

Abstract
Androgen receptor (AR) signaling is essential for the initial development and progression of prostate cancer (PCa) as well as the growth and survival of castration-resistant tumors. However, AR action may be opposed by estrogen receptor beta (ERβ) that responds to androgen metabolites produced in the prostate. The balance between the activity of these two receptors is not only influenced by the steroidogenic capacity of the prostatic microenvironment but also by its redox status and local paracrine signals such as transforming growth factor- beta (TGF-β). In this review, we highlight the studies that revealed select roles for AR and ERβ in distinct compartments of the prostate cancer microenvironment. We also discuss new work that identified stromal-epithelial crosstalk through TGF-β1 signaling that drives the production of reactive oxygen species in stromal cells thereby selectively limiting the antitumor activity of ERβ in cancer cells. Therefore, any new therapeutic approaches that seek to limit AR but enhance ERβ activity in PCa, must take into account potential adaptive changes in the tumor microenvironment that utilize paracrine signals and altered redox balance to divert local androgen metabolites towards AR at the expense of ERβ.
Keywords: androgen receptor, estrogen receptor-beta, prostate cancer, reactive oxygen species, transforming growth factor-beta, tumor microenvironment
Androgen signaling is crucial for the stimulation of new cell growth and development in prostate epithelial cells, which undergo a constant low level of turnover. Due to this growth and developmental function of androgen receptor (AR), it is to be expected that in prostate cancer (PCa) an abundance of AR signaling underlies the overgrowth of epithelial cells seen in carcinogenesis. During early stages of the disease, multiple factors can contribute to the hyperactivity of AR. Mutations within the AR gene can lead to a more promiscuous receptor that can be activated in the absence of ligand, or by additional growth factors such as epidermal growth factor and insulin-like growth factor-1 [1–5]. Increased expression of AR-specific co-activators and/or alterations in post-translational modifications can also be the source of aberrant AR signaling in early PCa [6].
While initial therapy for PCa is removal of the organ, recurrent disease is a common problem. Without a localized target tissue, the recurrent cancer cells, which are still dependent upon androgen signaling for continued growth, are typically targeted through systemic androgen deprivation [7]. This chemical castration is achieved by administration of a gonadotropin releasing hormone agonist or a specific AR-antagonist [8,9]. The former two lead to an overall decrease in endogenous production of androgens, whereas the latter targets the ability of circulating androgens to effectively activate the AR. This chemical castration is initially effective, but after prolonged androgen deprivation eventually the metastatic disease becomes castration resistant and unresponsive to this therapeutic approach [10]. Castration-resistant PCa (CRPC), however, is still dependent upon AR signaling; it is the therapeutic approach to ablating androgen activation that loses efficacy, as aberrant AR activation resumes despite the modalities employed for chemical castration [11]. Patients with CRPC still exhibit activation of AR target genes (e.g. prostate specific antigen or PSA), thus demonstrating that AR is still active in the course of the disease despite a therapeutic attempt to remove potent circulating androgens.
Recent studies using an intricate series of highly specific AR-knockouts in mice reveal the complicated the role of AR in PCa and generated some unresolved controversy. Specifically, Niu et al. have suggested that AR in the prostatic epithelium is tumor suppressive, whereas AR in prostatic stromal cells promotes invasion by a cancerous epithelium [12]. Additional follow-up of these results led to the hypothesis that one of the mechanisms through which AR alters epithelial invasive characteristics is through modulation of transforming growth factor-beta (TGF-β) signaling. A subset of TGF-β responsive genes, including the inducible pro-inflammatory enzyme cyclooxygenase-2 (COX-2), is not upregulated following TGF-β treatment when AR is stably re-expressed in PC3 PCa cells [12]. These TGF-β responsive genes act to promote tumor aggressiveness, thus inhibiting their induction via reexpression of AR provides tumor-suppressive activity. Androgen signaling, even in castration-resistant disease, appears to be a double-edged sword. Clearly, studies such as these would argue against global androgen deprivation therapy, again underscoring the need for a more thorough understanding of PCa growth and progression to develop more targeted and efficacious therapies.
