

Abstract
Ectoenzymes expressed on the surface of vascular cells and leukocytes modulate the ambient nucleotide milieu. CD73 is an ecto-5’ nucleotidase that catalyzes the terminal phosphohydrolysis of AMP and resides in the brain on glial cells, cells of the choroid plexus, and leukocytes. Though CD73 tightens epithelial barriers, its role in the ischemic brain remains undefined. When subjected to photothrombotic arterial occlusion, CD73−/− mice exhibited significantly larger (49%) cerebral infarct volumes than wild type (WT)4 mice, with concordant increases in local accumulation of leukocyte subsets (neutrophils, T lymphocytes, macrophages, microglia). CD73−/− mice were rescued from ischemic neurological injury by soluble 5’ nucleotidase. In situ, CD73−/− macrophages upregulated expression of costimulatory molecules far more than WT macrophages, with a sharp increase of the CD80:CD86 ratio. To define the CD73-bearing cells responsible for ischemic cerebroprotection, mice were subjected to irradiative myeloablation, marrow reconstitution, and then stroke following engraftment. Chimeric mice lacking CD73 in tissue had larger cerebral infarct volumes and more tissue leukosequestration than did mice lacking CD73 on circulating cells. These data show for the first time a cardinal role for CD73 in suppressing ischemic tissue leukosequestration. This underscores a critical role for CD73 as a modulator of brain inflammation and immune function.
INTRODUCTION
Cerebral ischemia elicits a strong inflammatory response (1) involving multiple cellular and humoral mediators. Little is known about humoral mediators whose catabolism in the extracellular intravascular milieu modulates cell-cell interactions that promulgate inflammation and ischemic tissue damage. Within the primary area of cerebral infarction, neurons and glial cells become damaged, resulting in extensive Wallerian and terminal degeneraton, loss of distal microvascular flow, and regional edema (2). These characteristic histopathological changes are accompanied or exacerbated by infiltration of lymphocytes, polymorphonuclear and mononuclear leukocytes, as well as by reactive astrocytosis, all of which can play a role in the development of secondary injury after acute brain infarction (3,4). Recruitment of inflammatory cells into infarcted tissue occurs by a stepwise process of homing, adhesion, and ultimately, diapedesis (5). Cells migrate between the endothelial cells that line the inner surface of blood vessels and astroglial feet that comprise the neurovascular unit, ultimately reaching brain parenchyma (6). Recent work has shown that transcellular metabolism by endothelial-surface ENTPDase1 (CD39) of extracellular ATP and ADP released by activated platelets can mitigate explosive amplification of thrombotic nidus formation, thereby reducing damage in ischemic/reperfused stroke (6).
CD73/ecto-5’ nucleotidase (ecto-5’-NT) is a GPI-anchored cell-surface glycoprotein, an ectoenzyme which colocalizes near CD39 at the endothelial surface. CD73 catalyzes hydrolysis of AMP, the terminal product resulting from CD39 activity, to generate the anti-inflammatory and vasodilator nucleoside adenosine. In the extracellular milieu, the CD73-mediated generation of adenosine serves as a key regulator of mucosal and endothelial properties critical to homeostasis, including dynamic processes such as mucosal integrity, vascular flow, and leukocyte traffic (7). Under basal conditions, ATP is continuously released into the extracellular space through regulated transport. Furthermore, at sites of hypoxic and/or ischemic injury, additional large boluses of ATP are liberated from apoptotic and necrotic cells (5). On resting vascular endothelium, extracellular ATP and ADP bind to purino-receptors of the P2X and P2Y families, thereby eliciting pro-thrombotic and pro-inflammatory cascades. CD73, which co-localizes with CD39, quickly degrades these nucleotide-mediated inflammatory stimuli to yield adenosine, which binds to the P1 type of purino-receptors. This sets off a secondary signaling cascade which promotes vascular flow via vasodilation and suppresses inflammation by inhibiting leukocyte extravasation (5).
To specifically evaluate the contribution of CD73 as an inflammatory modulator in the microenviroment of ischemic brain injury, experiments were performed using a modification of a recently described model of photothrombotic occlusion of the middle cerebral artery (8). The photothrombosis stroke model was employed because of its propensity to create intravascular thrombus similar to that seen in human stroke. It also produces cortical infarcts which are highly reproducible in location and size, which is essential for quantification of cellular response. Genetic, pharmacologic, and cellular approaches were used to study the contribution of CD73 to leukocyte trafficking and neurologic outcomes in the setting of stroke.
