) plays a key role in limiting the development of lung fibrosis. Loss of FRNK function in vivo leads to increased lung fibrosis in an experimental mouse model. The increase in lung fibrosis is confirmed at the histological, biochemical, and physiological levels. Concordantly, loss of FRNK function results in increased fibroblast migration and myofibroblast differentiation and activation of signaling proteins that drive these phenotypes. FRNK-deficient murine lung fibroblasts also have an increased capacity to produce and contract matrix proteins. Restoration of FRNK expression in vivo and in vitro reverses these profibrotic phenotypes. These data demonstrate the multiple antifibrotic actions of FRNK. More important, FRNK expression is down-regulated in human IPF, and down-regulation of FRNK in normal human lung fibroblasts recapitulates the profibrotic phenotype seen in FRNK-deficient cells. The effect of loss and gain of FRNK in the experimental model, when taken together with its down-regulation in human IPF, suggests that FRNK acts as an endogenous negative regulator of lung fibrosis by repressing multiple profibrotic responses.
Normal tissue repair and remodeling are tightly controlled and self-limited processes. However, exuberant tissue repair and remodeling have been associated with progressive tissue fibrosis in multiple organs, such as the lung and the kidney.1–3 Idiopathic pulmonary fibrosis (IPF) is one such example, a fatal and progressive fibrotic disease of the lungs.1,4 Medical therapy is ineffective in halting the progressive fibrotic response in human lungs with IPF.5–7 A better understanding of the molecular mechanism(s) contributing to the inexorable fibroproliferative process in lungs with IPF will ultimately lead to the identification of novel molecular targets.
During tissue repair, fibroblasts are activated, migrate into the wounded area, proliferate, and transdifferentiate into a myofibroblastic phenotype.2,8,9 Furthermore, their transdiffererentiation into myofibroblasts engenders an enhanced ability to produce several profibrotic mediators, including cytokines and extracellular matrix (ECM) proteins (collagen and fibronectin).8,10,11 Fibroblastic foci of lung tissue with IPF are enriched with fibroblasts and myofibroblasts, and the extent of the foci is a major prognostic factor for patients with IPF,4,12 supporting the importance of the fibroblast in IPF pathogenesis.
Transforming growth factor (TGF)-β1 is the most potent profibrotic cytokine identified and is accepted as a central mediator of the fibrotic responses in lung, liver, and kidney.2,13–15 Bidirectional cross talk between integrins and ECM proteins is critical for myofibroblast differentiation and, in turn, for activation of latent TGF-β1.14,16–18 Integrin engagement with ECM proteins activates focal adhesion kinase (FAK) through phosphorylation of Tyr397 (Y397).19–21 Activated FAK promotes cell migration and invasion and mediates myofibroblast differentiation and resistance to apoptosis,19,22–24 suggesting a potential role for FAK in the profibrotic actions of fibroblasts in lung fibrosis.
FAK-related nonkinase (FRNK) is an independently expressed cytoplasmic protein that is identical in sequence to the C-terminal region of FAK.25,26 FRNK overexpression in vitro has been used as a tool to characterize FAK-mediated signaling events, because its overexpression inhibits integrin-mediated FAK activation and cell migration.26–28 However, the function of endogenous FRNK itself, especially the effect of loss of FRNK function in disease pathobiological characteristics, has been largely neglected. We have shown that FRNK expression is down-regulated in human lung fibroblasts derived from patients with IPF, in a manner that tightly correlates with their migration rate.29
Small Rho GTPases (eg, Rac and Rho) regulate cell migration through modulating lamellipodia formation.30,31 Published work supports the importance of small GTPases to fibrotic responses. For example, Rac and Rho activation is increased in IPF patient–derived lung fibroblasts, Rac1-deficient mice show resistance to bleomycin-induced skin fibrosis, and Rho regulates myofibroblast differentiation.29,32–34 Another cell migration regulatory protein, S100A4 (aliases metastasin, fibroblast-specific protein 1, mts1, and calvasculin), is a member of a calcium-binding family of proteins.35–37 S100A4 can induce cell-protrusive activity and metalloproteinase expression, both key events that lead to migration.35–37 It is increased in fibrotic lungs38 and plays a role in epithelial-to-mesenchymal transition, and in lung fibrosis.39 We undertook this study to determine the functional role of FRNK in profibrotic molecular signaling, and in the development of in vivo lung fibrosis. The results demonstrate that FRNK is a multifunctional negative regulator of lung fibrosis, through altering profibrotic signaling, cell migration, ECM protein expression, and myofibroblast differentiation and contraction.
Materials and Methods
Reagents
TGF-β1 was obtained from R&D Systems (Minneapolis, MN). The following antibodies were purchased: phospho-FAK (pY397; Biosource, Camarillo, CA), procollagen α 1 type 1 (1A1) and fibronectin (Santa Cruz Biotechnology, Santa Cruz, CA), FAK (N-terminal domain) (Santa Cruz Biotechnology), FRNK (recognizes both FRNK and FAK C-terminal; Upstate Biotechnology, Lake Placid, NY), F4/80 (eBioscience, San Diego, CA), α-smooth muscle actin (SMA; American Research Products, Belmont, MA), S100A4, Ki-67 (Dako, Carpentaria, CA), von Willebrand factor (Abcam, Cambridge, MA), phospho-Smad3 and Smad3 (Cell Signaling Technology, Boston, MA), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Research Diagnostics, Flanders, NJ). Quantikine enzyme-linked immunosorbent assay (ELISA) kits were obtained from R&D Systems. Chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and Fisher Scientific (Waltham, MA).
Animal Model of Lung Fibrosis
All animal interventions were approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham. FRNK knockout mice were generously provided by Dr. J. Thomas Parsons (University of Virginia, Charlottesville, VA) and were generated as previously described.40 FRNK deficiency in these animals was confirmed by both Northern and Western blot analyses.40 The FRNK knockout mice were congenic with C57BL/6 wild-type (WT) mice because they were backcrossed with C57BL/6 mice for at least 12 generations.22 There is no overt phenotype in the FRNK knockout mice under unchallenged conditions.22,40 The animal lung fibrosis model of bleomycin, the administration of bleomycin, and the administration of adenoviral vectors were all previously described.22,41 Briefly, 8- to 11-week-old female animals were challenged with 1 U/kg body weight bleomycin or saline using an intratracheal catheter. Saline, with or without recombinant adenoviral vectors (50 μL, 108 plaque forming units), was instilled intratracheally at day 7 after bleomycin (or saline) challenge, as previously described.42
Lung Collagen Determination
The whole lung collagen level was determined by the whole lung hydroxyproline level. The harvested lungs were hydrolyzed in 6 mol/L HCl at 110°C for 24 hours, and the amount of hydroxyproline in the lung acid–hydrolysates was determined by colorimetric assay, as previously described.43 Collagen deposition in 5- to 10-μm lung tissue sections (paraffin-embedded tissues) was localized by Masson’s trichrome staining using a commercially available staining kit, according to the manufacturer’s instructions (Poly Scientific, Bay Shore, NY).
Lesional Density and Cell Density Assays
The lungs were inflated to a fixed pressure of 25 cm H2O with 10% buffered formalin, fixed in 10% buffered formalin for 24 to 48 hours at room temperature, and embedded in paraffin. Fibrotic lesional density was measured on H&E-stained sections by morphometric methods. Lesional density was calculated as percentage of lesional volume/total lung volume. Cell density is represented by the ratio of the number of cells in lesion/lesional area measured on the digitized images of the H&E-stained lung tissue sections. The density of a specific cell type was determined as previously described on the tissue sections stained with cell type–specific antibodies.
Lung Physiological Function
Lung compliance measurements and mechanical ventilation were performed using the FlexiVent machine (SCIREQ, Montreal, QC, Canada), according to the manufacturer’s instructions, with minor modifications. Mice were anesthetized, their tracheas were cannulated under direct vision, and they were attached to a computer-controlled ventilator (FlexiVent) for forced oscillation measurements. After 5 to 10 minutes of adjustment to the ventilator, physiological measures of airway and lung tissue compliance and resistance were performed according to the manufacturer’s instructions, using accompanying software (SCIREQ version 5.1). In addition to these measures, we performed static inflation-deflation pressure volume loops, in which the slope of the deflation limb was used to measure compliance. The respiratory system was open during the lung compliance measurement.
Human Lung Tissue
The studies and protocols have obtained approvals from the local Institutional Review Board. De-identified explanted lung tissues from patients with IPF and normal human controls were provided by the airway tissue procurement program of the University of Alabama at Birmingham. The explanted lung tissues were used for histological and biochemical studies and for isolation of lung fibroblasts.
Cells and Cell Culture
Isolation and propagation of primary lung fibroblasts were previously described.29 Adult normal human lung fibroblasts were also purchased from ATCC (Manassas, VA). Fibroblasts were maintained and propagated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and antibiotics, as previously described. Experiments were performed on early passages (passages 2 to 9) of primary lung fibroblasts.
Generation and Use of Adenoviral Vectors
The adenoviral vectors containing the FRNK cDNA (Ad-FRNK) and green fluorescent protein (GFP) cDNA (Ad-GFP) were generated and used as previously described.22,29 Cells in serum-free media [DMEM with 1% bovine serum albumin (BSA)] were infected with Ad-FRNK or control vectors (Ad-GFP) before TGF-β1 treatment.