As men age, serum testosterone levels naturally decline and the main source of androgens shifts to the adrenal gland, whose major androgen product is dehydroepiandrosterone (DHEA) [13]. However, a concomitant equilibrium of estrogen levels leads to decrease in the testosterone to estrogen ratio [14]. Thus, recent attention has been paid to understanding the balance between androgenic and estrogenic signaling in prostate biology (Figure 1). There are type distinct estrogen receptors (ERs), designated as ERα and ERβ. Unlike AR, whose activation is associated with prostate growth, signaling through ER appears to be more complex, due in large part to differential activities of the ER subtypes. ERα activation in the prostate is associated with three distinct responses: aberrant proliferation, inflammation, and cancer [15]. Aromatase knockout mice have lifelong elevated levels of androgens, yet the mice fail to develop PCa [16]. However, administration of synthetic estrogens early in development triggers abnormal prostate biology later in life in these knockout mice [17].
Figure 1. The predominant steroid receptors in the prostate are AR and ERβ.

AR acts to enhance cancer cell motility, while ERβ is inherently anti-motility. Systemic therapy targeting AR is used to slow down the progression of cancer, perhaps by tipping the balance towards ERβ signaling. However, local ROS production serves to inactivate ERβ, and thus allows unopposed AR activity. It is the balance in action of these 2 receptors that ultimately can determine the motility of the cancer cells. Thus, therapies targeted at swaying the balance towards ERβ activity may serve to limit cancer cell motility by utilizing an inherent regulatory pathway.
While these studies suggest that ERα signaling may be associated with the development of PCa, the prevailing ER subtype in the prostate is ERβ [18,19]. Knockout mice have been generated that are deficient only in the ERβ subtype, and extensive studies performed with these mice have shown that while ERα mediates estrogen-induced prostatic inflammation and pathologies, ERβ may confer a beneficial effect in maintenance of normal homeostasis [15]. Supporting epidemiological evidence also exists, as men who consume higher dietary intakes of phytoestrogens (weakly estrogenic compounds that exhibit a significantly higher affinity for ERβ over ERα) exhibit a lower incidence of PCa [20,21].
A unique feature of ERβ is its sensitivity to oxidation. This reportedly high redox sensitivity of ERβ may be important in tumors or tissues exposed to chronic inflammation. In particular, chronic inflammation leads to increased levels of reactive oxygen species (ROS) and subsequent development of an oxidizing milieu. Keeping this in mind, the oxidation-sensitive motifs in ERβ may be of particular relevance in PCa. Kumar et al. showed that oxidative stress in PCa cells is required for an aggressive phenotype, and that PCa cells are capable of generating high levels of ROS [22]. Recent work from our laboratory showed that prostate stromal cells are also capable of generating ROS in response to locally produced TGF-β1 [23].
Zinc-finger motifs containing 4 cysteine residues are an essential feature of the DNA-binding domain of members of the nuclear receptor superfamily of transcription factors [24]. In ERβ, as in other nuclear receptors, the first zinc-finger is necessary for DNA binding and the second zinc-finger contributes to receptor dimerization, an essential step for stabilization of DNA binding at the promoter [24,25]. ERβ oxidation can occur within the 2nd zinc finger motif, leading to a conformational change that destabilizes the receptor and ultimately prevents DNA binding [25,26]. Electrophoretic mobility shift assays have demonstrated a loss of DNA-binding when ERβ is subjected to oxidation by H2O2 [25]. As Figure 2 illustrates, more recent work from our laboratory using chromatin immunoprecipitation assays demonstrated a direct decrease in ERβ occupancy at the E-cadherin promoter in PCa cells exposed to H2O2 [23]. This is in accordance with ERβ work done in breast cancer that demonstrated a restoration of ERβ DNA-binding capacity following treatment with the thiol reducing agent dithiothreitol [27].
Figure 2. ERβ activity at the E-cadherin promoter in PCa cells.
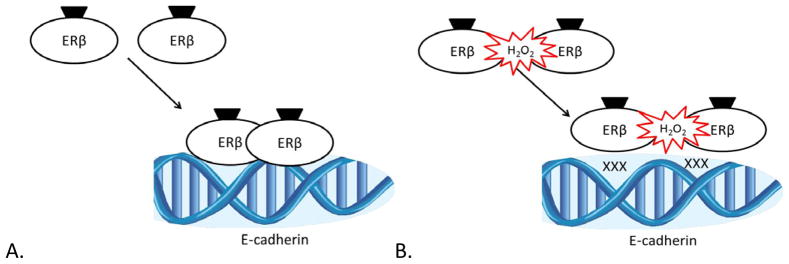
(A) In a redox neutral milieu, ligand binds ERβ and initiates receptor dimerization, occupancy of its cognate DNA-binding element in the E-cadherin promoter, and activation of transcription. (B) In the presence of H2O2, ERβ is oxidized at the 2nd zinc-finger motif and is unable to dimerize, thus destabilizing its DNA-binding capacity and leading to a loss of E-cadherin transcriptional activation.
