

Abstract
We report a 27-year-old previously healthy man, who presented with fever and painful swelling of both ankles for 6 months, and who had been jaundiced for 1 week. Clinical examination revealed diffuse macular rash, severe pallor, deep icterus, generalised lymphadenopathy and hepatosplenomegaly. Detailed evaluation revealed granulomata in bone marrow aspirate, and numerous acid fast bacilli in lymph node biopsy. Bone marrow PCR was also positive for Mycobacterium tuberculosis. A diagnosis of disseminated tuberculosis was made and antitubercular therapy was initiated. Investigation also showed features of haemophagocytosis within the bone marrow. Results of further tests satisfied the criteria for haemophagocytic lymphohistiocytosis, probably secondary to tuberculosis. However, rapid deterioration in his clinical condition led to his death within 5 days of diagnosis, before appropriate therapy for haemophagocytic lymphohistiocytosis could be instituted. This case report highlights an unusual and deadly presentation of tuberculosis in an immunocompetent individual.
Background
First described by Scott and Robb-Smith,1 haemophagocytic lymphohistiocytosis (HLH) is a disease characterised by impaired functioning of natural killer cells and cytotoxic T cells, resulting in uncontrolled immune activation and progressive hyperinflammatory cellular damage with multiorgan dysfunction. Simultaneously, abnormal activation and proliferation of benign macrophages produces the characteristic phenomenon of haemophagocytosis, yielding a clinical picture of pancytopenia, hepatosplenomegaly and lymphadenopathy.2 HLH has been broadly classified into primary cases of an idiopathic nature and secondary forms, typically triggered by infection. Inappropriate TH1 response to intracellular pathogens is one of the proposed pathogenetic mechanisms for infection-associated HLH.3 While a broad range of infections have been linked with HLH, most secondary cases can be attributed to infection with Ebstein-Barr virus. Other implicated pathogens include cytomegalovirus, HIV, parasites such as Leishmania donovani, fungi such as Candida and bacteria such as Mycobacterium tuberculosis. Lymphomas constitute an important non-infectious cause for HLH.2 Due to the disseminated nature of inflammation HLH carries a grim prognosis and is often fatal. Involvement of multiple organs and especially the reticuloendothelial system can also produce a clinical presentation similar to disseminated infections like tuberculosis; thus cases of HLH secondary to tuberculosis can easily be missed if the entire illness is attributed to tuberculosis alone.
This report outlines such a case wherein the presentation was entirely compatible with disseminated tuberculosis alone, and chance demonstration of haemophagocytosis revealed associated HLH. Although unusual, secondary HLH should be ruled out in patients with disseminated tuberculosis, especially in the presence of pancytopenia. The additional feature of possible Poncet's disease further adds to the novelty of this case.
Case presentation
A 27-year-old previously healthy man, presented with low-grade intermittent fever and painful swelling of both ankles for 6 months, and jaundice for 1 week. He had consulted several local physicians for his symptoms, and had received intermittent therapy with paracetamol, which afforded temporary relief. He had also noted weight loss, although he was unable to quantify it. His medical history was uneventful. He had never received corticosteroids or any other immunosuppressants. He was unaware of contact with any known cases of tuberculosis. He denied unprotected sexual exposure and any history of substance abuse.
At admission, he was haemodynamically stable and afebrile. Clinical examination revealed diffuse faint macular rash, severe pallor and deep icterus. Generalised lymphadenopathy involving bilateral cervical, axillary and inguinal lymph nodes was noted. The enlarged nodes were non-tender, firm and matted. There was no bony tenderness and no petechiae or ecchymoses. Examination of his ankles revealed swelling, warmth and tenderness suggestive of arthritis. Examination of other joints was normal. Abdominal examination showed mild hepatosplenomegaly. Respiratory examination was unremarkable.