Whole Lung and Cell Protein Extracts
Whole lung or cell homogenates were prepared in 1% NP-40 lysis buffer with the following inhibitors: 100 μmol/L phenylmethanesulfonyl fluoride, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 100 μmol/L sodium vanadate, and 20 μg/mL TLCK using a polytron (Brinkmann Instruments, Westbury, NY). The resultant supernatants after centrifugation (14,000 × g for 20 minutes at 4°C) were analyzed by immunoblotting immediately or stored at −80°C until used.44
Analysis of BAL Fluid
To collect bronchoalveolar lavage (BAL) fluid, the trachea was cannulated and the lungs were lavaged three times using 0.8 mL of sterile PBS. The recovery of the total BAL fluid exceeded 90%. Fractions (600 g) were centrifuged for 10 minutes, and the supernatant from the first fraction was collected and kept at −80°C for ELISA assay of total TGF-β1 level, according to manufacturer’s instructions (ELISA kit from R&D Systems). The cell pellets from all fractions were pooled and counted using a hemocytometer. BAL cell numbers were enumerated, and cytospin preparations were prepared (cells spun onto glass slides, 150 × g for 6 minutes) (Shandon, Fisher Scientific). Cytospin preparations were then stained with Diff-Quick solution (Fisher Scientific). A total of 250 to 350 cells were counted randomly under a microscope, and cell type identification was performed based on standard criteria.
Western Blot Analysis and Rac and Rho Activation Assays
Immunoblotting was performed on 1% NP-40 whole lung tissue lysates or whole cell lysates, as previously described.29 Equivalent amounts (mg) of lysates were electrophoresed using disulfide-reduced 8% to 12% SDS-PAGE, transferred to an Immobilon-P membrane (Millipore Corp, Bedford, MA) for probing, and developed with the enhanced chemiluminescence system (Fisher Scientific). Rac and Rho activation levels were determined by the level of GTP-bound forms of Rac and Rho per the instructions (kits from Millipore, Billerica, MA), as previously described.29
Cell Migration Assay
The wound closure monolayer/scratch motility assay was performed as previously described.29 Briefly, fibroblasts were plated in serum-free DMEM with 1% BSA for 24 hours. Mitomycin c was added to inhibit cell proliferation. The monolayer was scratched, and the wound area covered by cell migration over the indicated time on digital photomicrographic images was calculated.
FRNK Down-Regulation
FRNK expression was down-regulated by using FRNK-specific siRNAs. The siRNA-related experimental procedures were performed as previously described.45 Targeting and control siRNA duplexes were synthesized by and purchased from Dharmacon (Fisher Scientific). The FRNK siRNA (sequence, 5′-AAAGCGAGACTTTGCTAGTTT-3′) specifically targeted the unique leader sequence of FRNK mRNA.
IHC Data
Immunohistochemistry (IHC) was performed as previously described.44 Briefly, frozen tissue sections were permeabilized using 0.6% Triton X-100, blocked, incubated overnight in primary antibody at 4°C, then reacted with a horseradish peroxidase–linked secondary antibody (Jackson ImmunoResearch, West Grove, PA) for 1 hour at room temperature, and developed using a 3,3′-diaminobenzidine substrate kit. Normal, nonimmune IgG primary antibody was used as a negative control.
Collagen Gel Contraction Assay
As previously described, collagen gels were cast in 6-well plates from type I collagen/DMEM solution composed of collagen type I (Sigma-Aldrich), DMEM containing HEPES and gentamicin, 0.142 mol/L NaOH, and PBS.46 A total of 100,000 lung fibroblasts per well were seeded into the collagen gel and incubated at 37°C in 5% CO2. A fibroblast-gel complex contraction was monitored by standardized photography over time. The ratio of collagen gel area before and after contraction was calculated. The data from individual experiments were pooled and presented as the percentage of contraction relative to the vehicle-treated group.
LCM Data
Laser-capture microdissection (LCM) was performed to capture lesional tissues from frozen lung tissue sections in an RNase-free environment. Fibrotic lesional tissues were LCM captured by using a Pix cell II LCM (Arcturus, Grand Island, NY) at the LCM core facility of the University of Alabama at Birmingham, as previously described,47 followed by total RNA extraction and assay by quantitative real-time RT-PCR.
Quantitative Real-Time RT-PCR Analysis
Quantitative real-time RT-PCR was performed as previously described.48,49 Briefly, total RNA was extracted from lung fibroblasts or lung tissues using the RNeasy Mini Kit (Qiagen, Valencia, CA) or from LCM-captured lung tissues using RNAqueous-Micro (Ambion, Austin, TX), according to the manufacturer’s instructions. The following primers were used: S100A4, 5′-TTGTGTCCACCTTCCACAAAA-3′ (sense) and 5′-GCTGTCCAAGTTGCTCATCA-3′ (antisense); FRNK, 5′-GTGGCCTGTCTTCTGGACTC-3′ (sense) and 5′-AGGACGAGGGTTTCAAACTG-3′ (antisense); mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-AACTTTGGCATTGTGGAAGG-3′ (sense) and 5′-ACACATTGGGGGTAGGAACA-3′ (antisense); and human GAPDH, 5′-GAGTCAACGGATTTGGTCGT-3′ (sense) and 5′-TTGATTTTGGAGGGATCTCG-3′ (antisense). Total RNA (1 to 3 μg) was reverse transcribed to cDNA with Maloney Murine Leukemia Virus Reverse Transcriptase (Promega, Madison, WI). Quantitative RT-PCR analysis was performed with the SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) using the Roche Light Cycler 480 (Madison, WI). Samples were assayed in triplicate, and the values were normalized to the relative amounts of GAPDH.
Statistical Analysis
Data were analyzed using the Student’s t-test analysis (Sigma Plot; SPSS Inc., San Jose, CA) for differences between two groups, and expressed as means ± SE. For comparisons between multiple groups, a three-way analysis of variance test was performed, followed by t-tests with Bonferroni correction using SAS version 9.3 (SAS Institute Inc., Cary, NC). All experiments were repeated at least three times. P < 0.05 was considered statistically significant.
Results
Loss of FRNK Function in Vivo Leads to Increased Lung Fibrosis
To determine the functional role of FRNK in the development of lung fibrosis, we determined the effect of genetic deletion of FRNK on fibrosis after bleomycin challenge in mice. By using several measures, the fibrotic response to bleomycin (21 days) was greatly increased in FRNK knockout mice, when compared with that in WT littermates (Figure 1). Morphometric analysis of lung tissue sections revealed an approximately threefold increase (22.3% ± 6.6% versus 7.7% ± 2.7%; P < 0.01, H&E-stained sections) in fibrotic lesional density (total lesional areas/total parenchymal area) (Figure 1, A and B) and a fourfold increase (4.4 ± 1.3-fold; P < 0.01) (Figure 1, C and D) in collagen area in the bleomycin-challenged FRNK knockout mice, when compared with that in WT mice. There was no difference in the low-level collagen staining observed in the lung parenchyma between FRNK knockout mice and WT littermate controls in the unchallenged state (data not shown).
Figure 1.

FRNK deficiency promotes lung fibrosis in response to bleomycin in mice. A: FRNK knockout (FRNK−/−) and WT littermate mice were intratracheally instilled with saline (Sal) or 1 U/kg bleomycin (Bleo). Lungs were harvested at day 21 and stained with H&E. Original magnification, ×200. B: The severity of fibrosis was examined morphometrically and represented by lesional density (% of lesional area over total parenchymal area, excluding airway and vessel). C: Masson’s trichrome staining for collagen deposition. Original magnification, ×400. D: The trichrome-positive area was quantified morphometrically. E: Hydroxyproline level was measured from non-challenged (None) or Sal- or Bleo-instilled mice, and represented as %hydroxyproline normalized to that in non-challenged WT mice. F: Lungs were harvested at day 14 after bleomycin instillation. Whole lung lysates underwent Western blot analysis for procollagen 1α1 (Pro-Col) or fibronectin (FN). GAPDH was used as a loading control. G and H: Densitometric analysis of Pro-Col and FN expression. I: Stepwise pressure-volume (PV) loops captured the mechanical properties [quasi-static compliance (CST)] of the lung at day 21 after challenge. Lung compliance (CST-PV) was represented as mL/cm H2O. Data were represented as means ± SE (n = 8 animals per group). ∗P < 0.01.
By using the quantifiable measure of hydroxyproline (a surrogate for collagen) from whole lung lysates, bleomycin-challenged FRNK knockout mice showed a 1.8-fold (337 μg versus 183 μg per lung; P < 0.01) greater collagen content, when compared with that in WT mice (Figure 1E). Whole lung fibronectin and procollagen-1 levels after bleomycin challenge were similarly greater (by 2.8- and 3.4-fold, respectively) in lysates from FRNK knockout mice, when compared with that in WT controls (Figure 1, F–H). No differences were noted between FRNK knockout mice and WT controls in unchallenged/basal (or saline-challenged) conditions, in any of the fibrosis measures (Figure 1, A–H).
The effect of loss of FRNK on lung fibrosis was also measured physiologically by measuring static compliance (stepwise pressure volume loop) (Figure 1I), using the FlexiVent apparatus. Lung compliance was significantly decreased (more impaired) in bleomycin-challenged FRNK knockout mice, when compared with that in bleomycin-challenged WT mice (Figure 1I) (0.055 versus 0.08 mL/cm H2O; P < 0.01). In contrast, there were no differences in lung physiological characteristics, between FRNK knockout mice and WT mice under both unchallenged and saline-challenged conditions (Figure 1I). There were no significant differences in the basal inflammatory response to bleomycin between genotypes, as measured by lung lavage total protein, total and differential cell counts, or total TGF-β1 levels (Supplemental Figure S1). Taken together, these data demonstrate that FRNK knockout mice exhibit a profibrotic phenotype at the histological, biochemical, and physiological levels.
