

Abstract
Purpose
Increasing evidence indicates that enhanced intratumoral androgen synthesis contributes to prostate cancer progression after androgen deprivation therapy. This phase II study was designed to assess responses to blocking multiple steps in androgen synthesis with inhibitors of CYP17A1 (ketoconazole) and type I and II 5α-reductases (dutasteride) in patients with castration-resistant prostate cancer (CRPC).
Experimental Design
Fifty-seven men with CRPC were continued on gonadal suppression and treated with ketoconazole (400 mg thrice daily), hydrocortisone (30 mg/AM, 10 mg/PM), and dutasteride (0.5 mg/d).
Results
Prostate-specific antigen response rate (≥50% decline) was 56% (32 of 57; 95% confidence interval, 42.4–69.3%); the median duration of response was 20 months. In patients with measurable disease, 6 of 20 (30%) responded by the Response Evaluation Criteria in Solid Tumors. Median duration of treatment was 8 months; 9 patients remained on therapy with treatment durations censored at 18 to 32 months. Median time to progression was 14.5 months. Grade 3 toxicities occurred in 32% with only one reported grade 4 (thrombosis) toxicity. Dehydroepiandrosterone sulfate declined by 89%, androstenedione by 56%, and testosterone by 66%, and dihydrotestosterone declined to below detectable levels compared with baseline levels with testicular suppression alone. Median baseline levels and declines in dehydroepiandrosterone sulfate, androstenedione, testosterone, and dihydrotestosterone were not statistically different in the responders versus nonresponders, and hormone levels were not significantly increased from nadir levels at relapse.
Conclusion
The response proportion to ketoconazole, hydrocortisone, and dutasteride was at least comparable with previous studies of ketoconazole alone, whereas time to progression was substantially longer. Combination therapies targeting multiple steps in androgen synthesis warrant further investigation. (Clin Cancer Res 2009;15(22):7099–105)
Prostate cancer that progresses after androgen deprivation therapy (ADT), termed castration-resistant prostate cancer (CRPC), expresses androgen receptor (AR) and multiple androgen-regulated genes at high levels (including PSA and TMPRSS2:ERG fusion genes), indicating that AR transcriptional activity has been reactivated despite castrate serum androgens levels (1–3). Mechanisms that may contribute to this AR reactivation include increased AR expression (increased AR mRNA in most patients and AR gene amplification in ~30%; ref. 4), AR mutations (primarily in patients treated with an AR antagonist; refs. 5, 6), increased activity of transcriptional coactivator proteins (7, 8), and stimulation of kinases that directly or indirectly enhance AR responses to low androgen levels (9–12).
A further mechanism contributing to tumor progression after ADT is increased intratumoral androgen synthesis. CRPC tumors have increased expression of enzymes mediating testosterone and dihydrotestosterone (DHT) synthesis from weak adrenal androgens (dehydroepiandrosterone and androstenedione) and may also upregulate enzymes including CYP17A1 that are required for de novo steroid synthesis (3, 13, 14). Consistent with increased intratumoral androgen synthesis in CRPC, androgen levels in the prostates of men who recur locally after ADT are comparable with levels in the prostates of eugonadal men (15–17). Moreover, testosterone levels in metastatic CRPC samples are actually higher than in prostate before castration (13). Significantly, high intratumoral androgen levels, in addition to reactivating AR, may render tumor cells relatively resistant to available weak competitive AR antagonists and contribute to the modest efficacy of these antagonists when used initially in combination with castration (combined androgen blockade; ref. 18) or as secondary hormonal therapy in CRPC (19, 20).
The contribution of androgens produced by the adrenal glands to CRPC was suggested in early adrenalectomy studies (21). Ketoconazole, which inhibits several cytochrome P450 enzymes including CYP17A1 that is required for adrenal androgen synthesis, has reported response rates in CRPC ranging from 20% to 75% (22). The response rate in the largest study of ketoconazole/hydrocortisone (with simultaneous or sequential antiandrogen withdrawal) was 32% when used after antiandrogen withdrawal (23). Interestingly, this study found a positive correlation between responses and pre-therapy levels of androstenedione, and the mean adrenal androgen levels were partially restored at disease progression (24). These observations suggest that responses might be improved with more effective androgen synthesis inhibitors, and encouraging response rates (>50%) have been reported in phase I/II clinical trials of a more potent CYP17A1 inhibitor, abiraterone (22, 25).
