

Abstract
Development of allograft rejection continues to be the major determinant of morbidity and mortality post-lung transplantation. We have recently demonstrated that a population of donor-derived mesenchymal stem cells are present in human lung allografts and can be isolated and expanded ex vivo. In this study, we investigated the impact of lung resident mesenchymal stem cells (LR-MSCs), derived from allografts of human lung transplant recipients, on T cell activation in vitro. Similar to bone marrow derived MSCs, LR-MSCs did not express MHC II nor the co-stimulatory molecules CD80 or CD86. In vitro, LR-MSCs profoundly suppressed the proliferative capacity of T cells in response to a mitogenic or an allogeneic stimulus. The immunosuppressive function of LR-MSCs was also noted in absence of direct cell contact, indicating that LR-MSCs mediated their effect predominantly via a soluble mediator. LR-MSCs isolated from lung transplant recipients demonstrated PGE2 secretion at baseline (385 ± 375 pg/ml) which increased in response to IL-1β (1149 ± 1081 pg/ml). Addition of prostaglandin synthesis inhibitors (indomethacin and NS-398) substantially abrogated LR-MSC-mediated immunosuppression, indicating that PGE2 may be one of the major soluble mediators impacting T cell activity. This is the first report to demonstrate that human tissue-derived MSCs isolated from an allogeneic environment, have the potential to mediate immunological responses in vitro.
Keywords: Human, Lung, Mesenchymal Stem Cells, T Cells, Transplantation
Introduction
Allograft rejection continues to be the factor limiting successful outcomes following lung transplantation. Acute allograft rejection is the most common complication following lung transplantation, seen in up to 50-60% of lung transplant recipients by 1 year (1), and predisposes to development of chronic rejection or bronchiolitis obliterans syndrome (BOS) (2-5). BOS develops in 60% of transplant recipients by 5 years, causes fixed obstruction of the airways and is the major cause of mortality after 1 year (6). T cell based allorecognition is important in driving the development of acute lung rejection and BOS (7). Acute allograft rejection is a T cell mediated response against allo-antigens(1) and both CD4+ and CD8+ T cell populations are important in mediating the complex events leading to chronic allograft rejection (8, 9).
Mesenchymal stem cells (MSCs) are a unique subset of progenitor cells defined by their capacity to differentiate into multiple mesenchymal lineages (10, 11). MSCs isolated from bone marrow (BM-MSCs) have attracted interest as key modulators of local immune responses (12), having been reported to profoundly inhibit T cell proliferation in vitro (13-17). We have recently described a population of MSCs that can be isolated and expanded from bronchoalveolar lavage (BAL) fluid of human lung allograft patients (18). Studies of sex-mismatched lung allograft donors and recipients demonstrated that these cells are lung-resident and donor-derived. Furthermore, gene chip analysis of these lung resident mesenchymal stem cells (LR-MSCs) revealed that they possess a unique repertoire of cytokines and growth factors that differ from BM-derived MSCs (18).
Since LR-MSCs represent an endogenous population of lung progenitor cells which are distinctly different from BM-MSCs and are components of the transplanted lung cellular milieu, we aimed to investigate whether these cells possess immunoregulatory potential. This study demonstrates that, in vitro, LR-MSCs inhibit the proliferation of diverse subsets of allogeneic T cells both in response to mitogenic and antigenic stimuli, and they seem to mediate their effects both via contact and elaboration of soluble mediators. Our data indicate that one such mediator contributing to T cell unresthree or more groupsponsiveness is prostaglandin E2 (PGE2).
Materials and Methods
Isolation and Identification of Lung-Derived Mesenchymal Stem Cells
Mesenchymal stem cells were obtained from BAL obtained from lung transplant recipients as previously described under an IRB approved protocol (18). BAL samples were utilized only if bronchoscopy was performed for routine surveillance within 6 months of transplantation with no evidence of acute rejection or infection on transbronchial biopsies and microbiological cultures respectively. Cells were maintained in culture in DMEM with penicillin/streptomycin and 10% fetal calf serum at 37°C in 5% CO2 and used at passage 2-6. LR-MSCs obtained from individual BAL samples were treated as separate cell lines.
Peripheral Blood Mononuclear Cell Isolation
Peripheral blood mononuclear cells (PBMC) from healthy volunteers were fractionated by density centrifugation using Lymphoprep gradient (Axis-shield, Oslo, Norway). The buffy coat was isolated and subpopulations of T cells (Pan T cell, CD4+, CD4+CD25-, and CD8+) were purified using the relevant magnetic microbead kits (Miltenyi Biotech Inc., Auburn, CA, USA) according to manufacturer's instructions.
Proliferation Assays and cytokine measurement
2 × 104 LR-MSCs (30Gy irradiated) were plated per well in a 96-well flat-bottom culture-treated plates 4 hrs prior to the addition of Pan T cells or subpopulations of T cells (2 × 105 T cells/well) in complete DMEM supplemented with 10% FCS. For mitogenic stimulation, the cultures were stimulated with PHA (12.5 μg/ml, Sigma-Aldrich, St. Louis, MO, USA) for 5 days. Eighteen hours prior to harvest, [3H] thymidine (1μCi) was added. [3H] Thymidine incorporation is expressed as mean of triplicates in counts per minute (cpm). For dose response experiments 2 × 105 responder T cells were incubated with various numbers of MSCs. For allogeneic stimulation, 2 × 104 MSCs/well were plated in 96-well round bottom culture-treated plates 4 hrs prior to the addition of the stimulator (1 × 105 PBMC/well (15Gy irradiated)) and 1 × 105cells/well of 3rd party responder PBMCs or responder Pan T cells. Cells were co-cultured in complete RPMI supplemented with 10% FCS for 8 days, with addition of [3H] thymidine for the last 18 hrs of culture. To mimic antigen presenting cells, CD28/CD3/CD2 antibody coated beads (Miltenyi Biotech Inc., Auburn, CA, USA) were used to induce proliferation according to the manufacturer's instructions at a ratio of 1:2 bead to T cell. The cells were cultured in presence and absence of LR-MSCs and assessed for proliferation 3 days later. For cytokine measurement third-party 30Gy irradiated MSCs (8×104 MSC/well) were plated into 24-well flat bottom plates in DMEM 4 hrs prior to the addition of 4×105 responder T cells/well in the presence or absence CD28/CD3/CD2 antibody coated beads. After 72 hours, cell-free supernatants were collected and analyzed for IL-2 and IL-10 levels using a highly specific enzyme immunoassay technique (eBiosciences, San Diego, CA and R&D Systems, Minneapolis, USA respectively).