MATERIALS & METHODS
General
All animal experiments were performed according to protocols approved by the University Committee on the Use and Care of Animals at the University of Michigan. All reagents, unless stated otherwise, were obtained from Sigma (St. Louis, MO). Wild-type C57Bl/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME) and used as controls. As previously described, (9) mice deficient for CD73 were generated by gene targeting of exon 3 and insertion of a neomycin cassette.
Photothrombotic Model of Cerebral Ischemia
Permanent occlusion of middle cerebral artery (MCAO) was induced as previously described (8), by an operator blinded to the genotype of the experimental animal. Ten week old male mice were anesthetized with 2.5 mg ketamine and 0.25 mg xylazine given intraperitonealy (Phoenix Pharmaceutical, St. Joseph, MO). Body temperature was maintained during surgery at 37 °C and for 45 minutes thereafter using a temperature controlled circulating liquid heating pad. After opening a 2–3 mm diameter oval bony window using a dental drill (Foredom Electric, Bethel, CT), the distal part of left middle cerebral artery (MCA) was exposed. A laser Doppler flow probe (Type N, 18 gauge, Transonic Systems, Ithaca, NY) was attached to the surface of the cerebral cortex 1.5 mm dorsal median to the bifurcation of the distal MCA. The probe was connected to a flow meter (Transonic model BLF21) and flow recorded with a continuous data acquisition program (Windaq, DATAQ Instruments, Akron, OH). Rose Bengal was diluted to 10 mg/ml in phosphate-buffered saline and injected intravenously, to achieve a final concentration of 40 mg/kg of body weight. A 1.5 mW green neon laser (540 nm, CVI Melles Griot, Albuquerque, NM) was directed at the MCA from a distance of 6 cm, and occlusion monitored by a cerebral blood flow probe. Occlusion was defined as a >80% reduction in blood flow for approximately 10 minutes. After obtaining stable occlusion, the laser remained in place for 15 additional minutes. In a subset of experiments CD73−/− and WT mice were injected intraperitoneally with 7.5 U of soluble 5’nucleotidase purified from Crotalus atrox venom given 30 minutes before induction of brain ischemia, while controls were injected with an equal volume of saline. In another subset of experiments CD73 was inhibited using adenosine 5’(α, β-methylene) diphosphate (AOPCP; 20 mg/kg) injected intraperitoneally. All drugs were given 30 minutes before the experimental procedure.
Magnetic Resonance Imaging
Infarct volumes were measured using magnetic resonance imaging (MRI) and performed by the University of Michigan Small Animal Imaging Resource 48 h after induction of brain ischemia. Throughout the MRI scanning procedure, mice were anesthetized with a 2% isoflurane/air mixture. Mice were positioned prone, head first, in a 7.0T Varian MR scanner (183 mm horizontal bore, Varian, Palo Alto, CA), with their body temperatures maintained at 37 °C using circulating heated air. A double-tuned volume radiofrequency coil was used to scan the head region of the mice. Axial T2-weighted images were acquired using a spin-echo sequence using the following parameters: repetition time (TR)/effective echo time (TE), 4000/40 ms; field of view (FOV), 30x30 mm; matrix, 256x256; slice thickness, 0.5 mm; slice spacing, 0 mm; number of slices, 25; and number of scans, 1 (total scan time 8 min.). Cerebral infarct volumes were quantified at 48 hours using VOI-11 software, by an observer blinded to experimental conditions.
Neurologic Deficit Scoring
Forty-eight hours after stroke, mice were assessed for neurological deficit using previously described 5-tiered grading system (10), with measurements performed by an observer blinded to experimental conditions. A score of 1 was given if the animal demonstrated normal spontaneous movement; a score of 2 was given if the animal was circling clockwise when viewed from above while receiving a mildly noxious stimuli (tail pinch); a score of 3 was given if the animal was observed to spin clockwise on a longitudinal axis including the tail; a score of 4 was given if the animal fell down on the contralateral side; a score of 5 was given if the animal was crouched on all four paws unresponsive to noxious stimuli.