Although CYP17A1 inhibitors can markedly reduce levels of circulating and presumably intratumoral androgen precursors, tumor cells may still convert any available androstenedione to testosterone (mediated by the enzyme AKR1C3) and then to the higher-affinity ligand DHT (mediated by type I or II 5α-reductase). In normal prostate, testosterone is reduced to DHT primarily by the type II 5α-reductase (SRD5A2). Selective inhibition of type II 5α-reductase with finasteride can decrease prostate cancer incidence, but this drug does not have clear activity in CRPC (26). Studies from several groups have shown that expression of the type I 5α-reductase (SRD5A1) is increased in primary prostate cancer, and we found that SRD5A1 expression was further increased in CRPC (3, 27). A dual inhibitor of type I and II 5α-reductases, dutasteride, is an effective treatment for benign prostatic hyperplasia and is under investigation for prostate cancer prevention, but its efficacy in CRPC has not been determined (28).
Based on these observations and available drugs, we hypothesized that combined inhibition of CYP17A1 with ketoconazole and inhibition of type I and II 5α-reductases with dutasteride would be an effective therapy in CRPC. We present here results of a phase II trial of combined treatment with ketoconazole, hydrocortisone, and dutasteride (KHAD) in CRPC. The trial was designed as an exploratory study to determine whether addition of dutasteride to standard ketoconazole/hydrocortisone would improve responses to at least 50% from the 32% response in the Cancer and Leukemia Group B 9583 study (23) and would be therefore worthy of further study. Additional endpoints included time to progression (TTP) and correlations between androgen levels and responses.
Materials and Methods
Patients and eligibility
This open-label, single-arm multicenter phase II study was initiated at the Dana-Farber/Harvard Cancer Center (Beth Israel Deaconess Medical Center and Dana-Farber Cancer Institute) and conducted through the Department of Defense Prostate Cancer Clinical Trials Consortium (trial registration number NCT00673127). From June 2005 to April 2007, 57 patients were consented and started on therapy. Participating institutions included Dana-Farber/Harvard Cancer Center (n = 26), Sunnybrook Health Science Centre (n = 10), Oregon Health and Science University (n = 8), M. D. Anderson Cancer Center (n = 8), and Johns Hopkins University (n = 5). The institutional review board of each institution approved the trial.
Eligibility included progressive CRPC, defined as a prostate-specific antigen (PSA) increase over baseline of >25% or >5 ng/mL, or new lesions on bone/computed tomographic scan after conventional androgen deprivation and antiandrogen withdrawal. Metastatic disease was not required. Additional criteria included ongoing gonadal androgen ablation with serum testosterone <0.5 ng/mL, PSA ≥2 ng/mL, no prior therapy with ketoconazole or corticosteroids for prostate cancer, and Eastern Cooperative Oncology Group performance status of 0 to 2. Prior chemotherapy was allowed. Patients taking drugs that may prolong QT intervals or known to be narrow therapeutic index CYP3A4 substrates were excluded.
The treatment was ketoconazole 400 mg orally thrice daily, hydrocortisone (30 mg/AM and 10 mg/PM), and dutasteride (0.5 mg/d). Dose modifications for toxicity were specified. Patients were evaluated every 4 weeks, with history, physical examination, and laboratory analysis including liver function tests and PSA. Serum for hormone measurements was obtained every 4 weeks for the first 12 weeks and then every 12 weeks until progression (measured in duplicate by RIA, Diagnostic Systems Laboratories). Measurable disease was evaluated by computed tomography and bone metastasis by bone scan every 12 weeks. Toxicity was graded according to the National Cancer Institute Common Toxicity Criteria version 3.0.
Endpoints
The primary endpoint was PSA response defined as a decline of at least 50% from baseline confirmed by a second measurement at least 4 weeks later; the reference for these declines was measured within 2 weeks before starting therapy. PSA progression was defined according to PSA Working Group Criteria (29). Measurable disease response and progression were evaluated according to the Response Evaluation Criteria in Solid Tumors. Progressive nonmeasurable disease was defined as two or more new lesions on bone scan, appearance of new nonbony metastases, or development of an indication for radiation therapy. TTP was defined from the date of treatment initiation until the date that PSA progression criteria were first met or the date of measurable or nonmeasurable disease progression; otherwise, it was censored at the date of the last PSA measurement without evidence of disease progression. Among patients who achieved a ≥50% PSA decline, PSA response duration was defined from the date of the first 50% PSA decline until the date of PSA or disease progression or was censored at the date of the last PSA measurement without progression. Hormone levels were compared using Wilcoxon rank-sum tests.
Statistical considerations
Enrollment proceeded in a two-stage design to differentiate a response rate of ≥50% from a response rate of ≤32% with type I and II error of 0.10. On assessment that ≥8 of the initial 23 patients had responded, enrollment continued to a total of 57 patients. The PSA response rate and exact binomial 95% confidence interval (95% CI) are reported. All patients, regardless of their disease evaluations, are included in the assessment of response. The PSA response duration and TTP distributions were summarized using the Kaplan-Meier method with 95% CI.