Transwell Experiments
LR-MSCs (30 Gy irradiated) (2 × 105 cells/well) were plated in the upper chamber of a 96-well transwell plate with a 0.5-μm pore size membrane (Corning, Acton, MA, USA) 4 hrs prior to the addition of Pan T cells (2 × 10 4 cells/well) to the lower chamber. Cultures were stimulated by the addition of 12.5 μg/ml PHA and [3H] thymidine was added for the last 18 hrs of culture.
Determination of PGE2 Synthesis and Immunoblot Analysis
To measure PGE2 in BAL, lipids were extracted from BAL fluid samples via solid phase extraction using Sep-Pak C-18 cartridges (Waters, Milford, MA). Eluted sample was reconstituted in assay buffer and PGE2 was quantified in using a highly sensitive and specific enzyme immunoassay kit from Cayman Chemicals (Ann Arbor, MI) according to manufacturer's suggestions. For measurement of PGE2 synthesis by LR-MSCs, LR-MSCs were cultured in the absence or presence of interleukin-1β (IL-1β) (10 ng/ml) for 24 hours. PGE2 was quantified in the supernatant by ELISA. In separate experiments, protein was isolated and COX-1 and COX-2 protein analyzed by western blot as previously described (19).
Statistical Analysis
Data are presented as mean values ± SEM. Statistical significance was analyzed using the GraphPad Prism 3 Software. Significance was assessed with a Student's t test for comparisons of two groups, or with ANOVA and a post hoc Bonferroni test for three or more groups. P < 0.05 was considered significant.
Results
Lung resident MSCs derived from human lung allografts fail to stimulate allogeneic T cells
We have previously demonstrated that donor LR-MSCs can be isolated as late as eleven years post-lung transplantation, clearly indicating that these cells can survive in an allogeneic environment (18). Since donor LR-MSCs are exposed to recipient circulating cells, we first investigated whether LR-MSCs cultured with third party T cells lead to T cell proliferation in vitro. No proliferation was noted when pan T cells were cultured with irradiated allogeneic LR-MSCs (2,125 ± 178 cpm and 2,311 ± 211.9 cpm mean T cell [3H]thymidine incorporation in the absence and presence of MSCs, respectively; p=0.53; Figure 1A). Similar results were noted when LR-MSCs were co-cultured with peripheral blood PBMCs, and subpopulation of T cells (CD4 and CD8 T cells, data not shown). We have previously demonstrated that LR-MSCs express HLA-I but not HLA II (DR or DQ) (18). Examination of co-stimulatory molecules demonstrated that, in addition, LR-MSCs do not express CD80 or CD86, indicating that they do not function as classical antigen presenting cells (Figure 1B).
Figure 1. A. Lung-derived MSCs do not elicit a Pan T cell immunological response in vitro.

2×104 LR-MSCs/well (irradiated 30Gy) were plated into 96-well flat bottom plates in complete DMEM supplemented with 10% FCS 4 hrs prior to the addition of 2×105 responder cells/well (Pan T cells derived from healthy volunteers). Cells were co-cultured for 5 days with addition of [3H] thymidine in the last 18 hrs of culture. Values represent thymidine uptake by proliferating cells (average CPM). No significant difference was noted in the T cell vs. T + LR-MSC group (p=0.53). Data represent the mean ± SEM of 10 separate experiments with LR-MSCs derived from 10 lung transplant recipients and peripheral Pan T cell populations isolated from 5 healthy volunteers. B. Immunophenotyping of LR-MSCs by flow cytometric analysis demonstrates absence of co-stimulatory molecules CD80 and CD86. Histograms show specific mAbs in purple and control isotype-specific IgGs in green.
LR-MSCs inhibit T cell proliferation in response to mitogen stimulation in vitro in a dose-dependent manner
As LR-MSCs were not immunogenic, we next sought to determine whether, similar to bone-marrow derived MSCs, they had an immunosuppressive impact on lymphocytes in response to mitogenic stimulation in vitro. LR-MSCs were co-cultured for 4 days with peripheral blood T cells of third-party healthy volunteers in the presence of PHA at a ratio of 1:10 LR-MSCs to T cells. Upon examination of the co-culture under light microscopy, we observed that wells containing T cells alone in the presence of PHA displayed typical T-cell clustering, indicative of a robust proliferative response. In contrast, wells containing a monolayer of LR-MSCs, in addition to T cells and PHA, lacked T cell clustering, with the T cells appearing to be distributed singularly on top of the LR-MSC monolayer. Additionally, T cells in the co-culture appeared alive and healthy, and their viability was confirmed by trypan blue exclusion (not shown). On quantitative analysis obtained by [3H] thymidine incorporation there was a significant reduction in PHA-induced T-cell proliferation in the presence of LR-MSCs ((84%±7%; P=.0001; Figure 2A and B). Lower inhibition was noted with decreasing ratio of LR-MSCs to responder T cells (Figure 2C).