Investigations
Routine laboratory parameters showed severe pancytopenia (haemoglobin: 5.9 g/dl, total leucocyte count: 800 cells/µl, platelet count: 15 000 cells/µl) and an elevated erythrocyte sedimentation rate (140 mm/h). Blood glucose, serum electrolytes and renal function tests (serum creatinine: 1.3 mg/dl) were all normal. Liver function tests were grossly deranged (total bilirubin: 12.0 mg/dl, direct bilirubin: 10.8 mg/dl, aspartate aminotransferase: 60 IU/l, alanine aminotransferase: 52 U/l, alkaline phosphatase: 570 U/l, serum albumin: 1.8 g/dl, globulin 4.3 g/dl). Prothrombin time was normal (16.1 s, control: 15.0 s, international normalised ratio: 1.09). Peripheral smear showed a normochromic normocytic picture with no blasts. Serum B12 and folate levels were normal. Human parvovirus B-19 and human herpes virus-6 PCRs were negative. HIV serology by ELISA was negative. Random serum cortisol performed late in the course of illness was elevated (48.17 µg/dl), ruling out secondary hypoadrenalism.
Abdominal ultrasonography confirmed hepatosplenomegaly, and also showed multiple hypoechoic lymph nodes at the porta hepatis, peripancreatic and paraaortic regions. CT showed mediastinal lymphadenopathy in addition to the intra-abdominal lymph nodes (figure 1).
Figure 1.
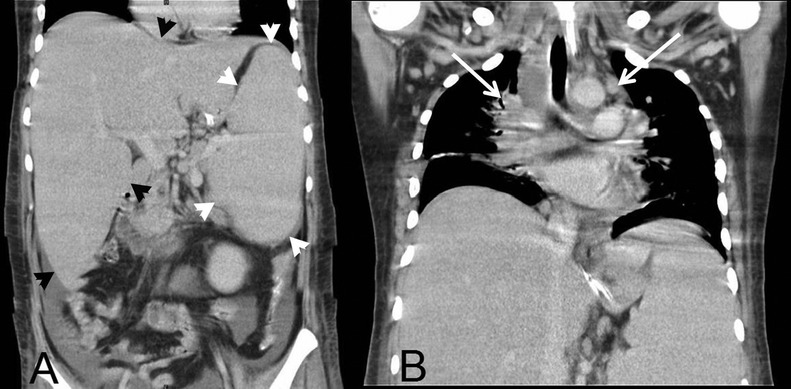
CT of chest and abdomen (coronal views) showing mediastinal lymphadenopathy (white arrows) and hepatosplenomegaly (black and white arrow heads).
Bone marrow examination revealed mild hypocellularity. A few well-formed granulomas composed of epithelioid cells and scattered giant cells were seen (figure 2). Haemophagocytosis was also evident (figure 3). Staining for acid fast bacilli was negative. Conventional aerobic culture was persistently sterile. Nested PCR for M tuberculosis complex (IS6110) was positive.
Figure 2.

Bone marrow biopsy showing granuloma formation (black arrow heads; H&E staining, magnification ×20).
Figure 3.
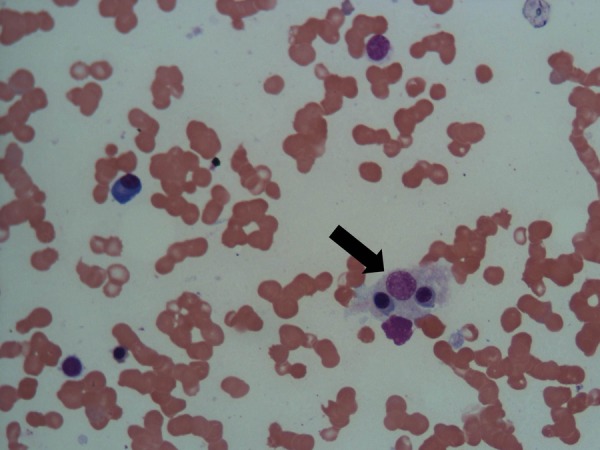
Bone marrow aspirate showing haemophagocytosis (black arrow; H&E staining, magnification ×40).
Axillary lymph node biopsy was also performed; histopathological examination showed focal necrosis with numerous acid fast bacilli (figure 4).
Figure 4.