Results
Patient characteristics
Patient and disease characteristics are in Table 1. The mean age at enrollment was 68 years, and the median duration of primary ADT was 3.4 years. Median baseline PSA was 24 ng/mL (range, 3.7–2,740 ng/mL), and median PSA doubling time before entry was 2.4 months. Measurable disease was present in 35% of patients, and 70% had bone metastases. Prior treatments included bicalutamide in 89% of patients and chemotherapy in 5% of patients.
Table 1.
Patient and disease characteristics
| Characteristics | n (%) of patients or median [range] of values |
|---|---|
| No. patients | 57 (100) |
| Non-Caucasian | 5 (9) |
| Hispanic or Latino | 1 (2) |
| At initial prostate cancer diagnosis | |
| Age (y) | 61 [44–80] |
| PSA (ng/mL), n = 46 | 16 [3–1,549] |
| Gleason score | |
| 4–6 | 14 (25) |
| 7 | 16 (28) |
| 8–9 | 20 (35) |
| Unknown | 7 (14) |
| Prior treatments | |
| Duration of primary ADT (y), n = 54 | 3.4 [0.2–15.5] |
| LHRH agonist | 51 (89) |
| Orchiectomy | 4 (7) |
| Antiandrogens (any) | 52 (91) |
| Bicalutamide | 51 (89) |
| Nilutamide | 6 (11) |
| Flutamide | 5 (9) |
| Finasteride | 2 (4) |
| Estrogens (including estramustine) | 8 (14) |
| Chemotherapy | 3 (5) |
| At registration | |
| Age (y) | 68 [48–90] |
| Years since diagnosis | 6.6 [0.3–20] |
| ECOG performance status 1 | 16 (28) |
| PSA (ng/mL) | 24 [3.7–2,740] |
| PSA doubling time (mo), n = 51* | 2.4 [1–24] |
| Alkaline phosphatase (units/L) | 84 [30–527] |
| Hemoglobin (g/dL) | 13.1 [10.7–15.6] |
| Type of disease | |
| Measurable disease | 20 (35) |
| Measurable only | 6 (11) |
| Measurable and nonmeasurable | 14 (25) |
| Nonmeasurable disease only | 28 (49) |
| PSA only | 9 (16) |
| Sites of disease (multiple) | |
| Viscera | 6 (11) |
| Soft tissue | 6 (11) |
| Lymph nodes | 23 (40) |
| Bone | 40 (70) |
PSA doubling time was calculated as natural log2 divided by the slope of the linear regression of natural log of PSA versus time using the three PSA values that confirmed progressive CRPC by PSA progression before study entry.
Abbreviation: ECOG, Eastern Cooperative Oncology Group.
Treatment activity
The PSA response rate (confirmed ≥50% decline in PSA from baseline) was 56% (32 of 57; 95% CI, 42.4–69.3%). PSA declined by ≥80% in 27 of 57 (47%) patients and by ≥90% in 16 of 57 (28%) patients (Fig. 1). Among the 32 PSA responders, the median PSA response duration was 20 months (95% CI, 13.5 to undetermined upper limit); 9 patients were continuing on treatment without progression at the time of this report with treatment durations censored at 18 to 32 months. PSA response rates according to type of disease at baseline (measurable, nonmeasurable, PSA only) are shown in Table 2 and were similar in all groups.
Fig. 1.
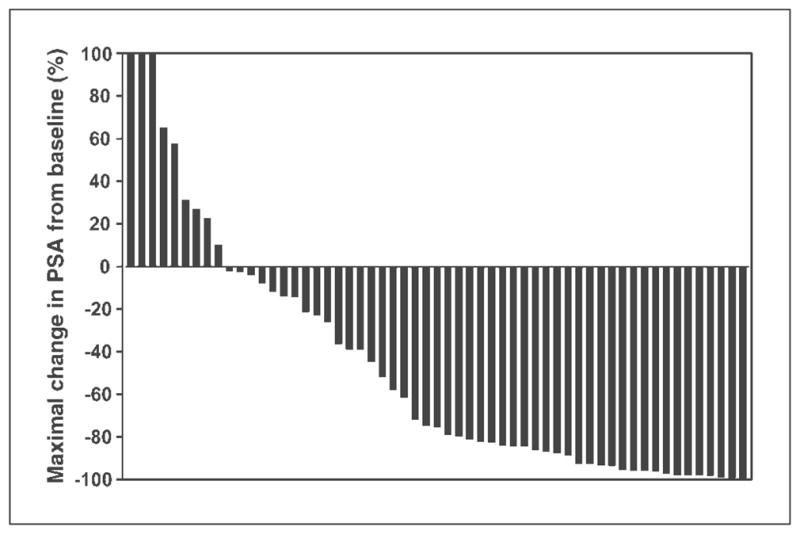
Maximal change in PSA from baseline among 57 CRPC patients treated with KHAD. Y axis is truncated at +100%, but 3 patients had ≥100% increase in PSA from baseline without decline. In total, 9 patients did not have any decline in PSA.