Exogenous FRNK Expression Reduces Lung Fibrosis in Vivo
To examine the effect of gain of FRNK function on lung fibrosis, FRNK expression was restored in vivo in FRNK knockout mice using FRNK-expressing adenoviral vector (Ad-FRNK), administered during the early inflammation phase (at 7 days) to improve targeting to cells actively involved in the fibrotic response. Successful expression of FRNK in murine lungs was validated by measuring adenovirally expressed hemagglutinin-tagged FRNK in lung lysates (Figure 2C) and by a demonstration that lesional cells were targeted and were capable of expression of adenovirally encoded GFP protein after intratracheal administration (data not shown). The basal (without challenge) lung collagen level (by hydroxyproline) and lung compliance were not different among FRNK knockout and WT mice (Figure 2, A and B). Bleomycin challenge increased lung collagen and reduced lung compliance in both groups, but to a greater extent in the FRNK knockout mice compared with that in the WT mice (P < 0.001) (Figure 2, A and B). Restoration of FRNK expression in bleomycin-challenged FRNK knockout mice abrogated the increased lung fibrosis. This was demonstrated by the reduction in physiologically, histologically, and biochemically detectable fibrosis (hydroxyproline level, lung compliance, matrix protein expression, and H&E/trichrome staining) on FRNK restoration (Figure 2). Forced FRNK expression in WT mice similarly ameliorated the fibrosis on bleomycin challenge (Figure 2). As a control, administration of a GFP-expressing adenovirus had no effect on fibrosis (Figure 2). At the level of key signaling proteins, FRNK overexpression (in WT mice) or FRNK restoration (in FRNK knockout mice) reduced the bleomycin-induced increment in FAK activation (pY397-FAK) (Figure 2D), α-SMA expression (Figure 2D), and Rac and Rho activation (data not shown) after bleomycin challenge. Thus, both gain- and loss-of-function approaches in vivo demonstrate that FRNK functions to limit the development of lung fibrosis, potentially via altering the profibrotic signaling and cellular response.
Figure 2.

Exogenous FRNK expression reduces lung fibrosis. A: FRNK−/− and WT mice were challenged with bleomycin (Bleo) or control saline (Sal), as in Figure 1. Recombinant adenoviral vectors (50 μL, 108 plaque forming units) containing Ad-FRNK or control Ad-GFP were instilled intratracheally at day 7 after bleomycin or saline challenge. Lungs were harvested at day 21 after challenge. The whole lung hydroxyproline level was measured and presented as % of hydroxyproline level in Sal-challenged WT mice. B and E: Lung compliance [quasi-static compliance–pressure-volume (PV)] was measured by stepwise PV curves at day 21 after challenge (B). Lung tissue sections were H&E stained, and representative images are shown (E). Original magnification, ×200 (E). C, D, and F: Lungs were harvested at day 14 after bleomycin installation (D and F) or day 3 after adenoviral vector installation (C). Equivalent amounts of whole lung lysate underwent Western blot analysis (one lane per individual animal). Data were represented as means ± SE (n = 10 animals per group). HA, hemagglutinin. ∗P < 0.01.
Loss of FRNK Function in Vivo Leads to Enhanced Activation and Expression of Proteins That Mediate Cell Migration and Myofibroblast Differentiation
Published studies demonstrate that FRNK inhibits cell migration when initiated by integrins and growth factors.26 The expression and/or activation of the well-characterized signaling molecules involved in migration (eg, FAK, Rac, and Rho) were measured in lung lysates to identify a potential mechanism(s) for the increased fibrosis seen in FRNK knockout mice. Although there was an increase in the activation of FAK and the small GTPases (Rac and Rho) in both genotypes in response to bleomycin, the increase in the activation of FAK (6.4- versus 2.7-fold; P < 0.01) (Figure 3A) and the activation of Rac and Rho in fibrotic lungs from FRNK knockout mice were approximately twice that of WT controls (Figure 3A). Furthermore, the proportion of cells in fibrotic lesions with active FAK was greater (3.8-fold, P < 0.01) (Figure 3, B and C) in bleomycin-challenged FRNK knockout mice, compared with WT controls. Cells with active FAK were only rarely detected in lung tissue from unchallenged or saline-challenged mice (data not shown). We also noted more total cells per unit area of lesion in the bleomycin-challenged FRNK knockout mice (Figure 3D), suggesting that the active FAK, Rac, and Rho may mediate enhanced cell migration in vivo into the lesions during lung repair.
Figure 3.
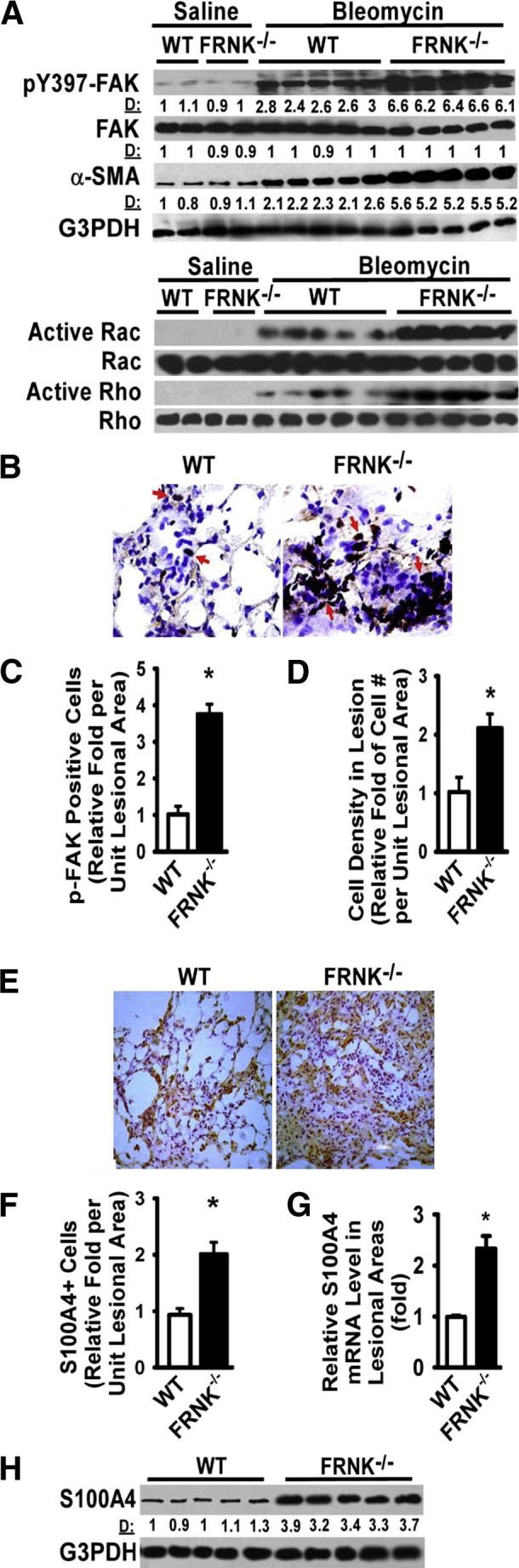
FRNK deficiency potentiates integrin/migration signaling and myofibroblast differentiation in fibrotic lungs. A: FRNK−/− and WT lungs were harvested at day 14 after bleomycin or saline instillation and lysed, and equivalent amounts of whole lung lysate were examined for indicated signaling and α-SMA expression. D: Relative band density by densitometry. B: Lesional cells containing active FAK (pY397-FAK) were detected by IHC on frozen lung tissue sections from WT and FRNK−/− mice. Arrows indicate pY397-FAK–positive cells. Original magnification, ×400. C: Lesional pY397-FAK–positive cells were quantified as the fold of pY397-FAK–positive cells in FRNK−/− mice relative to that in WT mice. D: The overall cell density in lesions was determined. E: S100A4-positive cells in fibrotic lesions were detected by IHC (brown). F: Lesional S100A4-positive cells were quantified as the fold of S100A4-positive cells in FRNK−/− mice relative to that in WT mice. G: Fibrotic/lesional tissues were captured by LCM from frozen lung sections. Total RNA was extracted, and the level of S100A4 mRNA was determined by quantitative real-time PCR (normalized to GAPDH) and represented as relative fold to that in WT mice. H: The S100A4 protein level in fibrotic lungs of WT and FRNK−/− mice was determined by using Western blot analysis. Data were represented as means ± SE (n = 8 animals per group). ∗P < 0.01.
In further support for FRNK’s role in modulating migration in vivo during fibrosis, fibrotic lungs from FRNK knockout mice exhibited more S100A4-expressing lesional cells, a higher S100A4 mRNA content in fibrotic lesions (by LCM and quantitative real-time PCR) (Figure 3, E–G), and a greater whole lung S100A4 protein content (Figure 3H), than that from fibrotic WT controls. S100A4 expression in saline-challenged or unchallenged mice from either genotype was not different (data not shown). Several cell types, such as fibroblasts, those derived from epithelial-mesenchymal transition, or a subpopulation of macrophages, have been reported to express S100A4 during fibrotic remodeling.50–52 We found no significant differences in percentages of fibrotic lesional cells that expressed a macrophage marker (F4/80) or an endothelial cell marker (von Willebrand factor) or in the proportion of proliferating cells (Ki-67) between genotypes (Supplemental Figure S1). Taken together, these data suggest that at least one mechanism of FRNK’s capacity to regulate in vivo fibrosis operates through altering cell migration, thereby increasing the cellular content of lesions.