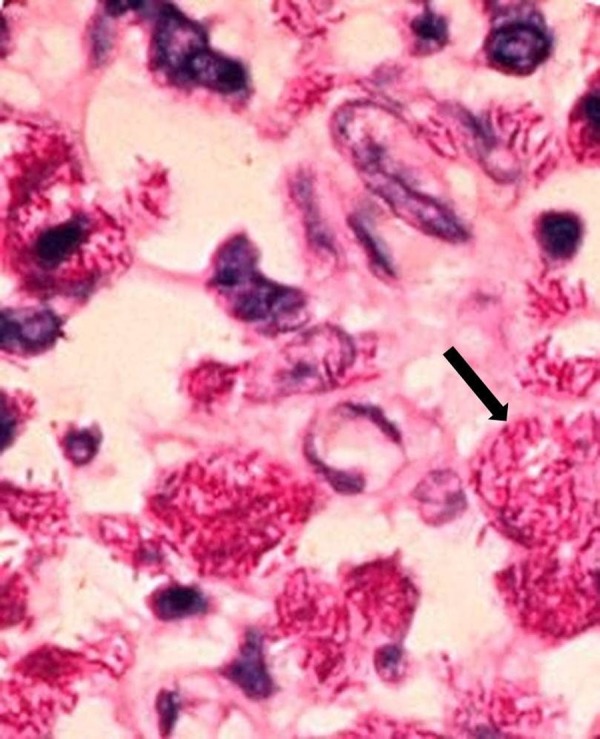
Lymph node biopsy showing numerous acid fast bacilli (black arrow; Ziehl-Neelsen staining, magnification ×20).
Serum ferritin levels were very high (>2000 ng/ml) and hypertriglyceridaemia was present (499 mg/dl).
Diagnostic aspiration of synovial fluid from ankle joints was advised, but refused by the patient. Serological evaluation including antinuclear antibodies, rheumatoid factor and anticyclic citrullinated peptide antibodies were negative. Serum uric acid level was normal (5.0 mg/dl).
Differential diagnosis
Disseminated tuberculosis with secondary HLH and probable Poncet's disease.
Treatment
The patient was started on antitubercular therapy with levofloxacin (750 mg orally q24 h), ethambutol (1200 mg orally q24 h) and streptomycin (1 g intramuscularly q24 h). Subsequently rifampicin (600 mg orally q24 h) was also added to the regimen with careful monitoring of liver function tests. Supportive care was administered with packed red cell and platelet concentrate transfusion.
Outcome and follow-up
Despite these measures, pancytopenia persisted and the patient developed high-grade fever. Considering possible neutropenic sepsis, the patient was also treated with meropenem (1 g intravenously q8 h) and teicoplanin (400 mg intravenously q24 h). Nevertheless, the patient deteriorated rapidly and succumbed to his illness within 3 days, before specific therapy for HLH could be instituted.
Discussion
Diagnostic guidelines for HLH are available4 and were applied in this patient. Fulfilled criteria included fever, splenomegaly, cytopenia, hypertriglyceridaemia, hyperferritinaemia and demonstrated haemophagocytosis without evidence of malignancy. It is also important to note that such criteria were primarily designed for familial HLH and are occasionally unsatisfied in cases with otherwise overt infection-associated HLH.3
Once a diagnosis of infection-associated HLH has been made, specific antibiotic therapy constitutes the mainstay of treatment. In addition, a number of immunomodulatory therapeutic regimens have been developed for HLH. These include therapy with dexamethasone, cyclosporine and etoposide, termed as the HLH-94 regimen,5 and a combination of intravenous immunoglobulin and a corticosteroid such as methylprednisolone. Plasmapheresis has also been used in the treatment of HLH.6 Bone marrow transplantation is largely limited as a therapeutic option to patients with familial HLH.5
Despite the availability of such regimens, HLH nevertheless carries a high death rate. An Asian study by Takahashi et al,6 for instance, reported a mortality of up to 33% in patients with bacterial infection-associated HLH. This study included two patients with underlying tuberculosis. A paediatric case of tuberculosis-associated HLH was described by Balasubramanian et al7; the infant in question received intravenous immunoglobulin as well as antitubercular drugs, but did not survive. Another case of tuberculosis with HLH was reported by Gupta et al8 This patient received dexamethasone along with antitubercular drugs and survived. Pertinently, the patient suffered only mild pancytopenia, and liver function was unaffected, permitting immediate institution of first-line antitubercular drugs. This was also the case with a patient described by No et al,9 who was administered full antitubercular treatment along with etoposide and dexamethasone, and subsequently made a complete recovery. Brastianos et al10 reviewed 36 cases of tuberculosis-associated HLH. In that series, patients who did not receive either antitubercular or immunomodulatory treatment uniformly succumbed, emphasising the deadly nature of this disease. Interestingly, survival was best in patients receiving antitubercular therapy alone; there might have been a selection bias, however, with critical patients receiving combination therapy more frequently than stable cases.