Figure 2. Lung-derived MSCs inhibit mitogen-driven proliferative response of Pan T cells in a dose-dependant manner.

A, 2×104 LR-MSCs/well (irradiated 30Gy) were plated into 96-well flat bottom plates 4 hrs prior to the addition of 2×105 responder cells/well (Pan T cells) in the presence or absence of PHA (12.5 μg/ml). The cells were cultured for 5 days with addition of [3H] thymidine in the last 18 hrs of culture. Values represent [3H] thymidine uptake by proliferating cells (average CPM). Data represent the mean ± SEM of experiments with LR-MSCs derived from 10 lung transplant recipients. *, p < 0.001 vs T cells; **, p < 0.001 vs T cells + PHA. B, Percentage suppression of T cell proliferation by LR-MSCs. Data for 10 LR-MSCs lines from figure 2A is presented here as a percentage of the T cell proliferation determined in the absence of LR-MSCs (100%). C. 30Gy irradiated MSCs were plated at 1:10, 1:10, 1:100, 1:500, and 1:1000 dilution of MSC:T cells in a 96-well plate 4 hrs prior to addition of 2×105 Pan T cells (constant in all wells) and PHA. Cells were co-cultured for 5 days with addition of [3H] thymidine in the last 18 hrs of culture. *, P<0.001 for all LR-MSC T cell ratios compared to T cells alone in presence of PHA. Data represent the mean ± SEM of 6 separate experiments.
LR-MSCs inhibit both CD4 and CD8 T cell populations in vitro and do this independently of pre-existing Treg populations
Since both CD4+ and CD8+ T cells have been shown to mediate allograft rejection, we aimed to determine whether LR-MSCs can block T cell proliferation of these individual cell populations in vitro. Both CD4+ and CD8+ T cells stimulated by PHA exhibited a markedly decreased proliferative capacity when co-cultured with LR-MSCs, with [3H]thymidine incorporation reduced to unstimulated baseline levels (p<0.001 and 0.002 respectively; Figure 3A and B). To determine whether LR-MSC require the presence of pre-existing CD4+CD25+ regulatory T cells to mediate this effect, peripheral blood CD4+ T cells populations were depleted of CD4+CD25+ T cells and were co-cultured with LR-MSCs and stimulated with PHA as previously described. LR-MSCs significantly inhibited proliferation of the CD4-CD25- population, indicating that pre-existing regulatory T cells are not necessary for LR-MSCs to inhibit T cell activation in vitro (p<0.001; Figure 3 C).
Figure 3. Lung-derived MSCs can inhibit the proliferative response of subpopulations of T cells and do not require pre-existing Tregs.

A and B, 2×104 LR-MSCs/well (irradiated 30Gy) were plated into 96-well flat bottom plates 4 hrs prior to the addition of 2×105 responder cells/well (CD4+ or CD8+ T cells) in the presence or absence of PHA (12.5μg/ml). The cells were cultured for 5 days with addition of [3H] thymidine in the last 18 hrs of culture. Values represent [3H] thymidine uptake by proliferating cells (average CPM). Data represent the mean ± SEM of experiments with LR-MSCs derived from 3 lung transplant recipients; C, CD4+CD25+ T cells were depleted using Miltenyi CD4+CD25+ isolation kit. The CD4+CD25- T cells were plated as described for CD4+T cells. Data represent the mean ± SEM of experiments with LR-MSCs derived from 3 lung transplant recipients.*, p< 0.05 vs. T cells stimulated with PHA in absence of LR-MSCs.
LR-MSCs suppress T cell proliferation in response to allogeneic stimuli
Although LR-MSCs were effective at regulating T cell activation in response to mitogenic stimulation, allogeneic stimulation may be more relevant to the transplanted lung. To examine this in vitro, we performed a mixed lymphocyte response (MLR) assay in which T cell-depleted and irradiated PBMCs were used to stimulate allogeneic Pan T cells in the presence or absence of third-party MSCs. The presence of LR-MSCs led to significant suppression of T cell proliferation in response to allogeneic stimulation (Figure 4A). LR-MSC inhibitory effects in an MLR could be exerted directly on effector T cells or mediated indirectly via effects on the antigen presenting cells (APCs). To differentiate between these possibilities, we examined the effect of LR-MSCs on T cell proliferation in response to CD28/CD3/CD2 antibody coated beads, circumventing the need for APCs. The presence of LR-MSCs caused significant inhibition of T cell proliferation in response to CD28/CD3/CD2 antibody coated beads (p=0.008; Figure 4B), demonstrating that the LR-MSC effect on T cells in an MLR is not mediated via its action on the APCs.
Figure 4. Third-party Lung-derived MSCs inhibit the proliferative capacity of T cells in response to allo-antigen.

Third-party 30Gy irradiated LR-MSCs (2×104 MSCs/well) were plated into 96-well flat bottom plates in DMEM 4 hrs prior to the addition of 1×105 responder T cells/well in the presence or absence of 15Gy irradiated allogeneic T-depleted PBMC (1×105 PBMC/well) or CD28/CD3/CD2 antibody coated beads. The cells were cultured for 8 days with addition of [3H] thymidine in the last 18 hrs of culture. Values represent thymidine uptake by proliferating cells (average CPM). Data represent the mean ± SEM of experiments with LR-MSCs derived from 5 lung transplant recipients.*, p < 0.001 vs T cells; **, p < 0.001 vs T cells + PBMC or beads.
