

Abstract
This review is on ultrastructure and subcellular physiology at normal and abnormal neuromuscular junctions. The clinical and electrophysiological findings in myasthenia gravis, Lambert-Eaton myasthenic syndrome (LEMS), congenital myasthenic syndromes, and botulinum intoxication are discussed. Single fiber electromyography (SFEMG) helps to explain the basis of testing neuromuscular junction function by repetitive nerve stimulation (RNS). SFEMG requires skill and patience and its availability is limited to a few centers. For RNS supramaximal stimulation is essential and so is display of the whole waveform of each muscle response at maximum amplitude. The amplitudes of the negative phase of the first and fourth responses are measured from baseline to negative peak, and the percent change of the fourth response compared with the first represents the decrement or increment. A decrement greater than 10% is accepted as abnormal and smooth progression of response amplitude train and reproducibility form the crux. In suspected LEMS the effect of fast rates of stimulation should be determined after RNS response to slow rates of stimulation. Caution is required to avoid misinterpretation of potentiation and pseudofacilitation.
Keywords: Acetylcholine, congenital myasthenic syndromes, Lambert–Eaton myasthenic syndrome, myasthenia gravis, repetitive nerve stimulation, single-fiber electromyography
Introduction
The neuromuscular (NM) junction is a simple relay between one of the nerve terminals of a motorneuron and the skeletal muscle fiber, which the nerve terminal innervates. Chemical transmission across the synaptic cleft overcomes the large electrical mismatch between the very fine nerve fiber and the large muscle fiber by introducing an amplification of an order larger than 100-fold.[1]
Presynaptic features
The chemical transmitter at the NM junction is acetylcholine (ACh), which is synthesized in the nerve terminal from acetyl coenzyme-A and choline by the enzyme choline acetyltransferase [Figure 1]. In the presynaptic terminal ACh is partitioned into at least three main compartments the largest of which is the ‘reserve’ ACh that is not directly available for release. A smaller compartment, called the ‘mobilization store,’ in turn supplies the smallest portion of ACh (immediate pool) available for release. This readily releasable portion functions as if it were released in units of several thousand molecules of ACh at a time as ‘quanta’. Vesicles attach to specific sites on the presynaptic membrane called active zones and release their contents into the primary synaptic cleft. In the resting state individual multimolecular packets or ‘quanta’ of ACh are released intermittently, and the frequency of this release is dependent upon extracellular calcium concentration and temperature.[2]
Figure 1.
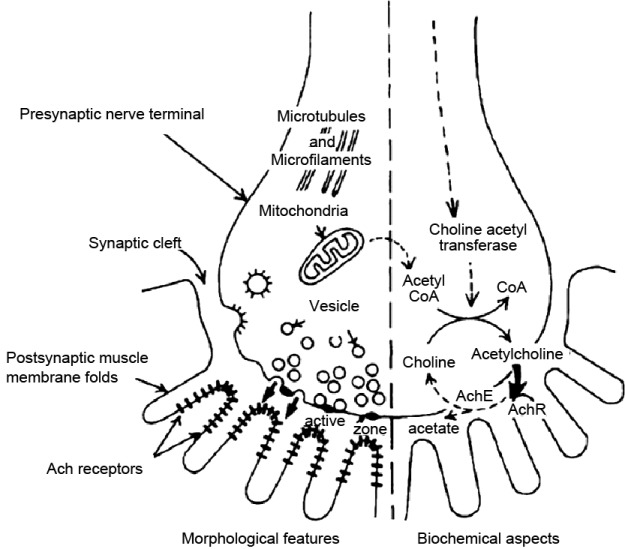
Morphological and biochemical aspects of the neuromuscular junction are presented to either side of the vertical dashed line
When the nerve action potential (AP) arrives at the axon terminal voltage-gated calcium channels open, and calcium ions enter the neuron. A biochemical cascade ensues causing the vesicles, which contain ACh, to fuse with the cell membrane, releasing ACh into the synaptic cleft. This cascade requires a synaptic fusion complex that is composed of (soluble N-ethylmaleimide sensitive fusion protein attachment factor (SNARE) proteins. For ACh release at the NM junction, some of the key SNARE proteins are synaptobrevin, synaptosomal-associated protein of 25 kd (SNAP-25), and syntaxin I.[3]
Postsynaptic features
The released ACh binds to transmembrane glycoproteins called acetylcholine receptors (AChRs) concentrated at the crests of postsynaptic folds opposite the presynaptic active zones [Figure 1].[4] The AChRs are sodium ion channels. When two molecules of ACh bind to specific extracellular sites on an AChR molecule, a funnel-shaped ion channel in the center of the receptor opens for a few milliseconds. The resulting fluxes of sodium down its electrochemical gradient, occurring through a few thousand AChR ion channels in response to a ‘quanta’ of ACh, briefly depolarizes the muscle membrane only at the junctional region. Each postsynaptic nonpropagating depolarization is called a miniature endplate potential (MEPP). Measured intracellularly by microelectrodes, MEPP amplitude provides a useful estimate of the response of the postsynaptic membrane to a single quantum of ACh. Several other nearby membrane proteins including agrin, rapsyn, and muscle-specific tyrosine kinase (MUSK) also influence the functioning of AChRs. This inflow of sodium depolarizes the muscle cell membrane and if sufficient, it triggers a regenerative muscle action potential that spreads throughout the cell into the transverse tubules releasing calcium from the sarcoplasmic reticulum and initiating the process of muscle contraction. After a certain time, the ACh comes off the AChR and acetylcholinesterase degrades it into an acetate group and choline. The choline is taken directly back into the neuron where it is combined with another acetate to reform ACh, which is repackaged into a vesicle.