Table 2.
PSA response rates by type of disease at baseline
| Enrolled, n | PSA response, n (%) | |
|---|---|---|
| Measurable disease ± nonmeasurable disease | 20 | 11 (55) |
| Measurable only | 6 | 3 (50) |
| Measurable and nonmeasurable | 14 | 8 (57) |
| Nonmeasurable only | 28 | 14 (50) |
| PSA only | 9 | 7 (78) |
| Overall | 57 | 32 (56) |
Among the 20 patients with measurable disease at baseline, 6 (30%) had complete or partial responses by the Response Evaluation Criteria in Solid Tumors, 5 (25%) had stable disease, and 1 had progressive disease. The remaining 8 patients discontinued participation in the study due to PSA progression (3 patients) or for other reasons (5 patients) before measurable disease responses could be assessed. The 1 patient with a complete response had baseline PSA of 2,740 ng/mL, a 1.5 cm lymph node metastasis, and bone metastases. The target lymph node metastasis was not present on 6-, 9-, 12-, or 15-month computed tomographic scans and nadir PSA was 2.1 ng/mL at 8 months. This patient stopped treatment after ~15 months by choice with an increasing PSA of 6 ng/mL.
There were 40 patients with bone metastases at baseline. Eighteen (40%) had stable disease (0 or 1 new lesions on bone scan) throughout the therapy, and 15 of these 18 had PSA responses. One patient had stable bone metastases for >1 year but then had bone progression resulting in treatment cessation. Eight patients had progression of bone metastases at the first bone scan (2 of these 8 patients had PSA responses within those first 3 months of treatment). The remaining 13 patients came off treatment within 3 months and did not have a follow-up bone scan (3 of these 13 had PSA progression).
The median time to disease progression was 14.5 months (95% CI, 11.0 to upper limit undetermined; Fig. 2). The median duration of treatment was 8 months, ranging from <1 to >32 months. Forty percent (22 of 57) continued treatment for ≥1 year. Ten patients stopped treatment with the reason specified as toxicity or side effects. An additional 10 patients stopped treatment by physician discretion or patient choice. One patient stopped treatment at 5 weeks for pain management and 1 patient stopped at 8 months for nerve root compression.
Fig. 2.
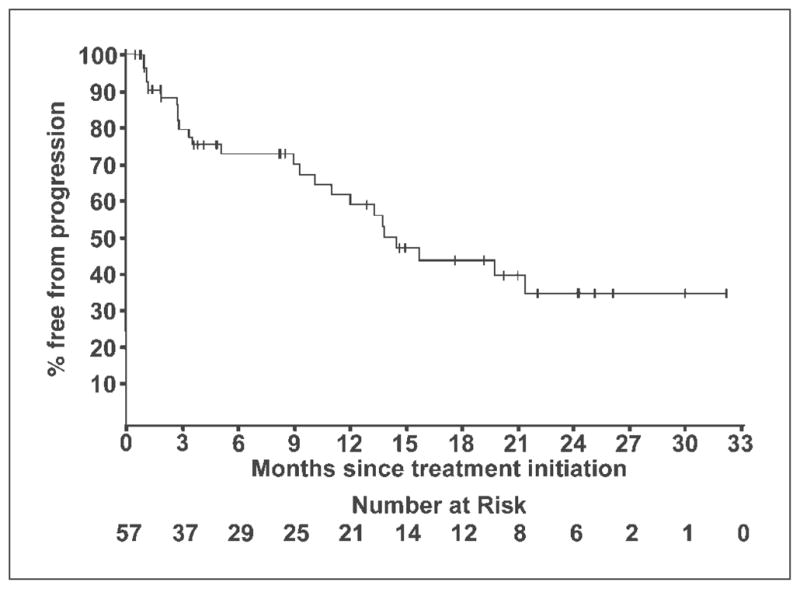
TTP among 57 CRPC patients treated with KHAD.