Loss of FRNK Enhances Lung Fibroblast Responses to the Profibrotic Effects of TGF-β1
Because FRNK can regulate both cell migration and myofibroblast differentiation in fibroblasts,22,29 and TGF-β is a central profibrotic cytokine that mediates key profibrotic processes, we studied the functional consequences of impaired FRNK function on TGF-β1–driven, in vitro surrogates of fibrosis. Although basal cell motility did not differ between lung fibroblasts derived from FRNK-deficient and WT mice, loss of FRNK leads to an induction of the migratory response to TGF-β1 in FRNK-deficient fibroblasts when compared with WT fibroblasts (Figure 4A and Supplemental Figure S2). The enhanced migration seen on TGF-β1 exposure was abrogated on restoration (or forced expression) of FRNK with FRNK-expressing adenovirus (Ad-FRNK) (Figure 4A). The basal migration is also reduced on exogenous FRNK expression in both cell types (Figure 4A). FRNK-deficient fibroblasts also demonstrated enhanced responses to TGF-β1 in their activation of FAK (Figure 4B), Rac, and Rho (data not shown), and expression of α-SMA (as a measure of myofibroblast differentiation), and collagen and fibronectin protein, when compared with WT fibroblasts (Figure 4, B and C). There was no difference in basal (no TGF-β conditions) gel contraction between FRNK-deficient and WT fibroblasts (Figure 4D). FRNK-deficient fibroblasts were able to contract collagen gels to a greater extent, in response to TGF-β1, than WT fibroblasts (Figure 4D). Concordantly, forced FRNK expression (by Ad-FRNK) abrogated TGF-β1–induced gel contraction in both FRNK-deficient and WT fibroblasts (Figure 4D and Supplemental Figure S3). All these TGF-β1 responses were similarly abrogated with restoration/forced expression of FRNK, whereas adenovirally mediated expression of GFP had no effect (Figure 4, B–D, and Supplemental Figure S3). We have confirmed and extended our prior observations of FRNK’s role in limiting myofibroblast formation,22 by showing in this study that α-SMA protein content in whole lung lysates is approximately twofold greater in bleomycin-challenged FRNK knockout mice, when compared with that in bleomycin-challenged WT controls (Figure 3A). Smad3 has been shown to play an essential role in in vivo lung fibrosis induced by TGF-β1.53 In our study, FRNK deficiency increased, whereas adenovirally mediated FRNK expression reduced, the Smad3 phosphorylation noted in response to bleomycin challenge in mice, or in response to TGF-β1 in lung fibroblasts (Supplemental Figure S4). Taken together, these data demonstrate that FRNK deficiency enhances, and the restoration of FRNK reduces, multiple profibrotic TGF-β1–driven responses in fibroblasts. They suggest that FRNK acts as a brake on in vivo fibrosis signals initiated by TGF-β1.
Figure 4.

FRNK deficiency potentiates, and FRNK restoration abrogates, TGF-β1–induced fibrotic responses in murine lung fibroblasts. A: Murine lung fibroblasts were derived from FRNK−/− and WT mice, and infected with adenoviral vector expressing either FRNK (Ad-FRNK) or control GFP (Ad-GFP). Cell migration in response to 10 ng/mL TGF-β1 in serum-free medium with 1% BSA (SFM) was examined by wound closure assay. Data are pooled and shown as % of wound area covered by cells over 24 hours, relative to that of uninfected WT fibroblasts in SFM medium. Data are given as means ± SE. B and C: FRNK−/− and WT lung fibroblasts were infected with Ad-FRNK or control Ad-GFP, followed by 10 ng/mL TGF-β1 treatment (for 36 hours) or vehicle. Equivalent amounts of whole cell lysates underwent Western blot analysis with indicated antibodies. FN, fibronectin; Pro-Col, procollagen 1. GAPDH was used as the loading control. D: FRNK−/− and WT lung fibroblasts were infected with Ad-FRNK or control Ad-GFP, followed by 10 ng/mL TGF-β1 treatment or vehicle and subjected to the collagen gel contraction assays for 50 hours. The ratio of collagen gel area after contraction against the original collagen gel area (before contraction) was calculated. Data are presented as the percentage of gel area relative to control (vehicle only, set as 100%). ∗P < 0.01.
FRNK Is a Negative Regulator of Profibrotic Effects of TGF-β1 in Normal Human Lung Fibroblasts, and Its Function Is Down-Regulated in Human IPF
To extend the findings to humans and give this work direct disease relevance, the effect of loss or gain of FRNK function on TGF-β1 responses was first examined in normal human lung fibroblasts. As with the mouse, several profibrotic TGF-β1 responses were enhanced in normal human lung fibroblasts on FRNK down-regulation (by >80% using siRNA, data not shown). These include TGF-β1–induced cell migration (Figure 5A), activation of FAK and Rac, and myofibroblast differentiation (expression of α-SMA) (Figure 5B). Conversely, gain of FRNK expression (by Ad-FRNK) in normal human lung fibroblasts abrogated the TGF-β1–induced signal for cell migration (Figure 5A) and the TGF-β1–induced activation of FAK, Rac, and myofibroblast differentiation (expression of α-SMA) (Figure 5B). As a control, adenoviral expression of control GFP (by Ad-GFP) had no noticeable effect in normal human lung fibroblasts treated with or without TGF-β1 (Figure 5). In summary, normal, non-transformed, human lung fibroblasts respond similarly to their murine lung cell counterparts on manipulation of FRNK.
Figure 5.

Down-regulation of FRNK expression potentiates TGF-β1–driven fibrotic responses in normal human lung fibroblasts. A: Serum-starved primary normal human lung fibroblasts were infected with siRNA toward FRNK or nontargeting control siRNA. FRNK down-regulation by FRNK siRNAs was confirmed by using Western blot analysis (data not shown). Fibroblasts were treated with or without 10 ng/mL TGF-β1 and subjected to a wound closure cell migration assay in serum-free medium with 1% BSA (SFM) for 24 hours. Exogenous FRNK expression was mediated by Ad-FRNK, and Ad-GFP was used as a control. Data are plotted as the percentage of wound area covered over 24 hours relative to that in untreated cells in SFM. Data are given as means ± SE. ∗P < 0.01. B: Primary normal human lung fibroblasts were treated as described in A, and equivalent amounts of whole cell lysate were examined for activation of FAK and Rac, and for myofibroblast differentiation (by α-SMA).
To extend our findings directly to diseased human lung tissue, devoid of the potential for in vitro artifact, we revealed that FRNK is down-regulated in IPF lung tissue compared with that from normal controls. This was documented at the mRNA level from fibrotic lesions (decreased by 67% ± 8.4%; P < 0.01) and at the protein level in lung lysates (Figure 6, A and B). The reduction in FRNK protein was physiologically significant in that it was associated with an increase in FAK activation (approximately threefold increase) (Figure 6B), which localized to cells within fibrotic lesions of IPF lung tissues (Figure 6C). When taken together with the gain/loss functional data in the mouse model of lung fibrosis, the observations in human IPF (fibroblasts and tissues) support the concept that FRNK acts as a brake to limit fibrosis through blockade of multiple TGF-β–mediated, disease-relevant signals, and that physiological FRNK deficiency promotes fibrosis.
Figure 6.

FRNK expression is impaired and FAK activation is increased in human IPF lung tissues. A: Lesional tissues were captured by LCM from frozen IPF lung sections (as in Figure 3G). Total RNA was extracted from captured IPF lung tissues and normal human lung tissue controls (Normal). FRNK mRNA levels were examined by quantitative real-time PCR. Data are normalized to GAPDH and represented as % of FRNK mRNA level relative to that in Normals. Data are given as means ± SE (n = 13 individual subjects with IPF and 6 individual normal human subjects). ∗P < 0.01. B: IPF and normal human lung tissues were lysed and underwent Western blot analysis with the indicated antibodies. C: Active FAK (pY397 of FAK) was examined in frozen normal (left panel) and IPF (middle panel) lung tissue sections IHC. Original magnification, ×400. Arrows indicate cells with FAK activation (middle panel). Right panel: Results from control IgG (cIgG) on IPF lung sections.
Discussion
The key findings herein are that FRNK deficiency potentiates, and FRNK expression abrogates, the development of lung fibrosis, through modulating TGF-β–driven, fibroblast profibrotic responses. These findings were substantiated in vivo through the increase in lung fibrosis measured at the histological, biochemical, and physiological levels in bleomycin-challenged FRNK knockout mice. Conversely, lung fibrosis was reduced on gain of exogenous FRNK expression in both FRNK knockout mice and WT mice. The data indicate that FRNK exerts its antifibrotic actions, in part, through blocking myofibroblast differentiation, matrix protein synthesis, and fibroblast migration, all TGF-β–driven responses. To our knowledge, this is the first time that FRNK has been directly implicated in the regulation of fibrosis in vivo, and provides evidence for the mechanism(s) of antifibrotic actions of FRNK.
TGF-β is a pleiotropic cytokine central to fibrogenesis in the lung. Reports clearly show that inhibition of TGF-β by antibodies, or inhibition of its activation, deletion of its receptor, or blockade of Smad signaling, attenuates experimental pulmonary fibrosis.53–56 The potent and consistent profibrotic actions of TGF-β on fibroblasts include induction of myofibroblast differentiation, matrix synthesis, and up-regulation of matrix protease inhibitors, whereas its effects on fibroblast migration and proliferation are context dependent.2,57 This study, combined with our prior in vitro work,22,29,49,58 reveals that impaired FRNK function amplifies the TGF-β signals, thereby allowing unbridled cell migration, matrix synthesis, and myofibroblast differentiation.
Ample evidence in cultured cells documents that FRNK inhibits cell migration when initiated by integrin activation and/or induced by growth factors, through its focal adhesion targeting domain.19 However, little is known regarding FRNK’s function in vivo. FRNK knockout mice develop normally, and have no detectable histological or physiological pulmonary abnormalities when unchallenged,40 suggesting that FRNK is not essential for normal lung development or homeostasis. Limited prior work has shed some light on the role of FRNK in tissue repair in vivo. These studies reveal that FRNK is up-regulated after balloon-induced vascular injury and mediates the vascular smooth muscle cell contractile phenotype in vivo, and that forced expression of FRNK blocks in vitro smooth muscle cell migration and proliferation.26,59 However, no prior work demonstrates any role for FRNK in the actual fibrotic response to tissue injury.