Local steroidogenesis is an intricate and tightly controlled process that provides another level of regulation to steroid hormone action beyond that occurring following the binding of ligand to steroid hormone receptors. Differential enzyme expression in various tissues allows metabolism that either activates or inactivates various steroid hormone precursors into ligands that will preferentially bind the steroid hormone receptor of importance in that tissue. In the prostate, for example, the enzyme 5α-reductase is highly expressed. 5α-reductase converts testosterone into dihydrotestosterone (DHT), a ligand much more potent for the AR than testosterone [28,29]. Unlike testosterone, DHT is unable to be converted to estradiol by the enzyme aromatase [30]. Thus, expression of 5α-reductase ensures adequate activation of AR in the prostate while limiting the production of the highest affinity ligand for ER.
Nonetheless, strong evidence exists for both the peripheral and intraprostatic production of estrogens in ageing males [14,15]. However, there is an increasing body of evidence supporting the role of androgen metabolites as the main ligand source for ER-dependent signaling in the prostate [18,31]. Specifically, 2 DHT metabolites, 5α-androstane-3α,17β-adiol (3α-Adiol) and 5α-androstane-3β,17β-adiol (3β-Adiol), have been shown to be potent ERβ ligands [31,32]. The importance of these metabolites as ERβ ligands is underscored by the fact that the local concentration of 3β-Adiol in the prostate is one hundred fold higher than that of estradiol [33].
Activation of ERβ in the prostate is ensured by local tissue expression of the enzymes AKR1C2 and AKR1C3, which are responsible for the metabolism of DHT into 3α-Adiol and 3β-Adiol, respectively [34,35]. Analysis of PCa specimens revealed limited expression of enzymes responsible for de novo steroidogenesis, indicating that the conversion of androgenic precursors is the predominant method of controlling the balance in androgenic and estrogenic signaling pathways [35]. These steroid-converting enzymes are of growing interest in PCa. Polymorphisms in the AKR1C enzymes have been correlated with PCa risk [36], and recent endeavors to identify small molecule modulators of these enzymes have been undertaken using modern high-throughput screening techniques [37].
Epithelial-to-mesenchymal transition (EMT) is a crucial early step in the acquisition of a more motile and invasive phenotype, hence permitting cancer cells to migrate and invade surrounding tissues. Loss of the epithelial marker E-cadherin is a known indicator of EMT, which is associated with loss of cell adhesion and a subsequent increase in cell motility [38]. As ERβ ligands, Adiols initiate a signaling pathway that inhibits cell motility by increasing cell adhesion through induction of E-cadherin expression [32,33,39]. An inverse relationship exists between ERβ expression and the progression of PCa to a high Gleason grade [33]. Tissue staining done by Mak et al. demonstrates that E-cadherin expression is directly correlated with ERβ expression in PCa [33]; thus, loss of ERβ expression is directly associated with increased motility and aggressiveness. Recently published work from our laboratory further supports the role of an Adiol-ERβ pathway in inducing expression of E-cadherin in PCa cells, but with the novel indication that this pathway is sensitive to inactivation via oxidation of ERβ by locally produced ROS [23]. Signaling through Adiol-ERβ appears to be one mechanism through which prostate growth is tightly regulated; hence, this pathway presents a putative target for therapeutic intervention.
One of the overarching themes reviewed herein is the crucial role of oxidative stress in altering steroid receptor signaling within the cancerous prostate. We propose that controlling the redox status of the local milieu may serve to limit the aggressiveness of PCa by influencing steroid receptor signaling. The role of the tumor microenvironment can change from inhibitive to permissive simply by paracrine signals generated by the cancer cells in an effort to escape intrinsic control [23]. Cancer cells themselves are capable of generating significant amounts of ROS that further alter the redox status of their microenvironment [50]. While multiple sources of ROS exist within the prostate, COX-2, a pro-inflammatory enzyme whose expression is generally labile and transient, has been shown to be capable of producing ROS as a byproduct of the 2-step oxidation process of arachidonic acid [40]. PCa cells overexpress COX-2, and consistently high levels of COX-2 are associated with worse prognosis [40,41]. Addition of selective pharmacologic inhibitors of COX-2 has been shown to decrease cancer cell growth, induce apoptosis, and decrease tumor size in animal models [40].