Toxicity
Toxicities were reported among all patients. Excluding impotence, 32% (18 of 57) of patients experienced at least one grade 3 or 4 toxicity. Grade 3 hypertension occurred in 4 patients, hyperglycemia in 3 patients, hypokalemia in 2 patients, and elevated alanine aminotransferase (serum glutamic oxaloacetic transaminase) in 2 patients. Grade 3 lymphopenia, rash, hot flashes, nausea, bladder hemorrhage, infection, aspartate aminotransferase (serum glutamic pyruvate transaminase), creatinine, nonneuropathic generalized weakness, cognitive disturbance, pain, dyspnea, and urinary frequency/urgency were reported each for 1 patient. There was one grade 4 thrombosis.
Hormone data
Baseline serum samples were available from 41 patients, and there was at least one subsequent on-therapy serum sample from 33 of these 41 patients. Median dehydroepiandrosterone sulfate (DHEAS) at baseline was 599 ng/mL and fell to 65 ng/mL after ~1 month on therapy (median, 89% decline; Table 3). Baseline median androstenedione was 0.84 ng/mL and dropped by a median of 56% to 0.22 ng/mL at 1 month. Baseline median testosterone was 0.37 ng/mL, and at 1 month, there was a 66% drop to a median of 0.13 ng/mL. Baseline median DHT was 2.6 pg/mL. This declined after 1 month in all patients, with the median at 1 month being below the level of sensitivity (<2.0 pg/mL). There was no further significant decline in any of these hormones after 2 to 3 months of therapy.
Table 3.
Hormone levels over time among all patients
| Hormone | Baseline value | Month 1
|
Month 2
|
Month 3
|
|||
|---|---|---|---|---|---|---|---|
| Value | Percentage change from baseline | Value | Percentage change from baseline | Value | Percentage change from baseline | ||
| Patients, n | 41 | 33 | 30 | 28 | 24 | 28 | 24 |
| Androstenedione (ng/mL) | 0.84 (0.61, 1.10) | 0.22 (0.20, 0.36) | −56 (−75, −43) | 0.27 (0.20, 0.32) | −68 (−74, −49) | 0.25 (0.20, 0.31) | −58 (−77, −49) |
| DHEAS (ng/mL) | 599 (340, 1206) | 65 (36, 125) | −89 (−94, −70) | 65 (37, 106) | −92 (−94, −77) | 65 (35, 100) | −90 (−94, −81) |
| Testosterone (ng/mL) | 0.37 (0.26, 0.47) | 0.13 (0.10, 0.15) | −66 (−72, −55) | 0.11 (0.9, 0.14) | −68 (−73, −54) | 0.12 (0.10, 0.19) | −64 (−72, −45) |
| DHT* (pg/mL) | 2.6 (<2.0, 3.8) | <2.0 (<2.0, <2.0) | |||||
NOTE: Median and interquartile range (25th, 75th percentiles).
For DHT, n = 33 at baseline, n = 32 at month 1, and n = 29 for percent change.
Baseline median levels of androstenedione, testosterone, and DHT were similar in the patients who responded to therapy (n = 20) versus the nonresponders (n = 21; Table 4). Baseline DHEAS was higher in the responders (840 ng/mL) versus the nonresponders (480 ng/mL), but this was not statistically significant. Median decline in DHEAS after 1 month of therapy was also greater in the responders (93%) versus the nonresponders (85%), but this difference did not reach significance (Table 4). The declines in androstenedione, testosterone, and DHT were comparable in the responders versus nonresponders. Among the responding patients who had progressed at the time of this analysis, paired serum samples after 1 month on therapy and at the time of progression were available from 15 cases. There was no significant increase in the median ratio at progression versus 1 month for DHEAS (1.17; 95% CI, 0.71–1.92), androstenedione (1.11; 95% CI, 0.65–1.92), testosterone (1.34; 95% CI, 0.89–1.99), or DHT (1.11; 95% CI, 0.66–1.82).
Table 4.
Hormone levels at baseline and percentage change at month 1 according to PSA response at 3 months
| Hormone | Baseline
|
Percentage change from baseline to month 1
|
||||
|---|---|---|---|---|---|---|
| PSA responders | PSA nonresponders | P* | PSA responders | PSA nonresponders | P* | |
| Patients, n | 20 | 21 | 14 | 16 | ||
| Androstenedione (ng/mL) | 0.82 (0.6, 1.18) | 0.85 (0.63, 0.98) | 0.98 | −58 (−73, −43) | −56 (−77, −45) | 0.82 |
| DHEAS (ng/mL) | 840 (397, 1,145) | 480 (311, 1,491) | 0.56 | −93 (−95, −85) | −85 (−91, −61) | 0.1 |
| Testosterone (ng/mL) | 0.33 (0.26, 0.50) | 0.39 (0.23, 0.47) | 0.91 | −66 (−74, −56) | −61 (−70, −49) | 0.29 |
| DHT† (pg/mL) | 2.3 (<2.0, 3.7) | 3 (<2.0, 4.0) | 0.63 | |||
NOTE: Median and interquartile range (25th, 75th percentiles).