Soluble mediators play an important role in mediating T cell suppression by LR-MSC
To determine whether MSCs require direct contact with effector T-cells to abrogate their proliferative response, we cultured the two populations in a transwell apparatus that separated the two populations by a porous membrane. In the absence of direct-cell contact, LR-MSCs were still able to inhibit T cell proliferation in response to PHA, albeit to a lesser extent (T cell proliferation reduced by 58% ± 10%, Figure 5 A). This inhibition was lower than what was seen with direct cell-cell contact (inhibition of 84%±7%). Transwell experiments were repeated with use of both irradiated and non-irradiated LR-MSCs (n=3) and no significant differences were noted (p=0.33, Figure 5 B). These data suggest that soluble mediators are important for the T cell-inhibitory affects of LR-MSCs but that direct cell contact further facilitates the suppressive ability of LR-MSCs.
Figure 5. Both soluble mediators and cellular contact mediate Inhibitory effect of LR-MSCs on T cells proliferation.

A. 2×104 LR-MSCs/well (irradiated 30Gy) were plated onto the upper chamber of a 96-transwell plate in complete DMEM 4 hrs prior to the addition of 2×105 responder cells/well to the lower chamber in the presence or absence of PHA (10 μg/ml). The cells were cultured for 5 days with addition of [3H] thymidine in the last 18 hrs of culture. Co-cultures of LR-MSCs and T cells were performed as described in Figure 2. Suppression of T cell proliferation by LR-MSCs is shown for transwell and direct co-culture conditions. Data is presented here as a percentage of the T cell proliferations in response to PHA in the absence of LR-MSCs (100%) in their respective conditions. Data is shown for 10 LR-MSC lines. Significant inhibition of T cell proliferation was seen both in absence and presence of direct contact.*, p <0.001 vs. T +PHA. B. Irradiation of LR-MSCs does not alter T cell suppressive ability of LR-MSCs. Figure demonstrates % suppression of T cells in a transwell in presence of non-irradiated and irradiated LR-MSCs derived from 3 lung transplant recipients. No significant difference was noted between the two conditions (p=0.33).
Prostanoid production by LR-MSCs contributes to their ability to inhibit T cell proliferative responses in vitro
Prostaglandin E2 is an immunomodulatory mediator that has been implicated as one of the potential candidates responsible for T cell inhibition by MSCs (16, 20). Like all prostanoids, PGE2 is synthesized from arachidonic acid via the actions of either the constitutive cyclooxygenase (COX)-1 or inducible COX-2 enzymes (21). PGE2 has an important role in pulmonary diseases and PGE2 has been demonstrated to be present in bronchoalveolar lavage fluid obtained from human lungs (22). PGE2 was quantitated in BAL obtained from lung transplant recipients (n=14) and measurable PGE2 was detected in all BAL samples (Mean = 420 pg/ml of BAL fluid, range= 142 to 1212 pg/ml). Next we examined the PGE2 synthetic capacity of lung allograft-derived MSCs isolated from BAL of lung transplant recipients. LR-MSCs (n=10) demonstrated PGE2 secretion at baseline (385 ± 375 pg/ml) with upregulation in response to IL-1β (1149 ± 1081 pg/ml) (Figure 6A). Upregulation of COX-2 in response to IL-1β was also demonstrated by western blot analysis (Figure 6B). As all co-culture experiments were performed with irradiated LR-MSCs, the effect of irradiation on LR-MSC PGE2 secretion was studied in 4 separate cell lines. Irradiation of LR-MSCs did not alter their PGE2 secretory ability at baseline (273 ± 81 and 258 ± 89 pg/ml in the absence and presence of irradiation, respectively; p=0.56) or in response to IL-1β stimulation (790 ± 102 and 783 ± 92 pg/ml in the absence and presence of irradiation, respectively; p=0.78).
Figure 6. Prostaglandin production by LR-MSCs contributes to their immunosuppressive potential.
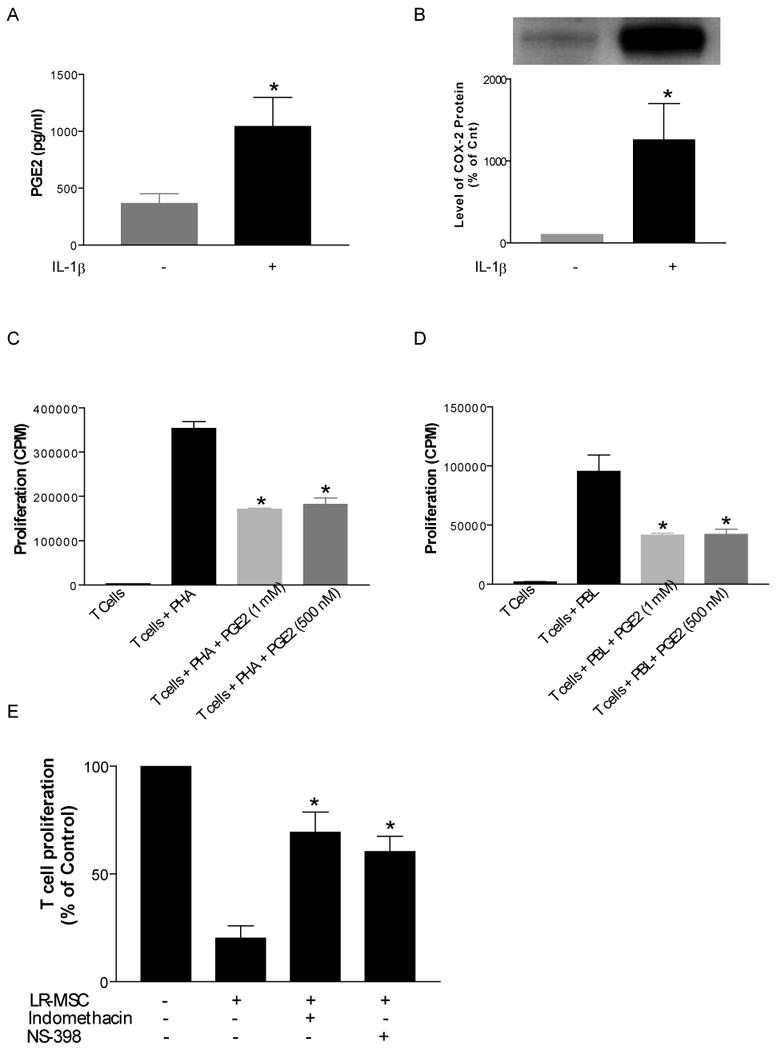
A. LR-MSCs demonstrate PGE2 synthetic capacity. LR-MSCs isolated from bronchoalveolar lavage of 15 lung transplant recipients were cultured in the presence or absence of IL-1β (10 ng/ml) for 24 h before cell-free supernatants were collected and analyzed for PGE2 levels using a highly specific enzyme immunoassay technique. Data represent mean ± SEM. B. LR-MSCs cultured in the presence or absence of IL-1β were scraped into lysis buffer and analyzed by Western blot for expression of COX-2. COX-2 was expressed at baseline and the expression was increased in the presence of IL-1β (*,p<0.05). C and D. Exogenous PGE2 inhibits T cell proliferation in response to both motogenic and allogeneic stimulation. E. LR-MSC mediated T cell suppression is reversed by inhibitors of prostaglandin synthesis. Irradiated LR-MSCs were co-cultured with PHA- stimulated T cells in presence of indomethacin (10μM) or NS398 (5μM) (*, p<0.05 compared to T cells +PHA) . Presence of indomethacin and NS398 significantly reversed suppression of T cell proliferation by LR-MSCs (*, p<0.05 compared to T cells + LR-MSC, n=4 experiments).