Response to nerve stimulation
In contrast to the intermittent random release of quanta of ACh at rest, up to 100 quanta of ACh are released into the primary synaptic cleft in response to a nerve impulse. This ACh diffuses across the synaptic cleft and binds to about 100,000 AChRs, which causes a larger nonpropagating depolarization of the postsynaptic membrane called the endplate potential (EPP). The amplitude of the endplate potential tends to be a multiple of the amplitude of a MEPP under similar conditions, which is one of the key pieces of evidence for the quantal theory of NM transmission.[2] If the EPP is large enough to reach a critical level of depolarization called the ‘threshold’ for activation, an all-or-none AP will be propagated in both directions along the nonjunctional muscle membrane. When this AP invades the transverse tubule system of the muscle, another voltage-sensitive calcium influx triggers mechanical contraction of the muscle fiber.
Safety factor
Normally an excess of ACh is released, and several times as many AChRs are activated, as would be necessary for an EPP to reach the threshold for AP propagation. This is called the ‘safety factor’ of NM transmission.[5] If the safety factor is greatly reduced, this may lead to blocking of NM transmission.
Conditions of Abnormal Neuromuscular Transmission
Myasthenia gravis
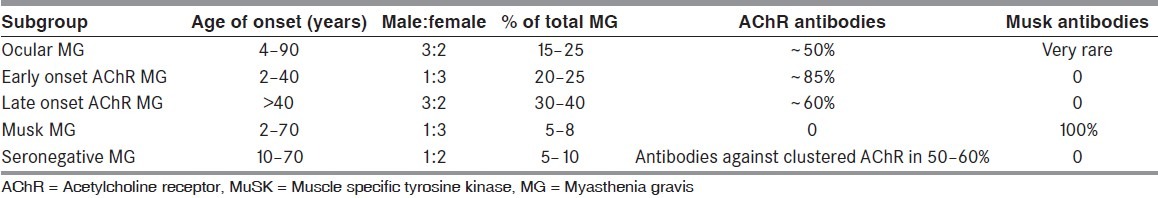
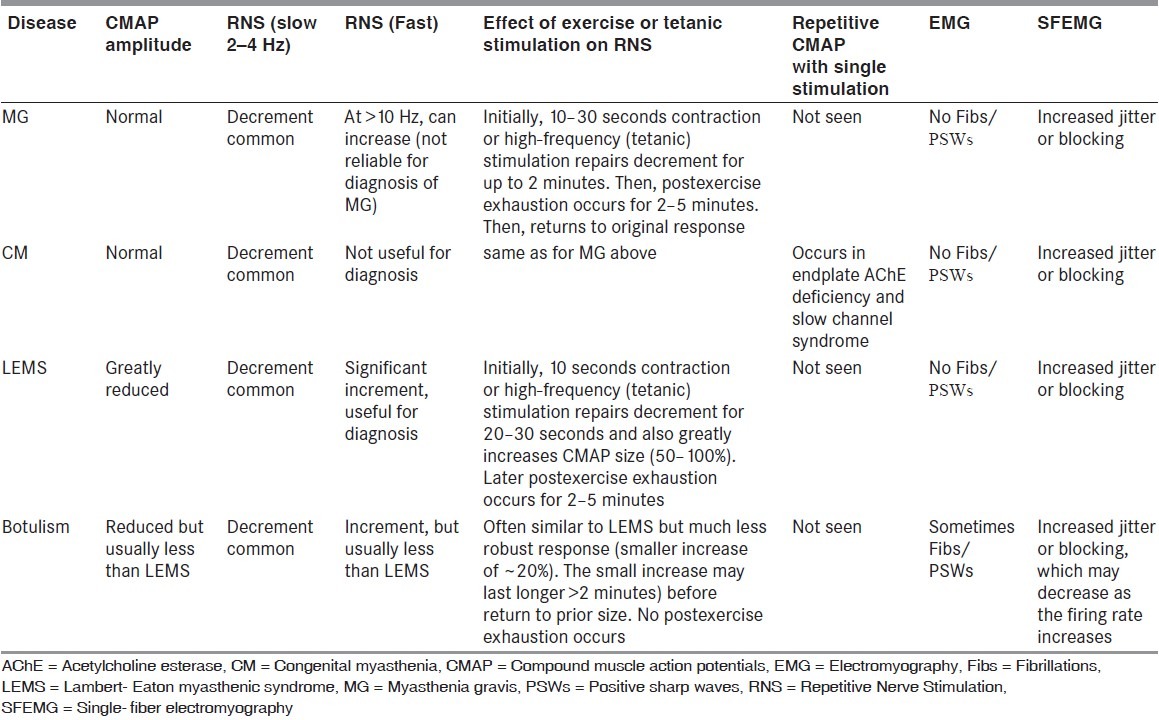
Myasthenia gravis (MG) is characterized clinically by asymmetric weakness and easy fatigability of extraocular, bulbar, nuchal, or limb muscles with normal reflexes and a 15% association with thymoma. MG affects approximately 100 patients per million population.[6] Loss of approximately 60% of AChRs is necessary to cause myasthenic weakness, and is seen in both the acquired and congenital forms of the disease. Efficient neuromuscular transmission depends on the tight clustering of AChRs at the neuromuscular junction, in which muscle specific tyrosine kinase (MuSK), Rapsyn and Dok-7 play key roles. The Simpson test is the simplest for clinical examination and is done by asking the patient to look up at a fixed point for an extended period which frequently brings out the ptosis in a patient with MG. If a patient has ptosis in one eye but not the other eye, hold the ptotic eye open (i.e., pull the lid up further than the patient can do on his or her own). Frequently, this will cause ptosis to appear in the other eye. The reason is that the central nervous system (CNS) is directing more effort to keeping the ptotic eye open. According to the Herring law, the CNS distributes the impetus to both eyes. Thus, by helping the patient keep the ptotic eye open, the CNS is tricked into reducing the effort and this brings out the ptosis in the other eye.[7,8] Cooling with an ice pack tends to ameliorate the deficits seen in MG by inhibiting acetylcholinesterase enzyme similar to tensilon test.[9] The various subtypes of MG are shown in Table 1.