P value by Wilcoxon rank-sum test comparing PSA responders versus PSA nonresponders.
For DHT, at baseline, n = 16 and 17 for PSA responders and nonresponders, respectively, and n = 16 and 15 for percent change from baseline to month 1, respectively.
Discussion
We conducted an exploratory phase II study to assess the efficacy of treatment with ketoconazole/hydrocortisone in combination with dutasteride in men with CRPC. The largest previous study of ketoconazole (Cancer and Leukemia Group B 9583) analyzed responses to ketoconazole/hydrocortisone given concurrently or subsequent to antiandrogen withdrawal (response rates of 27% and 32%, respectively), so the current study was powered to determine whether the response was >32% (23). Although response rates to ketoconazole in other smaller studies have ranged from 20% to 75% (22), the 56% response rate to KHAD indicates that 5α-reductase inhibition by dutasteride may enhance the response rate to ketoconazole/hydrocortisone. In contrast to the response rate, response durations in this study were markedly longer than those reported previously for ketoconazole/hydrocortisone. The median duration of response was 20 months, and 9 of the 32 responding patients had not yet progressed at the time of this analysis (at durations of 18–32 months). Among all patients, median TTP was 14.5 months. This is markedly longer than the median time to PSA progression of 8.6 months reported in Cancer and Leukemia Group B 9583 (23) and longer than the median response durations of between 3.3 and 9 months in seven other reported ketoconazole trials (22). It is also longer than the median TTP of 7.5 months in phase II studies of abiraterone (see below; ref. 30).
As expected, KHAD caused a median decline of ~90% in serum DHEAS levels and declines of ~50% to 70% in serum androstenedione and testosterone. Baseline median levels of androstenedione and testosterone were comparable among responders and nonresponders, whereas median DHEAS was higher in the responders (840 versus 480 ng/mL, respectively), although this difference was not statistically significant. In contrast, higher baseline androstenedione and DHEAS levels were correlated with responses to ketoconazole/hydrocortisone plus antiandrogen withdrawal in the Cancer and Leukemia Group B 9583 study, although only the androstenedione association was significant. A further finding in Cancer and Leukemia Group B 9583 was that median levels of DHEAS and androstenedione, which had declined after 1 month of therapy, were increased relative to nadir levels at the time of progression. We did not observe a consistent increase in any of the measured hormones at relapse, but samples from patients who are still responding remain to be examined.
DHT also declined in all patients, but basal levels were already at the limit of sensitivity and the median level after 1 month was below the sensitivity of the assay. Previous studies have not examined the effects of ketoconazole/hydrocortisone alone on DHT in castrate men, so we cannot determine the extent to which DHT levels were further suppressed by dutasteride or assess correlations between declines in serum DHT and responses. Moreover, concentrations of DHT (and of testosterone) in the tumor are likely higher than serum levels and may more closely correlate with responses (13, 16, 17).
Abiraterone is a more potent and specific CYP17A1 inhibitor than ketoconazole, with phase I studies showing >95% declines in serum testosterone to <0.01 ng/mL (25). Nonetheless, despite this more marked decrease in serum testosterone levels, the reported response rate to abiraterone in phase I/II studies (>50% PSA decline in 67% and >90% PSA decline in 19%) appears comparable with the KHAD response rate (25, 30). Moreover, as noted above, median time to PSA progression on abiraterone was 7.5 versus 14.5 months for KHAD. These observations suggest that intratumoral conversion of weak androgens to testosterone and DHT may still be contributing to treatment resistance and relapse in abiraterone-treated patients. In contrast, the addition of dutasteride to ketoconazole, by blocking intratumoral conversion of testosterone to DHT, may result in extremely low intratumoral DHT levels that compensate for the somewhat higher levels of residual testosterone. It also should be noted that dutasteride will presumably increase the aromatization of testosterone to estradiol and decrease the levels of DHT metabolites that may be estrogen receptor-β ligands, although the net effect of this altered testosterone metabolism on tumor growth is uncertain. In any case, the hypothesis that intratumoral DHT synthesis contributes to abiraterone resistance should be tested in a randomized phase II trial.