As has been demonstrated before, addition of PGE2 to T cells inhibited their proliferation in response to both mitogenic and allogeneic stimulation (Figure 6C and D). To determine if prostanoid secretion by LR-MSCs is important for mediating T cell suppression, irradiated LR-MSCs, T cells, or T cell/LR-MSC co-cultures stimulated with PHA were treated with either the nonselective COX inhibitor indomethacin (10 μM) or the selective COX-2 inhibitor NS-398 (5 μM). T cell suppression in the presence of LR-MSCs was substantially reversed by inhibition of prostaglandin generation by both of those drugs (p<0.05, n=3) demonstrating that an endogenous prostanoid, with PGE2 being a likely candidate, represents an important soluble mediator by which LR-MSCs exert T cell inhibition (Figure 6E).
Prostanoid production by LR-MSCs modulates T cells cytokine secretion
IL-2 and IL-10 are important cytokines secreted by T cells (23, 24) which can alter T cell proliferation and function. To determine if LR-MSCs modulate T cell secretory ability, pan T were activated by CD28/CD3/CD2 antibody coated beads in presence or absence of LR-MSCs. Cytokine determinations by ELISA in the supernatants from the co-cultures demonstrated a significant increased production of the regulatory cytokine IL-10 and a decreased production of the pro-inflammatory cytokine IL-2 in the presence of LR-MSCs. Addition of Cox inhibitor indomethacin completely reversed IL-2 inhibition and partially ameliorated IL-10 increase, demonstrating that PGE2 secreted by LR-MSCs contributes to modulation of cytokine secretion by T cells (Figure 7 A and B).
Figure 7. LR-MSCs modulate cytokine secretion by T cells.

Supernatants from T cells stimulated with CD28/CD3/CD2 antibody coated beads for 72 hours in presence and absence of LR-MSCs and indomethacin were analyzed for IL-2 and IL-10 by ELISA. A. Significant decrease in IL-2 was noted in presence of LR-MSCs (p=<0.001). Inhibition of prostaglandin secretion by addition of COX inhibitor indomethacin completely reversed IL-2 inhibition (p<0.001). B. Significant increase in IL-10 was noted in presence of LR-MSCs (p=<0.001) which was significantly but not completely ameliorated by indomethacin (p<0.01). Data represent the mean ± SEM of experiments performed in triplicates with 3 separate LR-MSC cell lines.
Discussion
In this study we examined interactions between T cells and donor-derived LR-MSCs isolated from the BAL of human lung transplants. We demonstrate that in vitro, LR-MSCs significantly inhibited proliferation of third party HLA-mismatched T cells. This effect was demonstrated in the contexts of both mitogen and alloantigen stimulation and across CD4 and CD8 T cell populations, and did not require the presence of preexisting CD4+CD25+ T cells. LR-MSC-mediated suppression of T cells was partially contact-dependent and largely explained by the actions of a soluble mediator. COX inhibitors blocked the immunosuppressive potential of LR-MSCs on T cells, suggesting that PGE2 may be a major mediator of the immunomodulatory effects of LR-MSC in vitro. This is the first report demonstrating that MSCs isolated from a transplanted lung have immunosuppressive capacity in vitro, a finding that contributes to our understanding of the immunosuppressive potential of tissue resident MSCs and provides hope for their potential as a therapeutic means of inducing long-lived allograft tolerance.
Transplantation offers the only definitive therapy for a variety of end stage lung diseases. However, long-term outcomes for lung transplant recipients are poor with the 10 year lung survival rate of only 26% (25). A major challenge in lung transplantation continues to be allograft rejection orchestrated by recipient derived inflammatory cells, primarily T cells, which infiltrate the graft during acute rejection (1). Recipient derived T lymphocytes also play a seminal role in the pathogenesis of BOS. Lymphocytic bronchitis, a condition characterized by lymphocytic infiltration of the small airways, is considered to be a harbinger of BOS in humans (26) and precedes development of luminal obliteration in animal models of tracheal transplantation (27-29). While host responses to allograft are very well characterized; the role of graft-derived cells in a transplant milieu remains to be elucidated. Our study suggests that resident donor-derived cells might have a role in modulating local micro-environment in the lung allograft, a novel paradigm which begets further investigation.