Brain Water Content
A separate cohort of mice not undergoing infarct volume or leukocyte trafficking measurements were euthanized, brains were removed rapidly, and divided into ischemic and nonischemic hemispheres. The samples were weighed and then dried at 95 °C for 24 hours to obtain the dry weight. The brain water content was calculated as (wet weight - dry weight)/dry weight.
Immunohistochemistry
Whole brains of mice that underwent photothrombotic occlusion were harvested forty-eight hours post-surgery, fixed with 4% paraformaldehyde and paraffin-embedded. Sections to be stained with CD73 then underwent deparaffinization, rehydration and the antigen was retrieved. Endogenous peroxidase was blocked in each of the five micrometer sections followed by a rodent serum block. Serial sections were then stained for CD73 (Abcam, Cambridge, MA) with a rabbit polyclonal antibody at a 1:200 dilution or von Willebrand (Abcam, Cambridge, MA) at a 1:500 dilution. Images were taken on an Eclipse TE2000-E microscope (Nikon Instruments Inc.).
Flow Cytometric Analysis of Inflammatory Cells
Forty-eight hours after surgery mice were euthanized and the brains removed and divided into ischemic and non-ischemic hemispheres. The cerebral hemispheres were then minced with a scalpel and the tissue pieces repeatedly drawn into a syringe through an 18 gauge needle to obtain single cell suspensions. A Percoll (GE Healthcare, Piscataway, NJ) gradient was then used for separation of infiltrating cells, discarding the myelin fractions and residual debris. FACs Lysis Buffer (BD, Franklin Lakes, NJ) was used to lyse red blood cell contaminants. Prior to flow cytometric analyses, live cells were counted using a hemocytometer and distinguished from dead cells by the absence of trypan blue (Invitrogen, Carlsbad, CA) staining. Nonspecific antibody binding was blocked using Fc Block (BD). Cell populations were purified and identified in three stages. First, the leukocyte/microglia population fraction of cells was isolated using an antibody to a common leukocyte antigen (CD45). Double staining pairings of CD45/CD4 and CD45/CD8 were used to identify CD4+ and CD8+ T cell subpopulations. Cells with high expression of CD45 (CD45hi) were further identified as neutrophils based on FITC-conjugated LY-6G (BD) positivity, (CD45hi LY6-Ghi) or as infiltrating macrophages by FITC-conjugated F4/80 (Serotec, Raleigh, NC, USA) positivity. Among CD45-bearing cells, cells with low expression of CD45 and also positive for F4/80 antibody were resident microglia. Dead cells were excluded with a propidium iodide gate. All samples were acquired on a FACS-Calibur Flow Cytometer and data was analyzed using CellQuest software (BD).
Bone-marrow Transplantation
For certain experiments, mice were myeloablated followed by bone marrow reconstitution (11). Ten week old male CD73−/− or wild-type (CD73+/+) littermate control mice were irradiated using 12.5 Gy radiation exposure, administered in three doses three hours apart. Mice were re-populated with CD73−/− bone marrow or wild-type bone marrow using approximately 4x106 bone marrow cells administered intravenously. Mice were allowed to recover for eight weeks before induction of brain ischemia and phenotype examination. For these experiments, four types of chimeric animals were generated; CD73−/− mice repopulated with (1) CD73−/− bone marrow to create global knock-out controls or (2) wild-type bone marrow to create tissue-only CD73-deficient mice; (3) wild-type mice reconstituted with wild-type bone marrow to create wild type controls or (4) CD73−/− bone marrow to create mice which lacked CD73 only on their leukocytes. These mice are denoted using the following abbreviation scheme: Bone Marrow Donor → Bone Marrow Recipient. Chimeras are referred to as KO → KO; WT → KO; WT → WT; and KO → WT.
Real-Time Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR) Assay
Total RNA in brain tissues at 48 hrs after photothrombotic MCAO was extracted via the RNAzol B method (Tel-Test, Friendswood, TX). Total RNA was reverse-transcribed into cDNA by using random primers (Life Technologies, Rockville, MD). To detect cerebral levels of interleukin-10 (IL-10), interleukin-6 (IL-6), KC, tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and VCAM-1 mRNA, real-time RT-PCR was performed by means of an ABI PRISM 7700 sequence detection system with TaqMan Universal PCR Master Mix and Assays-on-Demand gene expression probes (Applied Biosystems, Foster City, CA). TaqMan Rodent 18S Ribosomal RNA Control Regents VIC (Applied Biosystems) was used as an endogenous control gene. A standard curve for the serial dilution of murine brain cDNA was generated. The amplification cycle consisted of 2 min at 50°C, 10 min at 95°C, 15 s at 95°C and 1 min at 60°C. Relative quantitative values of targets were normalized according to the endogenous 18S ribosomal RNA gene control.