Table 1.
Comparison of different subgroups of myasthenia gravis[6]
Tensilon test
Administration of 10 mg Tensilon in fractional doses in an intensive care unit is commonly recommended. After an initial injection of 2 mg, the response in the selected muscles is monitored for 60 seconds. Subsequent injections of 3 and 5 mg are then given. If definite improvement is seen within 60 seconds after any dose, no further injections are necessary. Total dose recommended in children is 0.15 mg/kg. Worsening of weakness after administration of Tensilon (a paradoxical response) is also an indication that NM transmission is impaired. Unlike most patients with MG, many patients with MUSK-positive MG, a variant, do not improve with Tensilon. Strength after Tensilon may also improve in motor neuron disease and in lesions of the oculomotor nerves.[10]
Antibodies to human AChRs are detected in the blood of 80% of patients with generalized MG,[4] and immune complexes have been localized to the postsynaptic membrane.[11] The number of AChRs on the postsynaptic membrane in MG is decreased.[12] This correlates closely with the amplitude of MEPPs, which are on the average are one-fifth smaller than normal MEPPs.[13] The frequency of spontaneous quantal release (a presynaptic function) is normal in MG, as are presynaptic ACh content and synaptic vesicles.[14] Although the number of quanta released following a nerve impulse is normal, the EPP response to nerve stimulation is decreased in MG. Therefore some EPPs are too small to initiate a muscle AP, and weakness or fatigue results.[15] Anticholinesterases, by inhibiting the breakdown of ACh, increase the amplitude and duration of EPPs and increase its chance of reaching the threshold for propagation of muscle AP.
Lambert–Eaton myasthenic syndrome
Lambert–Eaton myasthenic syndrome (LEMS) is caused by auto-antibodies to the voltage-gated calcium channels (VGCC) on the presynaptic side of the NM junction. Frequently, it is a paraneoplastic disease, with the most common cancer being small-cell carcinoma of the lung (SCLC) followed by adenocarcinomas and lymphoproliferative disorders. Noncancer cases may be associated with other autoimmune diseases, such as insulin dependent diabetes mellitus and thyroid disease. Nonparaneoplastic patients can present in any age group while paraneoplastic patients are generally aged more than 50 years and are usually smokers. LEMS is characterized clinically by weakness and fatigability of proximal limb muscles with relative sparing of extraocular and bulbar muscles, hyporeflexia, and dry mouth. In addition, patients report an increase in strength with repeated exercise rather than a decrease as in MG. The tendon reflexes are commonly reduced or absent but strong contraction of the relevant muscle against resistance often allows the reflex to be elicited; this ‘potentiation’ of absent or hypoactive tendon reflexes is virtually diagnostic.[16] Pointers for paraneoplastic Lambert–Eaton myasthenic syndrome include rapid progression of symptoms, early involvement (within 6 months) of distal muscles, dysarthria, and impotence. Freeze-fracture electron microscopy of the presynaptic nerve terminal membranes of intercostal muscles from patients with LEMS demonstrate a disruption of the ‘active zones,’ the rows of large intramembranous particles arranged in parallel double rows on the presynaptic nerve terminal membrane where vesicles attach.[17] There is also a decrease in number of particles, which make up the active zones in LEMS presynaptic membrane.[18] Patients with LEMS have normal presynaptic ACh stores, normal postsynaptic response to individual ACh quanta (normal MEPPs and normal MEPP frequency), but reduced release of ACh quanta from the nerve terminal by a nerve impulse and therefore reduced EPPs.[15] The reduction in the size of the EEPs was proportional to the decreased number of active zone particles, which may represent the calcium channels for voltage-sensitive calcium-dependent ACh release.[19]
Congenital myasthenia
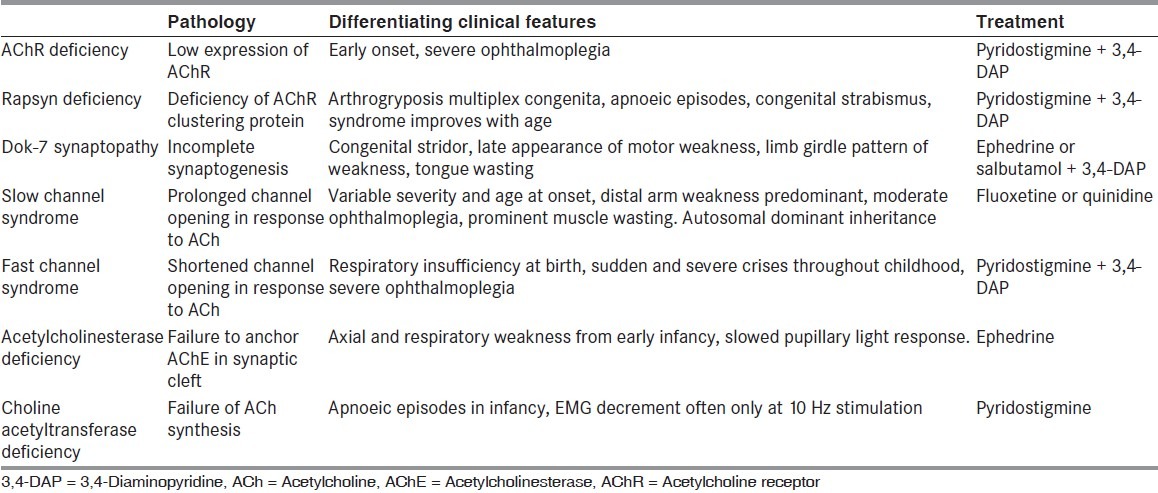
Congenital myasthenia comprises a heterogeneous group of disorders in which the symptoms of muscle weakness and fatigue have been present from birth, often with a positive family history of similar disorder but without circulating AChR antibody in the patients.[20] Immunotherapy is usually ineffective, and the response to anticholinesterase medication is variable.[21] Clinical features of the commonly encountered congenital myasthenic syndromes are shown in Table 2.[22]
Table 2.