We demonstrate that lung-resident MSCs derived from allografts possess immunosuppressive potential. MSCs are a unique, well characterized, population of progenitor cells characterized by their ability to differentiate into multiple mesenchymal lineages (30). Although an emerging body of data indicates that MSCs possess immunomodulatory properties both in vitro and in vivo (12, 31, 32), our study is unique is several ways. As we have clearly demonstrated that MSCs isolated from allografts are donor-derived and hence resident in a human adult lung (18), this is the first study to report that tissue-resident populations of MSCs possess immunoregulatory potential. It is important to note that we failed to isolate meaningful numbers of BM-derived MSCs from the BAL of lung allograft recipients indicating that they were not a major contributor to the lung MSC population (18). The fact t h a t L R-MSCs possess immunoregulatory potential, are the major population isolated from a lung allograft, and can be easily isolated from BAL makes them an attractive therapeutic option for prolonging lung allograft acceptance. Further, our study demonstrates for the first time that MSCs isolated from an allogeneic milieu retain their immunosuppressive potential. The majority of prior reports on the immunoregulatory role of BM-derived MSCs have utilized cells isolated from healthy volunteers. In a single report, Bacigalupo et al demonstrated that BM-derived MSCs from patients with sickle cell anemia were deficient in their ability to downregulate T cell proliferation (33). In this study we examined cells isolated from lungs post-transplant and demonstrate their ability to suppress T cell proliferation. Variability was noted in the degree of suppression by LR-MSCs isolated from different donors signifying the need to investigate further the mechanism of this variability and its clinical implications. Furthermore, it should be noted that the cells utilized in this study were isolated early post-transplant in absence of histological evidence of rejection. Whether the T cell suppressive ability of LR-MSCs changes over time and predicts development of acute and chronic allograft rejection in human lung transplant recipients remains to be established. The ability to isolate LR-MSCs with a minimally invasive procedure used routinely in the management of lung transplant recipients (bronchoscopy and BAL) offers a unique opportunity to study these cells in vitro and correlate their phenotypic properties to clinical outcomes in future studies.
The mechanisms by which MSCs mediate T cell suppression remain to be entirely elucidated (12, 31). Various studies, including our own, support the idea that a significant contributor to the immunosuppressive effects of MSC is the production of soluble mediators (13, 16). PGE2, an immunomodulatory lipid mediator, is presently one of the leading candidates for MSC-induced immune suppression (16, 20). PGE2 is a well established inhibitor of T cell proliferation (34, 35). It has been demonstrated that PGE2 inhibits IL-2 secretion and subsequent proliferation of T cells via a cAMP (protein kinase A) dependent mechanism (36, 37). Similarly by increasing intracellular cAMP, PGE2 has been demonstrated to augment IL-10 expression by T cells (38). G protein-coupled receptors EP2 and EP4 are thought to be important in modulation of cellular immune responses by PGE2 (39). Recently PGE2 has been recently demonstrated to both induce Tregs (40) and potentiate the action of T regs (41). Our results demonstrate that PGE2 is an important secretory product of LR-MSCs and inhibition of prostaglandin synthetic pathway attenuates LR-MSCs induced inhibition of T cells, pointing to a role for PGE2 in the immunosuppressive actions of LR-MSCs. Further our data on IL-2 and IL-10 modulation by LR-MSCs supports that LR-MSC derived PGE2 likely acts through similar pathways as those which have been characterized previously. However it should be noted that this reversal of inhibition by Indomethacin was partial (appx. 60%), and the remaining inhibition of T cells by LR-MSCs in presence of COX inhibitor could likely be secondary to either other soluble factors or a contact mediated interaction. It is interesting to note that this degree of reversal of inhibition is similar to the percentage inhibition of T cells in the transwell experiment indicating that among soluble mediators PGE2 is the likely dominant factor. The mechanism of increased inhibition in co-cultures with direct contact among LR-MSCs and T cells remains to be investigated and will be the focus of future studies.
The findings of this study also provide a possible explanation for our previous observation that donor MSCs can be isolated as far out as 11 years post-lung transplantation (18). Their lack of MHC-II and costimulatory molecules, failure to stimulate allogeneic T cells and ability to induce a suppressive local microenvironment by secretion of PGE2 likely explains the in vivo capacity of LR-MSCs to survive in an allogeneic environment. These results are similar to what have been described for BM-MSCs (42).
In summary, we demonstrate that resident MSCs derived from lung allografts inhibit T cell proliferation via elaboration of soluble mediators such as PGE2. These findings point to a possible important immunoinhibitory role of this novel resident endogenous progenitor cell population in a lung allograft milieu, demonstrate the need to study the contribution of local graft derived cells in the pathogenesis of allograft rejection, and provide hope for the potential use of LR-MSCs as a therapeutic means of inducing long-lived allograft tolerance.
Acknowledgments
Grant Support: NIH grants K23 HL077719 (to V. N. Lama), R01 HL085149-01 (to D.J. Pinsky), R01 HL55397 (to D.J. Pinsky), The American Society of Transplantation and Chest Foundation clinical research award in lung transplantation (to V. N. Lama), and Scleroderma Research Foundation award (to V.N. Lama and D.J. Pinsky).