Early studies employed adrenalectomy or hypophysectomy as therapies for CRPC, with about one-third of patients having objective responses in measurable disease and the majority having subjective improvement (21). Adrenalectomy clearly ablates adrenal androgen synthesis, but there are likely other sources of weak androgens, including CRPC cells that may express increased levels of CYP17A1, although it remains to be established whether CRPC cells synthesize substantial levels of androgens de novo from cholesterol (13, 14). In any case, we suggest that adrenalectomy does not reflect maximal androgen deprivation and that androgen levels (particularly intratumoral levels) may be further reduced by more potent inhibitors of androgen synthesis or by combinations of inhibitors that block at multiple steps.
In summary, this study indicates that dutasteride may enhance the response rate to ketoconazole/hydrocortisone and substantially increases the duration of response and overall TTP. This result supports the conclusion that DHT synthesis by the type I 5α-reductase, which is increased in CRPC, contributes to prostate cancer survival and progression after ADT. Further randomized trials of 5α-reductase inhibitors in conjunction with ketoconazole or more potent CYP17A1 inhibitors are warranted. Moreover, further efforts should be made to develop inhibitors of other enzymes mediating androgen synthesis. Finally, it will clearly be important to determine whether these efforts to more effectively suppress androgen synthesis and AR activity lead to improved survival.
Translational Relevance.
Androgen receptor is reactivated in prostate cancer that relapses after androgen deprivation therapy [castration-resistant prostate cancer (CRPC)], with one mechanism being increased expression of enzymes (AKR1C3 and SRD5A1, respectively) mediating the synthesis of testosterone and dihydrotestosterone from precursor steroids. Consistent with this mechanism, inhibitors of CYP17A1 (ketoconazole or the more potent abiraterone), the enzyme that mediates androgen precursor synthesis, are active in CRPC, but responses are partial and transient. In this exploratory single-arm phase II study, we assessed the efficacy of blocking two steps in androgen synthesis by combining a dual SRD5A1/SRD5A2 inhibitor (dutasteride) with a CYP17A1 inhibitor (ketoconazole) in CRPC. The response rate and marked prolongation in time to progression compared with previous single-agent trials indicate that intratumoral conversion of low levels of testosterone to dihydrotestosterone is a mechanism of resistance to CYP17A1 inhibitors and support further combination therapy trials.
Acknowledgments
Grant support: GlaxoSmithKline, Department of Defense Prostate Cancer Research Program grant PC060807, NIH grants R01CA111803 and P01CA89021, and Prostate Cancer Foundation Challenge grant.
We thank Erica Bloom and Geetha Mylvaganam (Beth Israel Deaconess Medical Center) for hormone assays.
Footnotes
Note Added in Proof
Plasma dutasteride concentrations measured in 6 patients by a validated LC/MS-MS method ranged from 80–140 ng/ml approximately 2–3 times higher than the mean 40 ng/ml levels in patients on dutasteride monotherapy at the same dose. This finding is in agreement with the Dutasteride product label describing the potential for CYP3A4 inhibitors such as ketoconazole to impair dutasteride metabolism.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–61. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 2.Holzbeierlein J, Lal P, LaTulippe E, et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol. 2004;164:217–27. doi: 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stanbrough M, Bubley GJ, Ross K, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–25. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 4.Visakorpi T, Hyytinen E, Koivisto P, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–6. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 5.Taplin ME, Bubley GJ, Shuster TD, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332:1393–8. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 6.Taplin ME, Bubley GJ, Ko YJ, et al. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999;59:2511–5. [PubMed] [Google Scholar]
- 7.Gregory CW, He B, Johnson RT, et al. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001;61:4315–9. [PubMed] [Google Scholar]
- 8.Agoulnik IU, Vaid A, Nakka M, et al. Androgens modulate expression of transcription intermediary factor 2, an androgen receptor coactivator whose expression level correlates with early biochemical recurrence in prostate cancer. Cancer Res. 2006;66:10594–602. doi: 10.1158/0008-5472.CAN-06-1023. [DOI] [PubMed] [Google Scholar]