Recent work has highlighted the significance of cell migration/invasion in the setting of lung injury/fibrosis that occurs in response to mediators, including lysophosphatidic acid or hyaluronan.58,60–63 The intracellular signaling pathways that govern cell migration are complex, and exhibit dynamic and reciprocal cross talk and positive/negative feedback loops and are context dependent.64–66 In most tested systems, FAK and the small GTPases (Rac and Rho) promote cell migration through modulating both focal adhesion turnover and the cytoskeleton reorganization necessary to generate the forces required for migration.19,66–68 Thus, their activation can be conceived as both a marker and an effector of cell migration. For example, inhibition of Rac blocks several fibrotic responses in scleroderma fibroblasts, and Rac1-deficient mice show resistance to bleomycin-induced skin fibrosis and inflammation.32,69 Rac1 may modulate asbestos-induced lung fibrosis through mitochondrial electron transfer.70 We demonstrate increased activation of FAK in fibrotic lesional cells in murine lung fibrosis, human IPF, and IPF fibroblasts.29 We also show enhanced activation of the small GTPases (Rac and Rho) in fibrotic lungs (Figure 3). There was no detectable effect of FRNK deficiency on inflammation and TGF-β1 expression in vivo after bleomycin instillation (Supplemental Figure S1). These data suggest that the pathophysiological effects of FRNK occur predominantly during the repair/fibrotic phase in a manner that is independent of inflammation or of TGF-β1 expression. Taken along with the in vitro data, it builds a strong case that one putative mechanism by which FRNK functions to limit exuberant fibrotic repair in vivo is through limiting FAK (and/or GTPase)-dependent cell migration.
Myofibroblasts have been implicated in the development of pathological fibrosis in multiple organs.2,8,10,11 We demonstrate that FRNK deficiency promotes myofibroblast differentiation in vivo and in vitro. These data confirm and extend our previous observation that α-SMA–expressing cells are increased in fibrotic lungs of FRNK knockout mice and that FRNK overexpression inhibits TGF-β1–induced α-SMA–containing cytoplasmic fiber formation in lung fibroblasts.22 Myofibroblast differentiation requires a signal from active TGF-β1, along with an integrin, to transduce matrix-derived stiffness and is marked by reorganization of focal adhesions and cytoskeletal structures.8 Although several distinct FRNK-dependent events can be discerned, we have shown that FRNK blocks FAK-extracellular signal-regulated kinase and p38 pathways in lung fibroblasts, and thereby inhibits myofibroblast differentiation.22
FRNK deficiency also increases the effect of TGF-β1 on matrix protein synthesis, in both murine and human lung fibroblasts. Thus, we speculate that the increased matrix protein production conspires with other enhanced TGF-β responses to increase the lung fibrosis seen in FRNK-deficient mice. The dysregulated FRNK expression noted in fibrotic lesional cells may similarly contribute to IPF pathogenesis, and contributes to the increased FAK activation in IPF.29,71 Recent studies support that FAK is a key signaling protein promoting TGF-β–induced myofibroblast differentiation,22,72,73 and is required for endothelin-1–mediated and JNK-mediated profibrotic signaling.71,74 FAK activation is increased in IPF and scleroderma,29,71,75 and, more important, it has been demonstrated that FAK inhibitors block bleomycin-induced lung fibrosis in mice.71 We introduced the concept that FRNK regulates lung fibrosis by acting as a brake, at least on FAK-mediated signaling, to ongoing fibrosis (Figure 7). We further suggest that FRNK deficiency promotes lung fibrosis, at least in part, because of the loss of its ability to halt the inexorable profibrotic effects of TGF-β1.
Figure 7.
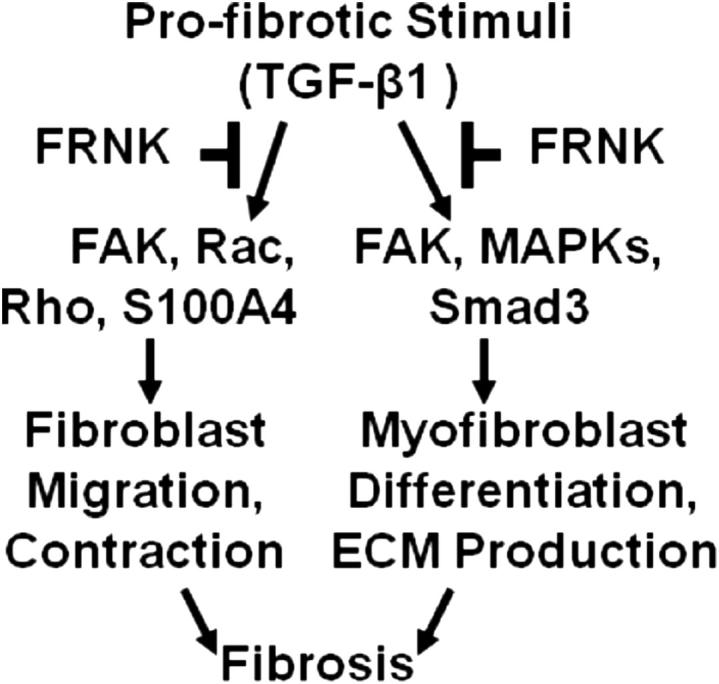
Model of mechanism of the antifibrotic function of FRNK. Injury or profibrotic stimuli, such as TGF-β1, activate fibroblasts, induce integrin-mediated and growth factor–initiated signaling, such as through FAK, Rac, Rho, and S100A4, promote cell migration and recruitment of cells to sites of injury, and promote cell contraction and fibrotic responses. These profibrotic signals also promote myofibroblast differentiation and ECM production, partly through FAK and mitogen-activated protein kinases (MAPKs; eg, ERK and p38). FRNK functions to limit these responses, including cell migration, myofibroblast differentiation, and ECM production. Under conditions of impaired FRNK function, as in down-regulated FRNK in IPF, these profibrotic responses are amplified, resulting in the development of an exuberant fibrotic process and expansion of fibrotic lesions.
Several important questions remain to be addressed by future experiments. For example, whether impaired FRNK modulates other profibrotic pathways (ie, epithelial cell injury/survival, expression and activation of matrix metalloproteinases and tissue inhibitors of metalloproteinases, TGF-β activation, and fibrocyte recruitment) should be determined.76–79 Although FRNK deficiency induces an increase in S100A4-expressing cells in fibrotic murine lungs (but not macrophages or endothelial cells), perhaps marking a motile cell phenotype, conclusive identification of the S100A4-expresing cell lineage(s) remains to be performed. Furthermore, S100A4 arguably may mark cells that undergo other profibrotic processes (ie, epithelial-to-mesenchymal transition, angiogenesis, and endoplasmic reticulum stress).39,55,76,80–84 Cell migration is generally necessary for the tissue repair process. Because FRNK expression inhibits basal cell migration (Figure 4), further studies are needed to understand the role of cell migration during different stages of lung injury and repair.
It is only through a complete understanding of the molecular mechanisms leading to persistent fibrotic response that we will be able to design strategies to mitigate persistent organ fibrosis. Our studies, for the first time to our knowledge, demonstrate that FRNK deficiency amplifies the fibrotic response, and that FRNK acts as a physiological brake to limit the extent of tissue remodeling through inhibiting multiple TGF-β–driven processes. Thus, FRNK and the FRNK-FAK pathway represent new therapeutic targets that may limit the extent of pathological fibrosis in human IPF. Small-molecule FAK inhibitors are being used in human clinical trials for cancer (http://www.clinicaltrials.gov); therefore, their use in fibrotic diseases might be feasible.
Acknowledgments
We thank Dr. Steven M. Rowe (University of Alabama at Birmingham Airway Tissue Procurement Program) for supplying the human lung tissues, Dr. Andra R. Frost for technical assistance of LCM and cell differential studies, and members of our laboratory for technical help and stimulating discussion.
Footnotes
Supported by NIH National Heart, Lung, and Blood Institute grants HL085324 and HL095451 (Q.D.); grants HL-58655, HL-103553, and Veterans Affairs (VA) MERIT Award (M.A.O.); grants CA127620 and CA109748 (C.L.G.); and a postdoctoral fellowship from the American Heart Association, Greater Southeast Affiliate (G.C.).
A guest editor acted as the editor-in-chief for this manuscript. No one at the University of Alabama at Birmingham was involved in the peer review process or final disposition of this article.
Contributor Information
Qiang Ding, Email: qding@uab.edu.
Mitchell A. Olman, Email: olmanm@ccf.org.
Supplemental Data
FRNK-knockout and WT mice have a similar early inflammatory response, total TGF-β1 level, cell proliferation rates, and percentage of lesional macrophages and endothelial cells in response to bleomycin. FRNK-knockout (FRNK−/−) and WT mice were intratracheally instilled with saline (Sal) or 1 U/kg body weight bleomycin (Bleo). BAL fluids were collected at day 7 after either saline or bleomycin challenge. A: BAL total protein level was determined by colorimetric assay (BCA kit; Pierce, Rockford, IL). B: BAL cell differential was determined on Diff-Quick–stained cytospins by standard techniques (Fisher Scientific). Lymphs, lymphocytes; MAC, macrophages; PMNs, polymorphonuclear neutrophils. C: BAL total cell number was counted in a hemocytometer. D: BAL total TGF-β1 level was measured by ELISA. All assays were performed according to manufacturer’s instructions. E: Frozen lung tissue sections (5 to 8 μm) from 1-U/kg bleomycin-instilled (day 14 after bleomycin) FRNK−/− and WT mice were IHC stained by antibodies specific toward Ki-67 (as a proliferation cellular marker), F4/80 (as a macrophage cellular marker), or von Willebrand factor (VWF; as an endothelial cellular marker) (data not shown). Positively stained cells were counted, and data are quantified as percentage of positively stained cells of total lesional cells. Data were pooled and presented as means ± SE (n = 8 to 10 animals per group).