Interestingly, it is not simply COX-2 expression by the cancer cells that is responsible for increased aggressiveness. A xenograft of Lewis lung carcinoma cells (positively expressing COX-2) was unable to form a tumor in a COX-2 −/− host, suggesting that stromal COX-2 is necessary for tumor development in this model. Upon further investigation, it was found that the COX-2 −/− fibroblasts were unable to secrete VEGF, an obligatory requirement for tumor formation [42]. Newer studies in PCa from our laboratory suggest that stromal COX-2 is capable of generating sufficient H2O2 to oxidize and inactivate ERβ in nearby cancer cells, ultimately leading to a disinhibition of the inherent motility-suppressing capacity of the prostate stroma on ERβ-expressing PCa cells [23]. In accordance with this molecular basis for stromal COX-2 in PCa, a study by Richardsen et al. showed that high expression of COX-2 in the prostate stroma correlates with decreased disease-specific survival [41]. Not surprisingly, these data have led to clinical trials of Cox-2 inhibitors (i.e. nonsteroidal anti-inflammatory drugs) in PCa, but the results of the trials remain inconclusive and further studies are needed [43,44].
Presumably, another target for therapeutic intervention is to directly target an endogenous regulatory mechanism capable of limiting cancer cell motility. As previously discussed, intraprostatic synthesis of Adiols serves to limit cancer cell motility through ERβ activation. Recent studies have demonstrated that synthetic ERβ ligands can act in a manner similar to endogenously produced Adiol in repressing a gene transcription pathway responsible for inducing an inflammatory response in the microglia [45]. However, while this suggests that synthetic Adiols may represent a potential therapy in the future, additional research is needed to ensure that ERβ is the target receptor.
Future therapies must also be mindful of potential crosstalk between tumor suppressive ERβ and tumor promoting AR. For example, mutated AR such as is often seen in CRPC, is capable of binding 3β-Adiol with a subsequent activation of AR target genes responsible for cancer cell proliferation [46]. Therefore, inactivation of ERβ via oxidation, as our work suggests, could disrupt its crosstalk with AR and tip the balance in favor of AR activation by weak ligands (i.e. Adiols) in vulnerable PCa cells. In this case, a unique cancer-promoting AR gene signature may be influenced by a combination of the steroidogenic capacity and redox status of various components of the tumor microenvironment. Therefore, future therapeutic interventions should seek to maintain ERβ activity in a hostile tumor microenvironment and limit the contributions of AR to PCa progression.
Highlights.
Estrogen receptor beta action limits the growth and migration of prostate cancer cells
Steroid ligands produced in prostatic tissue can activate estrogen receptor beta
TGF-β generates reactive oxygen species that inhibits estrogen receptor beta
Acknowledgments
The authors acknowledge the contributions of Eugenia Cifuentes-Pagano, Roberto Di Maio, Stephen Hammes, Teresa Liu, Patrick Pagano, Guillermo Romero and other members of the DeFranco laboratory for various contributions to this work. DBD was supported by NIH grant R01 DK078394 and MJG by a predoctoral fellowship from the Department of Defense.