Clinical features of the commonly encountered congenital myasthenic syndromes[6]
Botulism
Botulinum toxin a presynaptic blocker, can be taken up by endocytosis and cleaves some of the SNARE proteins required for synaptic fusion complex. In particular, botulinum toxins A, C, and E cleave SNAP-25 (the commercially available Botox® and Dysport® are both type A). Types B, D, F, and G cleave synaptobrevin. Type C also cleaves syntaxin. Any of the cleavages can block the formation of the complex, thus blocking ACh release and consequently blocking muscle contraction.
Electrophysiological studies in Neuromuscular Junction Disorders
Single-fiber electromyography
Single-fiber electromyography (SFEMG) is a selective electromyography (EMG) recording technique that allows identification of action potentials (APs) from individual muscle fibers. The selectivity of the technique results from the small recording surface (25 μm in diameter), which is exposed at a port on the side of the electrode, which is 3 mm from the tip [Figure 2].[23]
Figure 2.
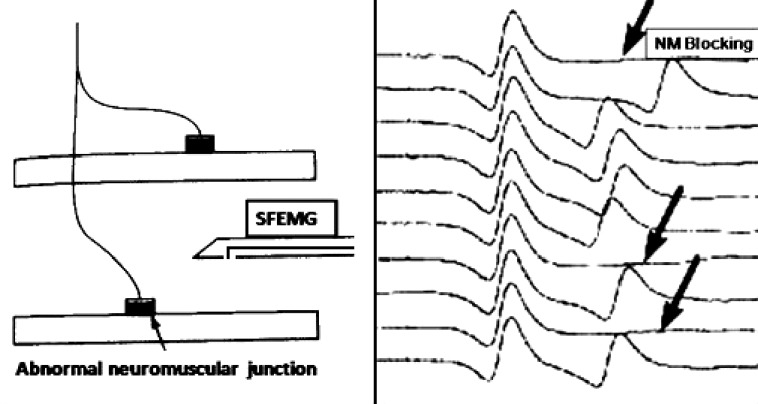
In the diagram on the left of the figure, SFEMG electrode can record electrical activity from two muscle fibers of the same motor unit. In the figure on right two muscle fiber action potentials, and variability in the interval between potentials is seen as a variable position of the second potential with increased jitter and occasional blockings (arrows) in myasthenic muscle
Neuromuscular jitter
When APs elicited by nerve stimulation are recorded with a SFEMG electrode, the latency from stimulus to response varies. This variation is the neuromuscular jitter, most of which is produced by fluctuations in the time for EPP at the NM junction to reach the AP threshold. When the SFEMG electrode is positioned to record from two or more muscle fibers in one voluntarily activated motor unit, the neuromuscular jitter is seen as variations in the time intervals between pairs of APs from these fibers.
Jitter may be measured either as the nerve is stimulated or as the patient voluntarily activates the muscle. The motor nerve may be stimulated proximally to its entry into the muscle, or individual motor nerve branches may be stimulated within the muscle. Axonal stimulation jitter studies are particularly useful in patients who have difficulty maintaining constant voluntary activation of the muscle or having tremor, in children who are too young to cooperate.
Jitter measurements performed during voluntary activation of the muscle are less subject to technical problems that can lead to misinterpretation of the results but requires more patient cooperation than stimulation jitter studies. As the patient slightly contracts the muscle, the SFEMG electrode is inserted into the muscle near the endplate zone; it is positioned to record two or more time-locked APs from the same motor unit [Figure 2]. The amplitudes of the APs are optimized by slightly adjusting the electrode position; in the best recording position for jitter measurements, all APs of interest should have sharply rising phases and adequate amplitudes. APs should be measured from 20 potential pairs and recorded from different portions of the muscle, using 3–4 skin insertions. The jitter is expressed as the mean value of consecutive differences of successive interpotential intervals (MCD) [Figure 3]. Jitter is increased whenever the ratio between the AP threshold and the EPP is greater than normal; thus, it is a sensitive measure of the safety factor of NM transmission. The normal mean MCD value varies from 10 to 50 μs among different muscles.[24] With more pronounced disturbances, impulses to individual muscle fibers intermittently fail to occur, producing NM blocking [Figure 2]. Only when blocking occurs is clinical weakness or a decrement on repetitive nerve stimulation (RNS) tests noted. In diseases of abnormal NM transmission, jitter may be increased in muscles that are clinically normal and showing no decrement on RNS.