References
- 1.Whelan TP, Hertz MI. Allograft rejection after lung transplantation. Clin Chest Med. 2005;26:599–612. vi. doi: 10.1016/j.ccm.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 2.Girgis RE, Tu I, Berry GJ, Reichenspurner H, Valentine VG, Conte JV, Ting A, Johnstone I, Miller J, Robbins RC, Reitz BA, Theodore J. Risk factors for the development of obliterative bronchiolitis after lung transplantation. J Heart Lung Transplant. 1996;15:1200–1208. [PubMed] [Google Scholar]
- 3.Hachem RR, Khalifah AP, Chakinala MM, Yusen RD, Aloush AA, Mohanakumar T, Patterson GA, Trulock EP, Walter MJ. The significance of a single episode of minimal acute rejection after lung transplantation. Transplantation. 2005;80:1406–1413. doi: 10.1097/01.tp.0000181161.60638.fa. [DOI] [PubMed] [Google Scholar]
- 4.Husain AN, Siddiqui MT, Holmes EW, Chandrasekhar AJ, McCabe M, Radvany R, Garrity ER. Analysis of risk factors for the development of bronchiolitis obliterans syndrome. Am J Respir Crit Care Med. 1999;159:829–833. doi: 10.1164/ajrccm.159.3.9607099. [DOI] [PubMed] [Google Scholar]
- 5.Khalifah AP, Hachem RR, Chakinala MM, Yusen RD, Aloush A, Patterson GA, Mohanakumar T, Trulock EP, Walter MJ. Minimal acute rejection after lung transplantation: a risk for bronchiolitis obliterans syndrome. Am J Transplant. 2005;5:2022–2030. doi: 10.1111/j.1600-6143.2005.00953.x. [DOI] [PubMed] [Google Scholar]
- 6.Estenne M, Hertz MI. Bronchiolitis obliterans after human lung transplantation. Am J Respir Crit Care Med. 2002;166:440–444. doi: 10.1164/rccm.200201-003pp. [DOI] [PubMed] [Google Scholar]
- 7.Wilkes DS, Egan TM, Reynolds HY. Lung transplantation: opportunities for research and clinical advancement. Am J Respir Crit Care Med. 2005;172:944–955. doi: 10.1164/rccm.200501-098WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Higuchi T, Jaramillo A, Kaleem Z, Patterson GA, Mohanakumar T. Different kinetics of obliterative airway disease development in heterotopic murine tracheal allografts induced by CD4+ and CD8+ T cells. Transplantation. 2002;74:646–651. doi: 10.1097/00007890-200209150-00010. [DOI] [PubMed] [Google Scholar]
- 9.Higuchi T, Maruyama T, Jaramillo A, Mohanakumar T. Induction of obliterative airway disease in murine tracheal allografts by CD8+ CTLs recognizing a single minor histocompatibility antigen. J Immunol. 2005;174:1871–1878. doi: 10.4049/jimmunol.174.4.1871. [DOI] [PubMed] [Google Scholar]
- 10.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 11.Gerson SL. Mesenchymal stem cells: no longer second class marrow citizens. Nat Med. 1999;5:262–264. doi: 10.1038/6470. [DOI] [PubMed] [Google Scholar]
- 12.Uccelli A, Pistoia V, Moretta L. Mesenchymal stem cells: a new strategy for immunosuppression? Trends Immunol. 2007;28:219–226. doi: 10.1016/j.it.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 13.Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, Grisanti S, Gianni AM. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. 2002;99:3838–3843. doi: 10.1182/blood.v99.10.3838. [DOI] [PubMed] [Google Scholar]
- 14.Bartholomew A, Sturgeon C, Siatskas M, Ferrer K, McIntosh K, Patil S, Hardy W, Devine S, Ucker D, Deans R, Moseley A, Hoffman R. Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp Hematol. 2002;30:42–48. doi: 10.1016/s0301-472x(01)00769-x. [DOI] [PubMed] [Google Scholar]
- 15.Krampera M, Glennie S, Dyson J, Scott D, Laylor R, Simpson E, Dazzi F. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood. 2003;101:3722–3729. doi: 10.1182/blood-2002-07-2104. [DOI] [PubMed] [Google Scholar]
- 16.Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105:1815–1822. doi: 10.1182/blood-2004-04-1559. [DOI] [PubMed] [Google Scholar]
- 17.Rasmusson I, Ringden O, Sundberg B, Le Blanc K. Mesenchymal stem cells inhibit lymphocyte proliferation by mitogens and alloantigens by different mechanisms. Exp Cell Res. 2005;305:33–41. doi: 10.1016/j.yexcr.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 18.Lama VN, Smith L, Badri L, Flint A, Andrei AC, Murray S, Wang Z, Liao H, Toews GB, Krebsbach PH, Peters-Golden M, Pinsky DJ, Martinez FJ, Thannickal VJ. Evidence for tissue-resident mesenchymal stem cells in human adult lung from studies of transplanted allografts. J Clin Invest. 2007;117:989–996. doi: 10.1172/JCI29713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lama V, Moore BB, Christensen P, Toews GB, Peters-Golden M. Prostaglandin E2 synthesis and suppression of fibroblast proliferation by alveolar epithelial cells is cyclooxygenase-2-dependent. Am J Respir Cell Mol Biol. 2002;27:752–758. doi: 10.1165/rcmb.4857. [DOI] [PubMed] [Google Scholar]
- 20.Cui L, Yin S, Liu W, Li N, Zhang W, Cao Y. Expanded adipose-derived stem cells suppress mixed lymphocyte reaction by secretion of prostaglandin E2. Tissue Eng. 2007;13:1185–1195. doi: 10.1089/ten.2006.0315. [DOI] [PubMed] [Google Scholar]
- 21.Ermert L, Ermert M, Goppelt-Struebe M, Walmrath D, Grimminger F, Steudel W, Ghofrani HA, Homberger C, Duncker H, Seeger W. Cyclooxygenase isoenzyme localization and mRNA expression in rat lungs. Am J Respir Cell Mol Biol. 1998;18:479–488. doi: 10.1165/ajrcmb.18.4.2939. [DOI] [PubMed] [Google Scholar]