Abbreviations
- 3α-Adiol
5α-androstane-3α,17β-adiol,
- 3β-Adiol
5α-androstane-3β,17β-adiol
- AR
androgen receptor
- CRPC
castrate-resistant prostate cancer
- COX-2
cyclooxygenase-2
- DHEA
dehydroepiandrosterone
- DHT
dihydrotestosterone
- EMT
epithelial-to-mesenchymal transition
- ER
estrogen receptor
- PCa
prostate cancer
- ROS
reactive oxygen species
- (TGF-β
transforming growth factor beta
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES CITED
- 1.Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, Bartsch G, Klocker H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994;54:5474–8. [PubMed] [Google Scholar]
- 2.Gioeli D, Ficarro SB, Kwiek JJ, Aaronson D, Hancock M, Catling AD, White FM, Christian RE, Settlage RE, Shabanowitz J, Hunt DF, Weber MJ. Androgen receptor phosphorylation. Regulation and identification of the phosphorylation sites. J Biol Chem. 2002;277:29304–14. doi: 10.1074/jbc.M204131200. [DOI] [PubMed] [Google Scholar]
- 3.Pollak M, Beamer W, Zhang JC. Insulin-like growth factors and prostate cancer. Cancer Metastasis Rev. 1998;17:383–90. doi: 10.1023/a:1006154108619. [DOI] [PubMed] [Google Scholar]
- 4.Wolk A, Mantzoros CS, Andersson SO, Bergstrom R, Signorello LB, Lagiou P, Adami HO, Trichopoulos D. Insulin-like growth factor 1 and prostate cancer risk: a population-based, case-control study. J Natl Cancer Inst. 1998;90:911–5. doi: 10.1093/jnci/90.12.911. [DOI] [PubMed] [Google Scholar]
- 5.Zhou ZX, Lane MV, Kemppainen JA, French FS, Wilson EM. Specificity of ligand-dependent androgen receptor stabilization: receptor domain interactions influence ligand dissociation and receptor stability. Mol Endocrinol. 1995;9:208–18. doi: 10.1210/mend.9.2.7776971. [DOI] [PubMed] [Google Scholar]
- 6.Reebye V, Frilling A, Habib NA, Mintz PJ. Intracellular adaptor molecules and AR signalling in the tumour microenvironment. Cell Signal. 2011;23:1017–21. doi: 10.1016/j.cellsig.2010.11.019. [DOI] [PubMed] [Google Scholar]
- 7.Bare RL, Torti FM. Endocrine therapy of prostate cancer. Cancer Treat Res. 1998;94:69–87. doi: 10.1007/978-1-4615-6189-7_5. [DOI] [PubMed] [Google Scholar]
- 8.Fluchter SH, Weiser R, Gamper C. The role of hormonal treatment in prostate cancer. Recent Results Cancer Res. 2007;175:211–37. doi: 10.1007/978-3-540-40901-4_13. [DOI] [PubMed] [Google Scholar]
- 9.Mezo G, Manea M, Szabi I, Vincze B, Kovacs M. New derivatives of GnRH as potential anticancer therapeutic agents. Curr Med Chem. 2008;15:2366–79. doi: 10.2174/092986708785909157. [DOI] [PubMed] [Google Scholar]
- 10.Cai C, Balk S. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr Relat Cancer. 2011 doi: 10.1530/ERC-10-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Attard G, Richards J, de Bono JS. New strategies in metastatic prostate cancer: targeting the androgen receptor signaling pathway. Clin Cancer Res. 2011;17:1649–57. doi: 10.1158/1078-0432.CCR-10-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niu Y, Altuwaijri S, Lai KP, Wu CT, Ricke WA, Messing EM, Yao J, Yeh S, Chang C. Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc Natl Acad Sci U S A. 2008;105:12182–7. doi: 10.1073/pnas.0804700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knudsen KE, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab. 2010;21:315–24. doi: 10.1016/j.tem.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ellem SJ, Schmitt JF, Pedersen JS, Frydenberg M, Risbridger GP. Local aromatase expression in human prostate is altered in malignancy. J Clin Endocrinol Metab. 2004;89:2434–41. doi: 10.1210/jc.2003-030933. [DOI] [PubMed] [Google Scholar]
- 15.Risbridger GP, Ellem SJ, McPherson SJ. Estrogen action on the prostate gland: a critical mix of endocrine and paracrine signaling. J Mol Endocrinol. 2007;39:183–8. doi: 10.1677/JME-07-0053. [DOI] [PubMed] [Google Scholar]
- 16.McPherson SJ, Wang H, Jones ME, Pedersen J, Iismaa TP, Wreford N, Simpson ER, Risbridger GP. Elevated androgens and prolactin in aromatase-deficient mice cause enlargement, but not malignancy, of the prostate gland. Endocrinology. 2001;142:2458–67. doi: 10.1210/endo.142.6.8079. [DOI] [PubMed] [Google Scholar]