Figure 3.
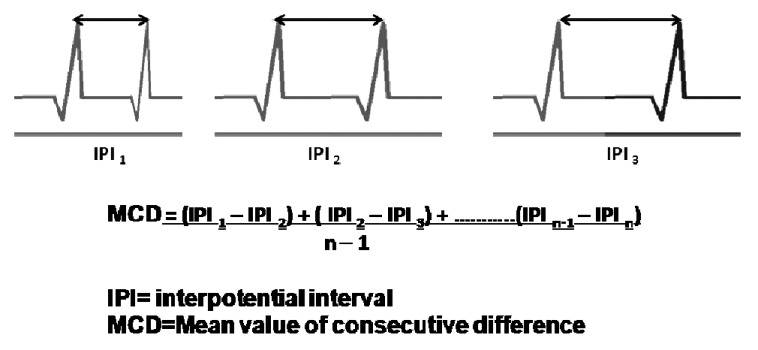
Jitter measurements after single-fiber electromyography. IPI is the interpotential interval or sti mulus-to-response latency during axonal stimulation jitter analysis and calculation of the mean difference between consecutive discharges (MCD)
Results of jitter measurements are recorded in each muscle in the following way:
The mean or median value of the MCD values in all the pairs or endplates that are measured (mean or median MCD).
The percentage of paired potentials or endplates in which blocking was seen (percent blocking).
The percentage of pairs or endplates in which jitter exceeded the normal limit for that muscle (percent abnormal pairs or endplates).
Normal jitter values
Reference jitter values have been determined for many muscles. Jitter increases slightly with age in normal subjects. A study is abnormal if the mean (or median) jitter exceeds the upper limit for the muscle, or if more than 10% of pairs or endplates have increased jitter or blocking. Jitter less than 5 μs is seen rarely in voluntarily activated SFEMG studies in normal muscles and more often in myopathies. These low values probably result from recordings that are made from split muscle fibers, both branches of which are activated by a single neuromuscular junction and should not be included in assessments of NM transmission. Reference values for jitter during axonal stimulation have been determined for the extensor digitorum communis (EDC) and orbicularis oculi muscles.[25] For other muscles, the normative values for stimulation jitter can be obtained by multiplying the values for voluntary activation by a conversion factor of 0.8.
SFEMG studies in myasthenia gravis
EDC is abnormal in most patients with generalized MG and is the muscle to be examined first.
If jitter is normal in a muscle with definite weakness, the weakness is not due to MG.
If the symptoms or weakness is limited to the ocular muscles, the orbicularis oculi or frontalis may be examined first.
If the EDC is examined first and is normal, the frontalis or orbicularis oculi muscles usually are examined next. If the first of these facial muscles is normal, the other should be examined before excluding the diagnosis of MG.
Jitter is less abnormal in patients with ocular myasthenia than in those with generalized disease. Jitter is increased in a limb muscle in more than half the patients with ocular myasthenia, demonstrating that the physiologic abnormality is more. Jitter also is increased in diseases of nerve and muscle; these must be excluded by other electrophysiologic and clinical examinations before concluding that the patient has MG.
SFEMG in LEMS or botulism demonstrate decreasing jitter with increasing firing rate.[26] These contrasting responses to repetitive stimulation reflect a differential effect of firing rate upon two main physiologic factors affecting transmitter release at the normal nerve terminal. These two factors are the amount of ACh transmitter available for release and the amount of available calcium affecting the probability of transmitter release.
Repetitive nerve stimulation
The most commonly used electrodiagnostic test of NM transmission involves repetitive stimulation of a motor nerve while recording compound muscle action potentials (CMAP) from a muscle innervated by that nerve. The result is abnormal if progressively fewer muscle fibers respond to nerve stimulation during a train of stimuli, producing a ‘decrementing’ pattern in the CMAP.
Transmitter depletion
During repetitive depolarization of the normal nerve, terminal quantal ACh (and the number of synaptic vesicles) is gradually depleted. This is most prominent during the first few stimuli at slow rates of repetition (3–4 Hz), after which ACh mobilization has responded to the stimulation by increasing ACh availability at nerve terminal, which matches ACh discharge. Because of the decreased safety factor for transmission in MG, however, a decrease in the size of the endplate potential with repeated stimulation may lead to subsequent blocking of AP production when the EPP falls below threshold. This normal process of quantal ACh depletion during slow RNS may not be as prominent in conditions such as LEMS or botulism in which transmitter release is defective.
Calcium facilitation
During RNS there is an increase of calcium influx during ACh depolarization caused by a nerve impulse. This increased calcium facilitates transmitter release for about 100–200 milliseconds following each impulse,[27] after which time the calcium have been sequestered by mitochondria. Repetitive stimuli occurring between 5 and 50 Hz may therefore produce a cumulative facilitation of transmitter release. Facilitation decreases above 50 Hz because impulses begin to overlap with each other. Facilitation by increased calcium is not so important in normal NM transmission, which is already operating optimally, as it is in conditions in which transmitter release is defective. In LEMS and botulism, facilitation by calcium influx into nerve terminals during 40–50 Hz stimulation may improve transmitter release such that NM transmission in some junctions will be restored to normal.