- 22.Borok Z, Gillissen A, Buhl R, Hoyt RF, Hubbard RC, Ozaki T, Rennard SI, Crystal RG. Augmentation of functional prostaglandin E levels on the respiratory epithelial surface by aerosol administration of prostaglandin E. Am Rev Respir Dis. 1991;144:1080–1084. doi: 10.1164/ajrccm/144.5.1080. [DOI] [PubMed] [Google Scholar]
- 23.Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annual review of immunology. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 24.Smith KA. Interleukin-2: inception, impact, and implications. Science. 1988;240:1169–1176. doi: 10.1126/science.3131876. [DOI] [PubMed] [Google Scholar]
- 25.Trulock EP, Christie JD, Edwards LB, Boucek MM, Aurora P, Taylor DO, Dobbels F, Rahmel AO, Keck BM, Hertz MI. Registry of the International Society for Heart and Lung Transplantation: twenty-fourth official adult lung and heart-lung transplantation report-2007. J Heart Lung Transplant. 2007;26:782–795. doi: 10.1016/j.healun.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 26.Sharples LD, McNeil K, Stewart S, Wallwork J. Risk factors for bronchiolitis obliterans: a systematic review of recent publications. J Heart Lung Transplant. 2002;21:271–281. doi: 10.1016/s1053-2498(01)00360-6. [DOI] [PubMed] [Google Scholar]
- 27.Boehler A, Chamberlain D, Kesten S, Slutsky AS, Liu M, Keshavjee S. Lymphocytic airway infiltration as a precursor to fibrous obliteration in a rat model of bronchiolitis obliterans. Transplantation. 1997;64:311–317. doi: 10.1097/00007890-199707270-00023. [DOI] [PubMed] [Google Scholar]
- 28.Hertz MI, Jessurun J, King MB, Savik SK, Murray JJ. Reproduction of the obliterative bronchiolitis lesion after heterotopic transplantation of mouse airways. Am J Pathol. 1993;142:1945–1951. [PMC free article] [PubMed] [Google Scholar]
- 29.Lama VN, Harada H, Badri LN, Flint A, Hogaboam CM, McKenzie A, Martinez FJ, Toews GB, Moore BB, Pinsky DJ. Obligatory role for interleukin-13 in obstructive lesion development in airway allografts. Am J Pathol. 2006;169:47–60. doi: 10.2353/ajpath.2006.050975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 31.Ozaki K, Sato K, Oh I, Meguro A, Tatara R, Muroi K, Ozawa K. Mechanisms of immunomodulation by mesenchymal stem cells. International journal of hematology. 2007;86:5–7. doi: 10.1532/IJH97.07003. [DOI] [PubMed] [Google Scholar]
- 32.Rasmusson I. Immune modulation by mesenchymal stem cells. Exp Cell Res. 2006;312:2169–2179. doi: 10.1016/j.yexcr.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 33.Bacigalupo A, Valle M, Podesta M, Pitto A, Zocchi E, De Flora A, Pozzi S, Luchetti S, Frassoni F, Van Lint MT, Piaggio G. T-cell suppression mediated by mesenchymal stem cells is deficient in patients with severe aplastic anemia. Exp Hematol. 2005;33:819–827. doi: 10.1016/j.exphem.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 34.Hilkens CM, Snijders A, Snijdewint FG, Wierenga EA, Kapsenberg ML. Modulation of T-cell cytokine secretion by accessory cell-derived products. Eur Respir J Suppl. 1996;22:90s–94s. [PubMed] [Google Scholar]
- 35.Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Prostaglandins as modulators of immunity. Trends Immunol. 2002;23:144–150. doi: 10.1016/s1471-4906(01)02154-8. [DOI] [PubMed] [Google Scholar]
- 36.Anastassiou ED, Paliogianni F, Balow JP, Yamada H, Boumpas DT. Prostaglandin E2 and other cyclic AMP-elevating agents modulate IL-2 and IL-2R alpha gene expression at multiple levels. J Immunol. 1992;148:2845–2852. [PubMed] [Google Scholar]
- 37.Minakuchi R, Wacholtz MC, Davis LS, Lipsky PE. Delineation of the mechanism of inhibition of human T cell activation by PGE2. J Immunol. 1990;145:2616–2625. [PubMed] [Google Scholar]
- 38.Benbernou N, Esnault S, Shin HC, Fekkar H, Guenounou M. Differential regulation of IFN-gamma, IL-10 and inducible nitric oxide synthase in human T cells by cyclic AMP-dependent signal transduction pathway. Immunology. 1997;91:361–368. doi: 10.1046/j.1365-2567.1997.00260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nataraj C, Thomas DW, Tilley SL, Nguyen MT, Mannon R, Koller BH, Coffman TM. Receptors for prostaglandin E(2) that regulate cellular immune responses in the mouse. J Clin Invest. 2001;108:1229–1235. doi: 10.1172/JCI13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baratelli F, Lin Y, Zhu L, Yang SC, Heuze-Vourc'h N, Zeng G, Reckamp K, Dohadwala M, Sharma S, Dubinett SM. Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. J Immunol. 2005;175:1483–1490. doi: 10.4049/jimmunol.175.3.1483. [DOI] [PubMed] [Google Scholar]
- 41.Mahic M, Yaqub S, Johansson CC, Tasken K, Aandahl EM. FOXP3+CD4+CD25+ adaptive regulatory T cells express cyclooxygenase-2 and suppress effector T cells by a prostaglandin E2-dependent mechanism. J Immunol. 2006;177:246–254. doi: 10.4049/jimmunol.177.1.246. [DOI] [PubMed] [Google Scholar]
- 42.Ryan JM, Barry FP, Murphy JM, Mahon BP. Mesenchymal stem cells avoid allogeneic rejection. J Inflamm (Lond) 2005;2:8. doi: 10.1186/1476-9255-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]