- 17.Bianco JJ, McPherson SJ, Wang H, Prins GS, Risbridger GP. Transient neonatal estrogen exposure to estrogen-deficient mice (aromatase knockout) reduces prostate weight and induces inflammation in late life. Am J Pathol. 2006;168:1869–78. doi: 10.2353/ajpath.2006.050623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Linja MJ, Savinainen KJ, Tammela TL, Isola JJ, Visakorpi T. Expression of ERalpha and ERbeta in prostate cancer. Prostate. 2003;55:180–6. doi: 10.1002/pros.10242. [DOI] [PubMed] [Google Scholar]
- 19.Pelletier G. Expression of steroidogenic enzymes and sex-steroid receptors in human prostate. Best Pract Res Clin Endocrinol Metab. 2008;22:223–8. doi: 10.1016/j.beem.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 20.Adlercreutz CH, Goldin BR, Gorbach SL, Hockerstedt KA, Watanabe S, Hamalainen EK, Markkanen MH, Makela TH, Wahala KT, Adlercreutz T. Soybean phytoestrogen intake and cancer risk. J Nutr. 1995;125:757S–770S. doi: 10.1093/jn/125.3_Suppl.757S. [DOI] [PubMed] [Google Scholar]
- 21.Adlercreutz H, Mazur W, Bartels P, Elomaa V, Watanabe S, Wahala K, Landstrom M, Lundin E, Bergh A, Damber JE, Aman P, Widmark A, Johansson A, Zhang JX, Hallmans G. Phytoestrogens and prostate disease. J Nutr. 2000;130:658S–9S. doi: 10.1093/jn/130.3.658S. [DOI] [PubMed] [Google Scholar]
- 22.Kumar B, Koul S, Khandrika L, Meacham RB, Koul HK. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008;68:1777–85. doi: 10.1158/0008-5472.CAN-07-5259. [DOI] [PubMed] [Google Scholar]
- 23.Grubisha MJ, Cifuentes ME, Hammes SR, Defranco DB. A local paracrine and endocrine network involving TGFbeta, Cox-2, ROS, and estrogen receptor beta influences reactive stromal cell regulation of prostate cancer cell motility. Mol Endocrinol. 2012;26:940–54. doi: 10.1210/me.2011-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yau C, Benz CC. Genes responsive to both oxidant stress and loss of estrogen receptor function identify a poor prognosis group of estrogen receptor positive primary breast cancers. Breast Cancer Res. 2008;10:R61. doi: 10.1186/bcr2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whittal RM, Benz CC, Scott G, Semyonov J, Burlingame AL, Baldwin MA. Preferential oxidation of zinc finger 2 in estrogen receptor DNA-binding domain prevents dimerization and, hence, DNA binding. Biochemistry. 2000;39:8406–17. doi: 10.1021/bi000282f. [DOI] [PubMed] [Google Scholar]
- 26.Atsriku C, Scott GK, Benz CC, Baldwin MA. Reactivity of zinc finger cysteines: chemical modifications within labile zinc fingers in estrogen receptor. J Am Soc Mass Spectrom. 2005;16:2017–26. doi: 10.1016/j.jasms.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 27.Liang X, Lu B, Scott GK, Chang CH, Baldwin MA, Benz CC. Oxidant stress impaired DNA-binding of estrogen receptor from human breast cancer. Mol Cell Endocrinol. 1998;146:151–61. doi: 10.1016/s0303-7207(98)00161-0. [DOI] [PubMed] [Google Scholar]
- 28.Andriole G, Bruchovsky N, Chung LW, Matsumoto AM, Rittmaster R, Roehrborn C, Russell D, Tindall D. Dihydrotestosterone and the prostate: the scientific rationale for 5alpha-reductase inhibitors in the treatment of benign prostatic hyperplasia. J Urol. 2004;172:1399–403. doi: 10.1097/01.ju.0000139539.94828.29. [DOI] [PubMed] [Google Scholar]
- 29.Nacusi LP, Tindall DJ. Targeting 5alpha-reductase for prostate cancer prevention and treatment. Nat Rev Urol. 2011;8:378–84. doi: 10.1038/nrurol.2011.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swerdloff RS, Wang C. Dihydrotestosterone: a rationale for its use as a nonaromatizable androgen replacement therapeutic agent. Baillieres Clin Endocrinol Metab. 1998;12:501–6. doi: 10.1016/s0950-351x(98)80267-x. [DOI] [PubMed] [Google Scholar]