Repetitive nerve stimulation in MG
For RNS, withdraw anticholinesterase medications at least 8 hours before the test if this is possible without compromising the patient's ability to breathe and swallow. Any accessible nerve–muscle combination may be used for testing, but weak muscles are preferred.
Stimulation technique
A decremental response in MG is best demonstrated at stimulation rates of 3–4 Hz. This initial decrement is the reflection of progressive blocking at the NM junctions of hundreds of muscle fibers causing EPPs to become sub-threshold. The initial fall in the size of the CMAP then levels off or even slowly increases during subsequent stimuli as ACh mobilization catches up with ACh quantal depletion. Stimulation at rates of 3–5 Hz may produce a decrement up to 8% in normal muscles. A decrement greater than 10% usually is abnormal [Figure 4]. The following observations help distinguish true decrements from artifactual changes in CMAP size:
Figure 4.
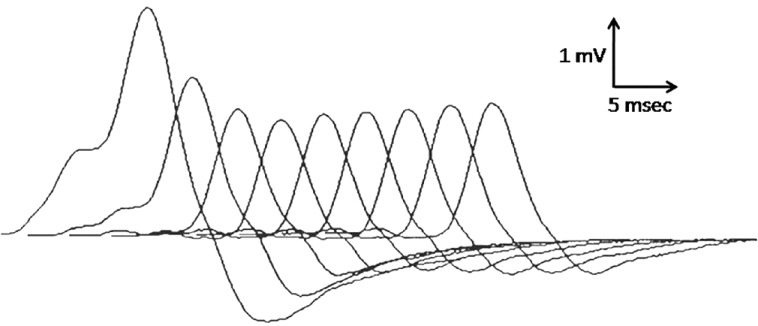
A prototype decrementing response to repetitive nerve stimulation in myasthenia gravis. The amplitude of the initial response is normal, and the decrement is maximal in the fourth response after which the responses may increase slightly, giving an envelope shape to the train of responses
Reproducibility: Decrement should be the same when stimulation is repeated after a period of rest.
Envelope shape: Changes during a train of stimuli should conform to a pattern seen in disease, without sudden or irregular variations between consecutive responses.
Potentiation and pseudofacilitation
Voluntary exercise of the measured muscle for several seconds produces a rapid NM stimulation at rates between 20 and 40 Hz. This is much less uncomfortable than tetanic 50 Hz external stimulation to the nerve and is the preferred method of rapid RNS. Immediately after such exercise, the size and area of the evoked myasthenic response may be larger. This is called postactivation potentiation. Potentiation may result from an increase in the number of ACh quanta released leading to more activated muscle fibers (i.e., facilitation) or from an increase in the amplitude or summation of action potentials of the individual muscle fibers (i.e., pseudofacilitation). Facilitation results when two or more nerve action potentials occur within a short time, producing a transient increase in the amount of ACh released from the motor nerve. Each nerve depolarization releases calcium into the periterminal space, which increases the local concentration of calcium for a short period of time. If the nerve depolarizes during this period, the increased calcium increases the number of ACh quanta released. In conditions of impaired NM transmission, this greater ACh release may improve synaptic transmission briefly, producing facilitation. By assessing the area under evoked responses before and after exercise, postactivation potentiation can be differentiated from ‘pseudofacilitation,’ in which the amplitude of the response is greater after exercise because of increased synchronization of its components but the area of the response remains the same.
A few minutes after exercise, the size of the evoked response in a myasthenic muscle may be smaller and the decrement greater than at rest. This is called postactivation exhaustion, the basis for which is unknown, although it has been attributed to receptor desensitization. Postactivation exhaustion may be the only abnormality to occur during repetitive testing of mildly involved myasthenic muscles. Therefore after 60 seconds of exercise a train of low-frequency repetitive stimulation every minute for up to 5 minutes may be useful to bring out a marginal decrement during postactivation exhaustion.
Diagnosis of Lambert–Eaton myasthenic syndrome by RNS
The CMAP amplitude in LEMS at rest is often 10% of normal, apparently because many muscle fibers are blocked as a result of decreased quantal release of transmitter on nerve stimulation.[28] At slow (2–3 Hz) rates of stimulation there may be a decremental response as additional transmitter depletion occurs. After a brief period of voluntary exercise (10-30 seconds), the CMAP in LEMS increases to almost normal size (usually over double what it was at rest),[29] presumably as a result of calcium facilitation of available transmitter release at rapid rates of stimulation. This significant postactivation potentiation is one of the electrical hallmarks of LEMS, the other being the low amplitude initial response. The development of potentiation at rapid rates of stimulation can also be displayed as an incremental response to rapid electrical stimulation in patients who are not able to activate the measured muscle voluntarily.[28] Postactivation exhaustion may occur in LEMS, but it is less important for potential diagnosis than it is in MG. Postactivation potentiation greater than 200% can also be seen in other presynaptic conditions such as botulism, hypermagnesemia, and hypocalcemia.[30,31]
Diagnosis of botulism by RNS
The electrical abnormalities in botulism are not found in all the muscles and it depends upon the severity of intoxication and evolve with time after absorption of the toxin.[32] Very mild cases may be normal on RNS, and a NM defect is detected only by increased jitter on SFEMG.
Findings typical of botulism include the following:[33]
Reduced CMAP amplitude in at least two muscles
At least 20% facilitation of CMAP amplitude during tetanic stimulation
Persistence of facilitation for at least 2 minutes after activation
No postactivation exhaustion
If none of the above are found, the diagnosis of botulism is unlikely. If all four are present, only hypermagnesemia is in the differential diagnosis. CMAP amplitudes usually are reduced, but the reduction is not so striking as is seen in LEMS. The reduction could easily be in the range seen in patients with axonal neuropathies.