In our own instance, rapid progression of the disease resulted in death prior to institution of immunomodulatory therapy. The obviously disseminated nature of tuberculosis also argued against empirical therapy with corticosteroids, without making a diagnosis. Most importantly, we believe that deranged liver functions interfered with early administration of potent antitubercular drugs, ultimately contributing to mortality.
It is also relevant to note that the entire gamut of findings in our patient could have been readily ascribed to disseminated tuberculosis alone. Together with the robust response to antitubercular therapy alone in the review by Brastianos et al,10 this finding raises the intriguing scenario of significant under-reporting of tuberculosis-associated HLH especially from resource-constrained regions lacking the necessary infrastructure to diagnose HLH.
Poncet's disease is a rare aseptic reactive arthritis that can accompany tubercular infection.11 A diagnosis of Poncet's disease is confirmed by negative serological evaluation, synovial fluid aspiration and/or synovial tissue biopsy. The principal differential diagnosis is direct tubercular infection of the affected joint. Although synovial fluid analysis could not be performed in our patient, symmetrical involvement of both ankles was clinically suggestive of Poncet's disease. A diagnosis of possible Poncet's disease was therefore made. Concomitant Poncet's disease with HLH in an immunocompetent individual has not been described previously and represents a new association.
In conclusion, HLH should be considered as a complication in all patients with disseminated tuberculosis with severe pancytopenia and poor response to antitubercular drugs. Physicians should be familiar with the diagnostic criteria as well as treatment modalities for this rare but deadly disease.
Learning points.
HLH can present with multiorgan involvement, which can be confusing or missed altogether in patients with underlying disseminated tuberculosis.
Presence of severe pancytopenia in such cases is an important clue to possible associated HLH.
Bone marrow examination is useful for ruling out associated HLH while simultaneously confirming a diagnosis of disseminated tuberculosis.
Treatment with antitubercular therapy alone might be ineffective in patients with tuberculosis-associated HLH.
Footnotes
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Scott R, Robb-Smith AH. Histiocytic medullary reticulosis. Lancet 1939;2013:194–8 [Google Scholar]
- 2.Rouphael NG, Talati NJ, Vaughan C, et al. Infections associated with haemophagocytic syndrome. Lancet Infect Dis 2007;2013:814–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yarali N, Yildrim I, Arik E, et al. Hemophagocytic syndrome associated with bacterial infections. J Pediatr Inf 2010;2013:162–4 [Google Scholar]
- 4.Henter JI, Horne A, Arico M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;2013:124–31 [DOI] [PubMed] [Google Scholar]
- 5.Fisman DN. Hemophagocytic syndromes and infection. Emerg Infect Dis 2000;2013:601–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi N, Chubachi A, Kume M, et al. A clinical analysis of 52 adult patients with hemophagocytic syndrome: the prognostic significance of the underlying diseases. Int J Hematol 2001;2013:209–13 [DOI] [PubMed] [Google Scholar]
- 7.Balasubramanian S, Kaarthigeyan K, Aparna V, et al. Tuberculosis associated hemophagocytic syndrome in infancy. Indian Pediatr 2008;2013:593–5 [PubMed] [Google Scholar]
- 8.Gupta AP, Parate SN, Bobhate SK, et al. Hemophagocytic syndrome: a cause for fatal outcome in tuberculosis. Indian J Pathol Microbiol 2009;2013:260–2 [DOI] [PubMed] [Google Scholar]
- 9.No JH, Kang JY, Lee BH, et al. A case of tuberculosis-associated hemophagocytic syndrome during antituberculosis medication for tuberculous pericarditis. Tuberc Respir Dis 2008;2013:522–6 [Google Scholar]
- 10.Brastianos PK, Swanson JW, Torbenson M, et al. Tuberculosis-associated haemophagocytic syndrome. Lancet Infect Dis 2006;2013:447–54 [DOI] [PubMed] [Google Scholar]
- 11.Kroot EJ, Hazes JM, Colin EM, et al. Poncet's disease: reactive arthritis accompanying tuberculosis. Two case reports and a review of the literature. Rheumatology (Oxford) 2007;2013:484–9 [DOI] [PubMed] [Google Scholar]