- 31.Dondi D, Piccolella M, Biserni A, Della Torre S, Ramachandran B, Locatelli A, Rusmini P, Sau D, Caruso D, Maggi A, Ciana P, Poletti A. Estrogen receptor beta and the progression of prostate cancer: role of 5alpha-androstane-3beta,17beta-diol. Endocr Relat Cancer. 2010;17:731–42. doi: 10.1677/ERC-10-0032. [DOI] [PubMed] [Google Scholar]
- 32.Guerini V, Sau D, Scaccianoce E, Rusmini P, Ciana P, Maggi A, Martini PG, Katzenellenbogen BS, Martini L, Motta M, Poletti A. The androgen derivative 5alpha-androstane-3beta,17beta-diol inhibits prostate cancer cell migration through activation of the estrogen receptor beta subtype. Cancer Res. 2005;65:5445–53. doi: 10.1158/0008-5472.CAN-04-1941. [DOI] [PubMed] [Google Scholar]
- 33.Mak P, Leav I, Pursell B, Bae D, Yang X, Taglienti CA, Gouvin LM, Sharma VM, Mercurio AM. ERbeta impedes prostate cancer EMT by destabilizing HIF-1alpha and inhibiting VEGF-mediated snail nuclear localization: implications for Gleason grading. Cancer Cell. 2010;17:319–32. doi: 10.1016/j.ccr.2010.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ji Q, Chang L, VanDenBerg D, Stanczyk FZ, Stolz A. Selective reduction of AKR1C2 in prostate cancer and its role in DHT metabolism. Prostate. 2003;54:275–89. doi: 10.1002/pros.10192. [DOI] [PubMed] [Google Scholar]
- 35.Hofland J, van Weerden WM, Dits NF, Steenbergen J, van Leenders GJ, Jenster G, Schroder FH, de Jong FH. Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Res. 2010;70:1256–64. doi: 10.1158/0008-5472.CAN-09-2092. [DOI] [PubMed] [Google Scholar]
- 36.Park JY, Tanner JP, Sellers TA, Huang Y, Stevens CK, Dossett N, Shankar RA, Zachariah B, Heysek R, Pow-Sang J. Association between polymorphisms in HSD3B1 and UGT2B17 and prostate cancer risk. Urology. 2007;70:374–9. doi: 10.1016/j.urology.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 37.Brozic P, Turk S, Rizner TL, Gobec S. Inhibitors of aldo-keto reductases AKR1C1–AKR1C4. Curr Med Chem. 2011;18:2554–65. doi: 10.2174/092986711795933713. [DOI] [PubMed] [Google Scholar]
- 38.Guarino M, Rubino B, Ballabio G. The role of epithelial-mesenchymal transition in cancer pathology. Pathology. 2007;39:305–18. doi: 10.1080/00313020701329914. [DOI] [PubMed] [Google Scholar]
- 39.Thomas C, Gustafsson JA. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer. 2011;11:597–608. doi: 10.1038/nrc3093. [DOI] [PubMed] [Google Scholar]
- 40.Kirschenbaum A, Liu X, Yao S, Levine AC. The role of cyclooxygenase-2 in prostate cancer. Urology. 2001;58:127–31. doi: 10.1016/s0090-4295(01)01255-9. [DOI] [PubMed] [Google Scholar]
- 41.Richardsen E, Uglehus RD, Due J, Busch C, Busund LT. COX-2 is overexpressed in primary prostate cancer with metastatic potential and may predict survival. A comparison study between COX-2, TGF-beta, IL-10 and Ki67. Cancer Epidemiol. 2010;34:316–22. doi: 10.1016/j.canep.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 42.Williams CS, Tsujii M, Reese J, Dey SK, DuBois RN. Host cyclooxygenase-2 modulates carcinoma growth. J Clin Invest. 2000;105:1589–94. doi: 10.1172/JCI9621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bucher C, Jordan P, Nickeleit V, Torhorst J, Mihatsch MJ. Relative risk of malignant tumors in analgesic abusers. Effects of long-term intake of aspirin. Clin Nephrol. 1999;51:67–72. [PubMed] [Google Scholar]
- 44.Norrish AE, Jackson RT, McRae CU. Non-steroidal anti-inflammatory drugs and prostate cancer progression. Int J Cancer. 1998;77:511–5. doi: 10.1002/(sici)1097-0215(19980812)77:4<511::aid-ijc6>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 45.Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK. An ADIOL-ERbeta-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell. 2011;145:584–95. doi: 10.1016/j.cell.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mizokami A, Koh E, Fujita H, Maeda Y, Egawa M, Koshida K, Honma S, Keller ET, Namiki M. The adrenal androgen androstenediol is present in prostate cancer tissue after androgen deprivation therapy and activates mutated androgen receptor. Cancer Res. 2004;64:765–71. doi: 10.1158/0008-5472.can-03-0130. [DOI] [PubMed] [Google Scholar]