In severe cases of botulism, transmitter release may be so severely blocked that no increment or potentiation occurs at rapid rates of stimulation, even though the initial response is small.[33] Decremental responses at slow rates of stimulation usually do not occur. The magnitude of the postactivation potentiation is usually less than that observed in LEMS, but it can last several minutes rather than the few seconds seen in LEMS.[34] Other features that may be observed include a decrement to slow rate stimulation, fibrillations and/or positive sharp waves, and motor unit action potentials, which are short, small, and polyphasic. Needle EMG is also a useful diagnostic indicator of botulism, particularly infantile botulism.[35]
Congenital Myasthenia
Slow channel syndrome
Slow RNS studies produce a decremental response as seen in low-affinity fast channel disease. Characteristic repetitive discharges are seen after single-nerve stimulation in most, but not all, muscles [Table 3]. Electrodiagnostic findings are similar to those seen with cholinesterase (ChE) inhibitor toxicity and congenital acetylcholinesterase (AChE) deficiency.
Table 3.
Electrophysiologic assessment of neuromuscular junction transmission defects[30]
Congenital acetylcholinesterase deficiency
RNS produces a decrement on 3-Hz stimulation in all muscles. Single-nerve stimulation produces repetitive CMAPs 6-10 milliseconds after the initial response, which fades quickly during repetitive stimulation, even at rates as low as 0.2 Hz. This is similar to findings in slow channel syndrome.
Conclusion
Appropriately selected nerve-muscle combinations are a must with preference for weak muscles for RNS testing. Proximal muscles are more likely to show decremental responses because they are warmer and testing of three muscles increase the chances of detecting an abnormal response. When properly done SFEMG and RNS facilitate in the diagnosis of neuromuscular transmission disorders.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil
References
- 1.Aminoff MJ, Layzer RE, Staya-Murti S, Faden AI. The declining electrical response of muscle to repetitive nerve stimulation in myotonia. Neurology. 1977;27:812–6. doi: 10.1212/wnl.27.9.812. [DOI] [PubMed] [Google Scholar]
- 2.Magleby KL. The effect of repetitive stimulation on facilitation of transmitter release at the frog neuromuscular junction. J Physiol. 1973;234:327–52. doi: 10.1113/jphysiol.1973.sp010348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prasarnpun S, Walsh J, Harris JB. Beta-bungarotoxin-induced depletion of synaptic vesicles at the mammalian neuromuscular junction. Neuropharmacology. 2004;47:304–14. doi: 10.1016/j.neuropharm.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 4.Herrmann C Jr, Lindstrom JM, Keesey JC, Mulder DG. Myasthenia gravis-current concepts. West J Med. 1985;142:797–80. [PMC free article] [PubMed] [Google Scholar]
- 5.Waud DR, Waud BE. In vitro measurement of margin of safety of neuromuscular transmission. Am J Physiol. 1975;229:1632–4. doi: 10.1152/ajplegacy.1975.229.6.1632. [DOI] [PubMed] [Google Scholar]
- 6.Jacob S, Viegas S, Lashley D, Hilton-Jones D. Myasthenia gravis and other neuromuscular junction disorders. Pract Neurol. 2009;9:364–71. doi: 10.1136/jnnp.2009.193912. [DOI] [PubMed] [Google Scholar]
- 7.Gorelick PB, Rosenberg M, Pagano RJ. Enhanced ptosis in myasthenia gravis. Arch Neurol. 1981;38:531. doi: 10.1001/archneur.1981.00510080093017. [DOI] [PubMed] [Google Scholar]
- 8.Gay AJ, Salmon ML, Windsor CE. Hering's law, the levators, and their relationship in disease states. Arch Ophthalmol. 1967;77:157–60. doi: 10.1001/archopht.1967.00980020159002. [DOI] [PubMed] [Google Scholar]
- 9.Sethi KD, Rivner MH, Swift TR. Ice pack test for myasthenia gravis. Neurology. 1987;37:1383–5. doi: 10.1212/wnl.37.8.1383. [DOI] [PubMed] [Google Scholar]
- 10.Oh SJ, Cho HK. Edrophonium responsiveness not necessarily diagnostic of myasthenia gravis. Muscle Nerve. 1990;13:187–91. doi: 10.1002/mus.880130302. [DOI] [PubMed] [Google Scholar]
- 11.Lang B, Willcox N. Autoantibodies in neuromuscular autoimmune disorders. Expert Rev Clin Immunol. 2006;2:293–307. doi: 10.1586/1744666X.2.2.293. [DOI] [PubMed] [Google Scholar]
- 12.Skeie GO, Apostolski S, Evoli A, Gilhus NE, Hart IK, Harms L, et al. Guidelines for the treatment of autoimmune neuromuscular transmission disorders. Eur J Neurol. 2006;13:691–9. doi: 10.1111/j.1468-1331.2006.01476.x. [DOI] [PubMed] [Google Scholar]