- 9.Craft N, Shostak Y, Carey M, Sawyers CL. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase [see comments] Nat Med. 1999;5:280–5. doi: 10.1038/6495. [DOI] [PubMed] [Google Scholar]
- 10.Weber MJ, Gioeli D. Ras signaling in prostate cancer progression. J Cell Biochem. 2004;91:13–25. doi: 10.1002/jcb.10683. [DOI] [PubMed] [Google Scholar]
- 11.Mellinghoff IK, Vivanco I, Kwon A, Tran C, Wongvipat J, Sawyers CL. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell. 2004;6:517–27. doi: 10.1016/j.ccr.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 12.Chen S, Xu Y, Yuan X, Bubley GJ, Balk SP. Androgen receptor phosphorylation and stabilization in prostate cancer by cyclin-dependent kinase 1. Proc Natl Acad Sci U S A. 2006;103:15969–74. doi: 10.1073/pnas.0604193103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Montgomery RB, Mostaghel EA, Vessella R, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–54. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Locke JA, Guns ES, Lubik AA, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407–15. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 15.Geller J, Albert JD, Nachtsheim DA, Loza D. Comparison of prostatic cancer tissue dihydrotestosterone levels at the time of relapse following orchiectomy or estrogen therapy. J Urol. 1984;132:693–6. doi: 10.1016/s0022-5347(17)49829-6. [DOI] [PubMed] [Google Scholar]
- 16.Mohler JL, Gregory CW, Ford OH, III, et al. The androgen axis in recurrent prostate cancer. Clin Cancer Res. 2004;10:440–8. doi: 10.1158/1078-0432.ccr-1146-03. [DOI] [PubMed] [Google Scholar]
- 17.Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res. 2005;11:4653–57. doi: 10.1158/1078-0432.CCR-05-0525. [DOI] [PubMed] [Google Scholar]
- 18.Eisenberger MA, Blumenstein BA, Crawford ED, et al. Bilateral orchiectomy with or without flutamide for metastatic prostate cancer. N Engl J Med. 1998;339:1036–42. doi: 10.1056/NEJM199810083391504. [DOI] [PubMed] [Google Scholar]
- 19.Joyce R, Fenton MA, Rode P, et al. High dose bicalutamide for androgen independent prostate cancer: effect of prior hormonal therapy. J Urol. 1998;159:149–53. doi: 10.1016/s0022-5347(01)64039-4. [DOI] [PubMed] [Google Scholar]
- 20.Scher HI, Liebertz C, Kelly WK, et al. Bicalutamide for advanced prostate cancer: the natural versus treated history of disease. J Clin Oncol. 1997;15:2928–38. doi: 10.1200/JCO.1997.15.8.2928. [DOI] [PubMed] [Google Scholar]
- 21.Mahoney EM, Harrison JH. Bilateral adrenalectomy for palliative treatment of prostatic cancer. J Urol. 1972;108:936–8. doi: 10.1016/s0022-5347(17)60911-x. [DOI] [PubMed] [Google Scholar]
- 22.Yap TA, Carden CP, Attard G, de Bono JS. Targeting CYP17: established and novel approaches in prostate cancer. Curr Opin Pharmacol. 2008;8:449–57. doi: 10.1016/j.coph.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 23.Small EJ, Halabi S, Dawson NA, et al. Antiandrogen withdrawal alone or in combination with ketoconazole in androgen-independent prostate cancer patients: a phase III trial (CALGB 9583) J Clin Oncol. 2004;22:1025–33. doi: 10.1200/JCO.2004.06.037. [DOI] [PubMed] [Google Scholar]
- 24.Ryan CJ, Halabi S, Ou SS, Vogelzang NJ, Kantoff P, Small EJ. Adrenal androgen levels as predictors of outcome in prostate cancer patients treated with ketoconazole plus antiandrogen withdrawal: results from a Cancer and Leukemia Group B study. Clin Cancer Res. 2007;13:2030–7. doi: 10.1158/1078-0432.CCR-06-2344. [DOI] [PubMed] [Google Scholar]
- 25.Attard G, Reid AH, Yap TA, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563–71. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- 26.Thompson IM, Goodman PJ, Tangen CM, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003;349:215–24. doi: 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
- 27.Thomas LN, Douglas RC, Lazier CB, Too CK, Rittmaster RS, Tindall DJ. Type 1 and type 2 5α-reductase expression in the development and progression of prostate cancer. Eur Urol. 2008;53:244–52. doi: 10.1016/j.eururo.2007.10.052. [DOI] [PubMed] [Google Scholar]
- 28.Rittmaster R, Hahn RG, Ray P, Shannon JB, Wurzel R. Effect of dutasteride on intraprostatic androgen levels in men with benign prostatic hyperplasia or prostate cancer. Urology. 2008;72:808–12. doi: 10.1016/j.urology.2008.06.032. [DOI] [PubMed] [Google Scholar]
- 29.Bubley GJ, Carducci M, Dahut W, et al. Eligibility and response guidelines for phase II clinical trials in androgen-independent prostate cancer: recommendations from the Prostate-Specific Antigen Working Group. J Clin Oncol. 1999;17:3461–7. doi: 10.1200/JCO.1999.17.11.3461. [DOI] [PubMed] [Google Scholar]
- 30.Attard G, Reid AH, A’Hern R, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009;27:3742–8. doi: 10.1200/JCO.2008.20.0642. [DOI] [PMC free article] [PubMed] [Google Scholar]