FRNK deficiency potentiates, and FRNK restoration abrogates, TGF-β1–induced fibrotic responses in murine lung fibroblasts. A:FRNK−/− and WT lung fibroblasts were infected with Ad-FRNK or control Ad-GFP, followed by 10 ng/mL TGF-β1 treatment, for 36 hours, or vehicle. Equivalent amounts of whole cell lysates underwent Western blot analysis with antibodies directed toward the hemagglutinin tag of FRNK and GFP. GAPDH was used as the loading control. B: Murine lung fibroblasts were derived from FRNK−/− and WT mice, and infected with adenoviral vectors expressing either FRNK (Ad-FRNK) or control GFP (Ad-GFP). Cell migration in response to 10 ng/mL TGF-β1 in serum-free medium with 1% BSA (SFM) was examined by wound closure assay for 24 hours. Representative images are shown.
FRNK deficiency potentiates, and FRNK restoration abrogates, TGF-β1–induced collagen gel contraction in murine lung fibroblasts. Murine lung fibroblasts were derived from FRNK−/− and WT mice, and infected with adenoviral vector expressing either FRNK (Ad-FRNK) or control GFP (Ad-GFP). Cells were treated with 10 ng/mL TGF-β1 or vehicle, followed by the collagen gel contraction assays for 50 hours at 37°C and 5% CO2, as described in Materials and Methods. Representative digital images are shown.
FRNK deficiency promotes Smad3 phosphorylation in bleomycin-challenged mice and TGF-β1–treated lung fibroblasts. A: Mice were challenged with bleomycin and treated with or without adenoviral vectors, as described in the legend to Figure 2. Lungs were harvested at day 14, and equivalent amounts of whole lung lysate underwent Western blot analysis (one lane per individual animal). B:FRNK−/− and WT lung fibroblasts were infected with Ad-FRNK or control Ad-GFP, followed by 10 ng/mL TGF-β1 treatment or vehicle. Equivalent amounts of whole cell lysates underwent Western blot analysis with the indicated antibodies.
References
- 1.American Thoracic Society (ATS), European Respiratory Society (ERS) American Thoracic Society: idiopathic pulmonary fibrosis: diagnosis and treatment: international consensus statement. Am J Respir Crit Care Med. 2000;161(Pt 1):646–664. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- 2.Chapman H.A. Disorders of lung matrix remodeling. J Clin Invest. 2004;113:148–157. doi: 10.1172/JCI20729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hewitson T.D. Renal tubulointerstitial fibrosis: common but never simple. Am J Physiol Renal Physiol. 2009;296:F1239–F1244. doi: 10.1152/ajprenal.90521.2008. [DOI] [PubMed] [Google Scholar]
- 4.King T.E., Jr., Tooze J.A., Schwarz M.I., Brown K.R., Cheniak R.M. Predicting survival in idiopathic pulmonary fibrosis: scoring system and survival model. Am J Respir Crit Care Med. 2001;164:1171–1181. doi: 10.1164/ajrccm.164.7.2003140. [DOI] [PubMed] [Google Scholar]
- 5.Ley B., Collard H.R., King T.E., Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183:431–440. doi: 10.1164/rccm.201006-0894CI. [DOI] [PubMed] [Google Scholar]
- 6.Ding Q., Luckhardt T., Hecker L., Zhou Y., Liu G., Antony V.B., de Andrade J., Thannickal V.J. New insights into the pathogenesis and treatment of idiopathic pulmonary fibrosis. Drugs. 2011;71:981–1001. doi: 10.2165/11591490-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noth I., Anstrom K.J., Calvert S.B., de Andrade J., Flaherty K.R., Glazer C., Kaner R.J., Olman M.A., Idiopathic Pulmonary Fibrosis Clinical Research Network (IPFnet) A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186:88–95. doi: 10.1164/rccm.201202-0314OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hinz B., Phan S.H., Thannickal V.J., Galli A., Bochaton-Piallat M.L., Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Desmouliere A., Gabbiani G. Myofibroblast differentiation during fibrosis. Exp Nephrol. 1995;3:134–139. [PubMed] [Google Scholar]
- 10.Olman M.A. Mechanisms of fibroproliferation in acute lung injury. In: Matthay M.A., Lenfant C., editors. Acute Respiratory Distress Syndrome. Marcel Dekker; New York: 2003. pp. 313–354. [Google Scholar]
- 11.Phan S.H. The myofibroblast in pulmonary fibrosis. Chest. 2002;122:286S–289S. doi: 10.1378/chest.122.6_suppl.286s. [DOI] [PubMed] [Google Scholar]
- 12.King T.E., Jr., Schwarz M.I., Brown K., Tooze J.A., Colby T.V., Waldron J.A., Jr., Flint A., Thurlbeck W., Chemiack R.M. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med. 2001;164:1025–1032. doi: 10.1164/ajrccm.164.6.2001056. [DOI] [PubMed] [Google Scholar]
- 13.Desmouliere A., Darby I., Gabbiani G. Normal and pathologic soft tissue remodeling: role of the myofibroblast, with special emphasis on liver and kidney fibrosis. Lab Invest. 2003;83:1689–1707. doi: 10.1097/01.lab.0000101911.53973.90. [DOI] [PubMed] [Google Scholar]
- 14.Jenkins R.G., Su X., Su G., Scotton C.J., Camerer E., Laurent G.J., Davis G.E., Chambers R.C., Matthay M.A., Sheppard D. Ligation of protease-activated receptor 1 enhances alpha(v)beta6 integrin-dependent TGF-beta activation and promotes acute lung injury. J Clin Invest. 2006;116:1606–1614. doi: 10.1172/JCI27183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X., Hu H., Yin J.Q. Therapeutic strategies against TGF-beta signaling pathway in hepatic fibrosis. Liver Int. 2006;26:8–22. doi: 10.1111/j.1478-3231.2005.01192.x. [DOI] [PubMed] [Google Scholar]
- 16.Wipff P.J., Rifkin D.B., Meister J.J., Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. 2007;179:1311–1323. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muro A.F., Moretti F.A., Moore B.B., Yan M., Atrasz R.G., Wilke C.A., Flaherty K.R., Martinez F.J., Tsui J.L., Sheppard D., Baralle F.E., Toews G.B., White E.S. An essential role for fibronectin extra type III domain A in pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177:638–645. doi: 10.1164/rccm.200708-1291OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hagood J.S., Olman M.A. Muscle fatigue: MK2 signaling and myofibroblast differentiation. Am J Respir Cell Mol Biol. 2007;37:503–506. doi: 10.1165/rcmb.2007-0005ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parsons J.T. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- 20.Cary L.A., Chang J.F., Guan J.L. Stimulation of cell migration by overexpression of focal adhesion kinase and its association with Src and Fyn. J Cell Sci. 1996;109(Pt 7):1787–1794. doi: 10.1242/jcs.109.7.1787. [DOI] [PubMed] [Google Scholar]
- 21.Hauck C.R., Hsia D.A., Schlaepfer D.D. The focal adhesion kinase: a regulator of cell migration and invasion. IUBMB Life. 2002;53:115–119. doi: 10.1080/15216540211470. [DOI] [PubMed] [Google Scholar]
- 22.Ding Q., Gladson C.L., Wu H., Hayasaka H., Olman M.A. FAK-related non-kinase inhibits myofibroblast differentiation through differential MAPK activation in a FAK-dependent manner. J Biol Chem. 2008;283:26839–26849. doi: 10.1074/jbc.M803645200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horowitz J.C., Rogers D.S., Sharma V., Vittal R., White E.S., Cui Z., Thannickal V.J. Combinatorial activation of FAK and AKT by transforming growth factor-beta1 confers an anoikis-resistant phenotype to myofibroblasts. Cell Signal. 2007;19:761–771. doi: 10.1016/j.cellsig.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horowitz J.C., Ajayi I.O., Kulasekaran P., Rogers D.S., White J.B., Townsend S.K., White E.S., Nho R.S., Higgins P.D., Huang S.K., Sisson T.H. Survivin expression induced by endothelin-1 promotes myofibroblast resistance to apoptosis. Int J Biochem Cell Biol. 2012;44:158–169. doi: 10.1016/j.biocel.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schaller M.D., Borgman C.A., Parsons J.T. Autonomous expression of a noncatalytic domain of the focal adhesion-associated protein tyrosine kinase pp125FAK. Mol Cell Biol. 1993;13:785–791. doi: 10.1128/mcb.13.2.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor J.M., Mack C.P., Nolan K., Regan C.P., Owens G.K., Parsons J.T. Selective expression of an endogenous inhibitor of FAK regulates proliferation and migration of vascular smooth muscle cells. Mol Cell Biol. 2001;21:1565–1572. doi: 10.1128/MCB.21.5.1565-1572.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayasaka H., Simon K., Hershey E.D., Masumoto K.H., Parsons J.T. FRNK, the autonomously expressed C-terminal region of focal adhesion kinase, is uniquely regulated in vascular smooth muscle: analysis of expression in transgenic mice. J Cell Biochem. 2005;95:1248–1263. doi: 10.1002/jcb.20501. [DOI] [PubMed] [Google Scholar]
- 28.Hauck C.R., Hsia D.A., Puente X.S., Cheresh D.A., Schlaepfer D.D. FRNK blocks v-Src-stimulated invasion and experimental metastases without effects on cell motility or growth. EMBO J. 2002;21:6289–6302. doi: 10.1093/emboj/cdf631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cai G.Q., Zheng A., Tang Q., White E.S., Chou C.F., Gladson C.L., Olman M.A., Ding Q. Downregulation of FAK-related non-kinase mediates the migratory phenotype of human fibrotic lung fibroblasts. Exp Cell Res. 2010;316:1600–1609. doi: 10.1016/j.yexcr.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hildebrand J.D., Taylor J.M., Parsons J.T. An SH3 domain-containing GTPase-activating protein for Rho and Cdc42 associates with focal adhesion kinase. Mol Cell Biol. 1996;16:3169–3178. doi: 10.1128/mcb.16.6.3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iwanicki M.P., Vomastek T., Tilghman R.W., Martin K.H., Banerjee J., Wedegaertner P.B., Parsons J.T. FAK, PDZ-RhoGEF and ROCKII cooperate to regulate adhesion movement and trailing-edge retraction in fibroblasts. J Cell Sci. 2008;121(Pt 6):895–905. doi: 10.1242/jcs.020941. [DOI] [PubMed] [Google Scholar]