- 13.Lindstrom JM, Lambert EH. Content of acetylcholine receptor and antibodies bound to receptor in myasthenia gravis, experimental autoimmune myasthenia gravis, and Eaton-Lambert syndrome. Neurology. 1978;28:130–8. doi: 10.1212/wnl.28.2.130. [DOI] [PubMed] [Google Scholar]
- 14.Das A, Cherian A, Chemmanam T. A fisherman who could not row. Lancet. 2009;373:432. doi: 10.1016/S0140-6736(09)60138-0. [DOI] [PubMed] [Google Scholar]
- 15.Elmqvist D, Quastel DM. A quantitative study of endplate potentials in isolated human muscle. J Physiol. 1965;178:505–29. doi: 10.1113/jphysiol.1965.sp007639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanders DB. Lambert–Eaton myasthenic syndrome: Diagnosis and treatment. Ann N Y Acad Sci. 2003;998:500–8. doi: 10.1196/annals.1254.065. [DOI] [PubMed] [Google Scholar]
- 17.Fukunaga H, Engel AG, Osame M, Iamhert EH. Paucity and disorganization of presynaptic membrane active zones in the Lambert-Eaton myasthenic syndrome. Muscle Nerve. 1982;5:686–97. [Google Scholar]
- 18.Lang B, Newsom-Davis J, Prior C, Wray D. Antibodies to motor nerve terminals: An electrophysiological study of a human myasthenic syndrome transferred to mouse. J Physiol. 1983;344:335–45. doi: 10.1113/jphysiol.1983.sp014943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engel AG. Myasthenia gravis and myasthenic syndromes. Ann Neurol. 1984;16:519–34. doi: 10.1002/ana.410160502. [DOI] [PubMed] [Google Scholar]
- 20.Engel AG, Ohno K, Sine SM. The spectrum of congenital myasthenic syndromes. Mol Neurobiol. 2002;26:347–67. doi: 10.1385/MN:26:2-3:347. [DOI] [PubMed] [Google Scholar]
- 21.Harper CM, Fukodome T, Engel AG. Treatment of slow-channel congenital myasthenic syndrome with fluoxetine. Neurology. 2003;60:1710–3. doi: 10.1212/01.wnl.0000061483.11417.1b. [DOI] [PubMed] [Google Scholar]
- 22.Palace J, Beeson D. The congenital myasthenic syndromes. J Neuroimmunol. 2008;201-202:2–5. doi: 10.1016/j.jneuroim.2008.05.030. [DOI] [PubMed] [Google Scholar]
- 23.Sanders DB, Stålberg EV. AAEM minimonograph #25: Single-fiber electromyography. Muscle Nerve. 1996;19:1069–83. doi: 10.1002/(SICI)1097-4598(199609)19:9<1069::AID-MUS1>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 24.Gilchrist JM. Ad hoc Committee. Single fiber EMG reference values: A collaborative effort. Ad Hoc Committee of the AAEM Special Interest Group on Single Fiber EMG. Muscle Nerve. 1992;15:151–61. doi: 10.1002/mus.880150205. [DOI] [PubMed] [Google Scholar]
- 25.Kouyoumdjian JA, Stalberg EV. Reference jitter values for concentric needle electrodes in voluntarily activated extensor digitorum communis and orbicularis oculi muscles. Muscle Nerve. 2008;37:694–9. doi: 10.1002/mus.21043. [DOI] [PubMed] [Google Scholar]
- 26.Sanders DB, Howard JF., Jr AAEE Minimonograph #25: Single-fiber electromyography in myasthenia gravis. Muscle Nerve. 1986;9:809–19. doi: 10.1002/mus.880090904. [DOI] [PubMed] [Google Scholar]
- 27.Cherian A, Baheti NN, Kuruvilla A. Muscle channelopathies and electrophysiological approach. Ann Indian Acad Neurol. 2008;11:20–7. doi: 10.4103/0972-2327.40221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jablecki C. Lambert-Eaton myasthenic syndrome. Muscle Nerve. 1984;7:250–7. doi: 10.1002/mus.880070311. [DOI] [PubMed] [Google Scholar]
- 29.Sanders DB, Kim YI, Howard JF, Goetsch CA. Eaton-Lambert syndrome: A clinical and electrophysiological study of a patient treated with 4-aminopyridine. J Neurol Neurosurg Psychiatry. 1980;43:978–85. doi: 10.1136/jnnp.43.11.978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keesey JC. AAEE Minimonograph #33: Electrodiagnostic approach to defects of neuromuscular transmission. Muscle Nerve. 1989;12:613–26. doi: 10.1002/mus.880120802. [DOI] [PubMed] [Google Scholar]
- 31.Gutmann L, Takamori M. Effect of Mg++ on neuromuscular transmission in the Eaton-Lambert syndrome. Neurology. 1973;23:977–80. doi: 10.1212/wnl.23.9.977. [DOI] [PubMed] [Google Scholar]
- 32.De Jesus PV, Jr, Slater R, Spitz LK, Penn AS. Neuromuscular-physiology of wound botulism. Arch Neurol. 1973;29:425–31. doi: 10.1001/archneur.1973.00490300087012. [DOI] [PubMed] [Google Scholar]
- 33.Oh SJ. Botulism: Electrophysiological studies. Ann Neurol. 1977;1:481–5. doi: 10.1002/ana.410010514. [DOI] [PubMed] [Google Scholar]
- 34.Fakadej AV, Gutmann L. Prolongation of post-tetanic facilitation in infant botulism. Muscle Nerve. 1982;5:727–9. [Google Scholar]
- 35.Pickett J, Berg B, Chaplin E, Brunstetter-Shafer M-A. Syndrome of botulism in infancy: Clinical and electrophysiologic study. N Engl J Med. 1976;295:770–2. doi: 10.1056/NEJM197609302951407. [DOI] [PubMed] [Google Scholar]