- 32.Liu S., Kapoor M., Shi-Wen X., Kennedy L., Denton C.P., Glogauer M., Abraham D.J., Leask A. Role of Rac1 in a bleomycin-induced scleroderma model using fibroblast-specific Rac1-knockout mice. Arthritis Rheum. 2008;58:2189–2195. doi: 10.1002/art.23595. [DOI] [PubMed] [Google Scholar]
- 33.Huang X., Yang N., Fiore V.F., Barker T.H., Sun Y., Morris S.W., Ding Q., Thannickal V.J., Zhou Y. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am J Respir Cell Mol Biol. 2012;47:340–348. doi: 10.1165/rcmb.2012-0050OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akhmetshina A., Dees C., Pileckyte M., Szucs G., Spriewald B.M., Zwerina J., Distler O., Schett G., Distler J.H. Rho-associated kinases are crucial for myofibroblast differentiation and production of extracellular matrix in scleroderma fibroblasts. Arthritis Rheum. 2008;58:2553–2564. doi: 10.1002/art.23677. [DOI] [PubMed] [Google Scholar]
- 35.Boye K., Maelandsmo G.M. S100A4 and metastasis: a small actor playing many roles. Am J Pathol. 2010;176:528–535. doi: 10.2353/ajpath.2010.090526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garrett S.C., Varney K.M., Weber D.J., Bresnick A.R. S100A4, a mediator of metastasis. J Biol Chem. 2006;281:677–680. doi: 10.1074/jbc.R500017200. [DOI] [PubMed] [Google Scholar]
- 37.Ai K.X., Lu L.Y., Huang X.Y., Chen W., Zhang H.Z. Prognostic significance of S100A4 and vascular endothelial growth factor expression in pancreatic cancer. World J Gastroenterol. 2008;14:1931–1935. doi: 10.3748/wjg.14.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lawson W.E., Polosukhin V.V., Zoia O., Stathopoulos G.T., Han W., Plieth D., Loyd J.E., Neilson E.G., Blackwell T.S. Characterization of fibroblast-specific protein 1 in pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171:899–907. doi: 10.1164/rccm.200311-1535OC. [DOI] [PubMed] [Google Scholar]
- 39.Degryse A.L., Tanjore H., Xu X.C., Polosukhin V.V., Jones B.R., Boomershine C.S., Ortiz C., Sherrill T.P., McMahon F.B., Gleaves L.A., Blackwell T.S., Lawson W.E. TGF{beta} signaling in lung epithelium regulates bleomycin-induced alveolar injury and fibroblast recruitment. Am J Physiol Lung Cell Mol Physiol. 2011;300:L887–L897. doi: 10.1152/ajplung.00397.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hayasaka H., Martin K.H., Hershey E.D., Parsons J.T. Disruption of FRNK expression by gene targeting of the intronic promoter within the focal adhesion kinase gene. J Cell Biochem. 2007;102:947–954. doi: 10.1002/jcb.21329. [DOI] [PubMed] [Google Scholar]
- 41.Olman M.A., Mackman N., Gladson C., Moser K., Loskutoff D. Changes in procoagulant and fibrinolytic gene expression during bleomycin induced lung injury in the mouse. J Clin Invest. 1995;96:1621–1630. doi: 10.1172/JCI118201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simmons W.L., Rivera K.E., Curiel D.T., Williams W.F., Olman M.A. Adenovirally mediated gene transfer of functional human tissue-type plasminogen activator to murine lungs. Am J Respir Cell Mol Biol. 1998;18:307–314. doi: 10.1165/ajrcmb.18.3.2892. [DOI] [PubMed] [Google Scholar]
- 43.Berg R.A. Determination of 3- and 4- hydroxyproline. Methods Enzymol. 1982;82(Pt A):372–398. doi: 10.1016/0076-6879(82)82074-0. [DOI] [PubMed] [Google Scholar]
- 44.Ding Q., Grammer J.R., Nelson M.A., Guan J.L., Stewart J.E., Jr., Gladson C.L. p27Kip1 and cyclin D1 are necessary for focal adhesion kinase regulation of cell cycle progression in glioblastoma cells propagated in vitro and in vivo in the scid mouse brain. J Biol Chem. 2005;280:6802–6815. doi: 10.1074/jbc.M409180200. [DOI] [PubMed] [Google Scholar]
- 45.Ding Q., Stewart J., Jr., Olman M.A., Klobe M.R., Gladson C.L. The pattern of enhancement of Src kinase activity on platelet-derived growth factor stimulation of glioblastoma cells is affected by the integrin engaged. J Biol Chem. 2003;278:39882–39891. doi: 10.1074/jbc.M304685200. [DOI] [PubMed] [Google Scholar]
- 46.Cai G.Q., Chou C.F., Hu M., Zheng A., Reichardt L.F., Guan J.L., Fang H., Luckhardt T.R., Zhou Y., Thannickal V.J., Ding Q. Neuronal Wiskott-Aldrich syndrome protein (N-WASP) is critical for formation of alpha-smooth muscle actin filaments during myofibroblast differentiation. Am J Physiol Lung Cell Mol Physiol. 2012;303:L692–L702. doi: 10.1152/ajplung.00390.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eltoum I.A., Siegal G.P., Frost A.R. Microdissection of histologic sections: past, present, and future. Adv Anat Pathol. 2002;9:316–322. doi: 10.1097/00125480-200209000-00006. [DOI] [PubMed] [Google Scholar]
- 48.Zhan S., Cai G.Q., Zheng A., Wang Y., Jia J., Fang H., Yang Y., Hu M., Ding Q. Tumor necrosis factor-alpha regulates the Hypocretin system via mRNA degradation and ubiquitination. Biochim Biophys Acta. 2011;1812:565–571. doi: 10.1016/j.bbadis.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.White K.E., Ding Q., Moore B.B., Peters-Golden M., Ware L.B., Matthay M.A., Olman M.A. Prostaglandin E2 mediates IL-1beta-related fibroblast mitogenic effects in acute lung injury through differential utilization of prostanoid receptors. J Immunol. 2008;180:637–646. doi: 10.4049/jimmunol.180.1.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanjore H., Cheng D.S., Degryse A.L., Zoz D.F., Abdolrasulnia R., Lawson W.E., Blackwell T.S. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem. 2011;286:30972–30980. doi: 10.1074/jbc.M110.181164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rock J.R., Barkauskas C.E., Cronce M.J., Xue Y., Harris J.R., Liang J., Noble P.W., Hogan B.L. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 2011;108:E1475–E1483. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Osterreicher C.H., Penz-Osterreicher M., Grivennikov S.I., Guma M., Koltsova E.K., Datz C., Sasik R., Hardiman G., Karin M., Brenner D.A. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc Natl Acad Sci U S A. 2011;108:308–313. doi: 10.1073/pnas.1017547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bonniaud P., Kolb M., Galt T., Robertson J., Robbins C., Stampfli M., Lavery C., Margetts P.J., Roberts A.B., Gauldie J. Smad3 null mice develop airspace enlargement and are resistant to TGF-beta-mediated pulmonary fibrosis. J Immunol. 2004;173:2099–2108. doi: 10.4049/jimmunol.173.3.2099. [DOI] [PubMed] [Google Scholar]
- 54.Sime P.J., Xin Z., Graham F.L., Csaky K.G., Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest. 1997;100:768–776. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li M., Krishnaveni M.S., Li C., Zhou B., Xing Y., Banfalvi A., Li A., Lombardi V., Akbari O., Borok Z., Minoo P. Epithelium-specific deletion of TGF-beta receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J Clin Invest. 2011;121:277–287. doi: 10.1172/JCI42090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Munger J.S., Huang X., Kawakatsu H., Griffiths M.J., Dalton S.L., Wu J., Pittet J.F., Kaminski N., Garat C., Matthay M.A., Rifkin D.B., Sheppard D. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 57.Sheppard D. Transforming growth factor beta: a central modulator of pulmonary and airway inflammation and fibrosis. Proc Am Thorac Soc. 2006;3:413–417. doi: 10.1513/pats.200601-008AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu S., Gladson C.L., White K.E., Ding Q., Stewart J., Jr., Jin T.H., Chapman H.A., Jr., Olman M.A. Urokinase receptor mediates lung fibroblast attachment and migration toward provisional matrix proteins through interaction with multiple integrins. Am J Physiol Lung Cell Mol Physiol. 2009;297:L97–L108. doi: 10.1152/ajplung.90283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sayers R.L., Sundberg-Smith L.J., Rojas M., Hayasaka H., Parsons J.T., Mack C.P., Taylor J.M. FRNK expression promotes smooth muscle cell maturation during vascular development and after vascular injury. Arterioscler Thromb Vasc Biol. 2008;28:2115–2122. doi: 10.1161/ATVBAHA.108.175455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tager A.M., LaCamera P., Shea B.S., Campanella G.S., Selman M., Zhao Z., Polosukhin V., Wain J., Karimi-Shah B.A., Kim N.D., Hart W.K., Pardo A., Blackwell T.S., Xu Y., Chun J., Luster A.D. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med. 2008;14:45–54. doi: 10.1038/nm1685. [DOI] [PubMed] [Google Scholar]
- 61.Li Y., Jiang D., Liang J., Meltzer E.B., Gray A., Miura R., Wogensen L., Yamaguchi Y., Noble P.W. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J Exp Med. 2011;208:1459–1471. doi: 10.1084/jem.20102510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.White E.S., Thannickal V.J., Carskadon S.L., Markwart S., Toews G.B., Arenbert D.A. Integrin alpha4beta1 regulates migration across basement membranes by lung fibroblasts: a role for phosphatase and tensin homologue deleted on chromosome 10. Am J Respir Crit Care Med. 2003;168:436–442. doi: 10.1164/rccm.200301-041OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Suganuma H., Tamura R., Chida K. Enhanced migration of fibroblasts derived from lungs with fibrotic lesions. Thorax. 1995;50:984–989. doi: 10.1136/thx.50.9.984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cukierman E., Pankov R., Stevens D.R., Yamada K.M. Taking cell-matrix adhesions to the third dimension. Science. 2001;294:1708–1712. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- 65.Hynes R.O. Integrins: versatility, modulation, and signating in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 66.Giancotti F.G., Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 67.Lawson C., Lim S.T., Uryu S., Chen X.L., Calderwood D.A., Schlaepfer D.D. FAK promotes recruitment of talin to nascent adhesions to control cell motility. J Cell Biol. 2012;196:223–232. doi: 10.1083/jcb.201108078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bishop A.L., Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348(Pt 2):241–255. [PMC free article] [PubMed] [Google Scholar]
- 69.Xu S.W., Liu S., Eastwood M., Sonnylal S., Denton C.P., Abraham D.J., Leask A. Rac inhibition reverses the phenotype of fibrotic fibroblasts. PLoS One. 2009;4:e7438. doi: 10.1371/journal.pone.0007438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Osborn-Heaford H.L., Ryan A.J., Murthy S., Racila A.M., He C., Sieren J.C., Spitz D.R., Carter A.B. Mitochondrial Rac1 GTPase import and electron transfer from cytochrome c are required for pulmonary fibrosis. J Biol Chem. 2012;287:3301–3312. doi: 10.1074/jbc.M111.308387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lagares D., Busnadiego O., Garcia-Fernandez R.A., Kapoor M., Liu S., Carter D.E., Abraham D., Shi-Wen X., Carreira P., Fontaine B.A., Shea B.S., Tager A.M., Leask A., Lamas S., Rodriguez-Pascual F. Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis Rheum. 2012;64:1653–1664. doi: 10.1002/art.33482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thannickal V.J., Lee D.Y., White E.S., Cui Z., Larios J.M., Chacon R., Horowitz J.C., Day R.M., Thomas P.E. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278:12384–12389. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
- 73.Shi-Wen X., Parapuram S.K., Pala D., Chen Y., Carter D.E., Eastwood M., Denton C.P., Abraham D.J., Leask A. Requirement of transforming growth factor beta-activated kinase 1 for transforming growth factor beta-induced alpha-smooth muscle actin expression and extracellular matrix contraction in fibroblasts. Arthritis Rheum. 2009;60:234–241. doi: 10.1002/art.24223. [DOI] [PubMed] [Google Scholar]
- 74.Liu S., Xu S.W., Kennedy L., Pala D., Chen Y., Eastwood M., Carter D.E., Black C.M., Abraham D.J., Leask A. FAK is required for TGFbeta-induced JNK phosphorylation in fibroblasts: implications for acquisition of a matrix-remodeling phenotype. Mol Biol Cell. 2007;18:2169–2178. doi: 10.1091/mbc.E06-12-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mimura Y., Ihn H., Jinnin M., Asano Y., Yamane K., Tamaki K. Constitutive phosphorylation of focal adhesion kinase is involved in the myofibroblast differentiation of scleroderma fibroblasts. J Invest Dermatol. 2005;124:886–892. doi: 10.1111/j.0022-202X.2005.23701.x. [DOI] [PubMed] [Google Scholar]
- 76.Kim K.K., Wei Y., Szekeres C., Kugler M.C., Wolters P.J., Hill M.L., Frank J.A., Brumwell A.N., Wheeler S.E., Kreidberg J.A., Chapman H.A. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest. 2009;119:213–224. doi: 10.1172/JCI36940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moore B.B., Murray L., Das A., Wilke C.A., Herrygers A.B., Toews G.B. The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am J Respir Cell Mol Biol. 2006;35:175–181. doi: 10.1165/rcmb.2005-0239OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Strieter R.M., Keeley E.C., Burdick M.D., Mehrad B. The role of circulating mesenchymal progenitor cells, fibrocytes, in promoting pulmonary fibrosis. Trans Am Clin Climatol Assoc. 2009;120:49–59. [PMC free article] [PubMed] [Google Scholar]
- 79.Willis B.C., Liebler J.M., Luby-Phelps K., Nicholson A.G., Crandall E.D., du Bois R.M., Borok Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166:1321–1332. doi: 10.1016/s0002-9440(10)62351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moore B.B. Fibrocytes as potential biomarkers in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:524–525. doi: 10.1164/rccm.200812-1847ED. [DOI] [PubMed] [Google Scholar]
- 81.Hasimoto N., Jin H., Liu T., Chensue S.W., Phan S.H. Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest. 2004;113:243–252. doi: 10.1172/JCI18847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Keeley E.C., Mehrad B., Strieter R.M. The role of fibrocytes in fibrotic diseases of the lungs and heart. Fibrogenesis Tissue Repair. 2011;4:2. doi: 10.1186/1755-1536-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hagood J.S., Prabhakaran P., Kumbla P., Salazar L., MacEwen M.W., Barker T.H., Ortiz L.A., Schoeb T., Siegal G.P., Alexander C.B., Pardo A., Selman M. Loss of fibroblast Thy-1 expression correlates with lung fibrogenesis. Am J Pathol. 2005;167:365–379. doi: 10.1016/S0002-9440(10)62982-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhong Q., Zhou B., Ann D.K., Minoo P., Liu Y., Banfalvi A., Krishnaveni M.S., Dubourd M., Demaio L., Willis B.C., Kim K.J., duBois R.M., Crandall E.D., Beers M.F., Borok Z. Role of endoplasmic reticulum stress in epithelial-mesenchymal transition of alveolar epithelial cells: effects of misfolded surfactant protein. Am J Respir Cell Mol Biol. 2011;45:498–509. doi: 10.1165/rcmb.2010-0347OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FRNK-knockout and WT mice have a similar early inflammatory response, total TGF-β1 level, cell proliferation rates, and percentage of lesional macrophages and endothelial cells in response to bleomycin. FRNK-knockout (FRNK−/−) and WT mice were intratracheally instilled with saline (Sal) or 1 U/kg body weight bleomycin (Bleo). BAL fluids were collected at day 7 after either saline or bleomycin challenge. A: BAL total protein level was determined by colorimetric assay (BCA kit; Pierce, Rockford, IL). B: BAL cell differential was determined on Diff-Quick–stained cytospins by standard techniques (Fisher Scientific). Lymphs, lymphocytes; MAC, macrophages; PMNs, polymorphonuclear neutrophils. C: BAL total cell number was counted in a hemocytometer. D: BAL total TGF-β1 level was measured by ELISA. All assays were performed according to manufacturer’s instructions. E: Frozen lung tissue sections (5 to 8 μm) from 1-U/kg bleomycin-instilled (day 14 after bleomycin) FRNK−/− and WT mice were IHC stained by antibodies specific toward Ki-67 (as a proliferation cellular marker), F4/80 (as a macrophage cellular marker), or von Willebrand factor (VWF; as an endothelial cellular marker) (data not shown). Positively stained cells were counted, and data are quantified as percentage of positively stained cells of total lesional cells. Data were pooled and presented as means ± SE (n = 8 to 10 animals per group).
FRNK deficiency potentiates, and FRNK restoration abrogates, TGF-β1–induced fibrotic responses in murine lung fibroblasts. A:FRNK−/− and WT lung fibroblasts were infected with Ad-FRNK or control Ad-GFP, followed by 10 ng/mL TGF-β1 treatment, for 36 hours, or vehicle. Equivalent amounts of whole cell lysates underwent Western blot analysis with antibodies directed toward the hemagglutinin tag of FRNK and GFP. GAPDH was used as the loading control. B: Murine lung fibroblasts were derived from FRNK−/− and WT mice, and infected with adenoviral vectors expressing either FRNK (Ad-FRNK) or control GFP (Ad-GFP). Cell migration in response to 10 ng/mL TGF-β1 in serum-free medium with 1% BSA (SFM) was examined by wound closure assay for 24 hours. Representative images are shown.
FRNK deficiency potentiates, and FRNK restoration abrogates, TGF-β1–induced collagen gel contraction in murine lung fibroblasts. Murine lung fibroblasts were derived from FRNK−/− and WT mice, and infected with adenoviral vector expressing either FRNK (Ad-FRNK) or control GFP (Ad-GFP). Cells were treated with 10 ng/mL TGF-β1 or vehicle, followed by the collagen gel contraction assays for 50 hours at 37°C and 5% CO2, as described in Materials and Methods. Representative digital images are shown.
FRNK deficiency promotes Smad3 phosphorylation in bleomycin-challenged mice and TGF-β1–treated lung fibroblasts. A: Mice were challenged with bleomycin and treated with or without adenoviral vectors, as described in the legend to Figure 2. Lungs were harvested at day 14, and equivalent amounts of whole lung lysate underwent Western blot analysis (one lane per individual animal). B:FRNK−/− and WT lung fibroblasts were infected with Ad-FRNK or control Ad-GFP, followed by 10 ng/mL TGF-β1 treatment or vehicle. Equivalent amounts of whole cell lysates underwent Western blot analysis with the indicated antibodies.