Statistical analyses
Values are reported as mean ± SEM, with the number of experiments performed provided in the figure legends. The significance of differences between groups with multiple comparisons was estimated by one-way ANOVA followed by Newman-Keuls test. Statistical significance was confirmed at p< 0.05.
RESULTS
Effect of CD73 gene absence on stroke outcome
Forty-eight hours after induction of permanent MCAO, cerebral infarct volumes were assessed in both CD73-deficient and wild-type mice using T2 weighted cortical MRIs (Figure 1a). Total infarct volumes were increased by 49% in CD73−/− mice compared with their WT littermates (71.2 ± 0.8 mm3, compared to 36.6 ± 1.7 mm3, respectively, p<0.001) (Figure 1b). The larger infarct volumes in CD73−/− mice corresponded with a functional outcome after MCAO, as the CD73−/− mice had greater neurological deficits when compared to WT mice (Figure 1c). Given the known role for CD73 in maintaining epithelial and endothelial barrier properties (9), a comparative analysis of cerebral edema was performed forty eight hours after induction of brain ischemia. In comparison with WT controls, brain water content was significantly increased (~30%) in the infarcted hemisphere of CD73−/− mice (4.53 ± 0.01 ml/g dry wt tissue vs. 6.5 ± 0.07 ml/g dry wt tissue (Figure 1d).
Figure 1.

Effect of CD73−/− genotype on cerebral infarct volume and functional outcome. A) MR images from representative mice (genotype indicated) 48 hours following photothrombotic MCA occlusion. B) Quantitative analysis of cerebral infarct volumes by MR at 48 hrs after MCA occlusion, with genotype indicated (n=6 per group). C) Neurologic deficit scores shown for individual animals of the indicated genotype. All 6 animals from the (A) and (B) panels are included, as well as data from another 4 animals which did not undergo infarct volume analysis by MR (n=10 per group). D) Brain water content 48 h after induction of photothrombotic MCA in contralateral (C) and ischemic (I) hemispheres in WT and CD73 null mice (n=5 per group). ***P<0.001.
Since inflammation can worsen the outcome of brain ischemia, experiments were performed to assess whether CD73 modulates leukocyte trafficking into ischemic cerebral tissue. Analysis of brain tissue was conducted using multiparameter flow cytometry 48 hours after induction of brain ischemia to quantify leukocyte populations in ischemic hemispheres. Ischemic hemispheres of CD73−/− animals had a more than 30% increase in the total numbers of infiltrating nucleated cells when compared to wild type ischemic hemispheres (data not shown). Since the non-ischemic hemispheres showed no significant differences in terms of infiltrating cell numbers between the two genotypes, we concluded that CD73 does not affect basal levels of cerebral inflammation (data not shown). To identify the mononuclear fraction more precisely, we considered CD45-positive cells which expressed the F4/80 mononuclear cell surface marker (CD45hiF4/80+), to be blood-derived macrophages. F4/80 surface marker was used because it is more specific for macrophages than CD11b, since CD11b is also found on some B and polymorphonuclear cells (12). The second cell population examined was CD45lowF4/80+ cells, which expressed 10 to 15 times less CD45 antigen than macrophages and hence were considered to be resident microglial cells. Two additional antibodies against B7-1 (CD80) and B7-2 (CD86) antigens were used in order to examine the degree of activation of mononuclear cells within the inflamed brain.
Analysis of cells infiltrating the ischemic brain showed a shift toward the mononuclear cellular fraction in CD73−/− mice compared with wild-type controls (Figure 2a-c). In the ischemic hemispheres of CD73−/− animals, 13.2 ± 1.2% of cells were macrophages vs. 7.1 ± 0.7% in wild-type mouse ischemic hemispheres (Figure 2b, c). This reflects a relative enrichment of the infiltrating macrophage infiltration, as well as an increase in their total numbers from 3.8x104 ± 0.4x104 in wild-type mice to 10.5x104 ± 1.1x104 in CD73−/− mice (Figure 2a).
Figure 2.
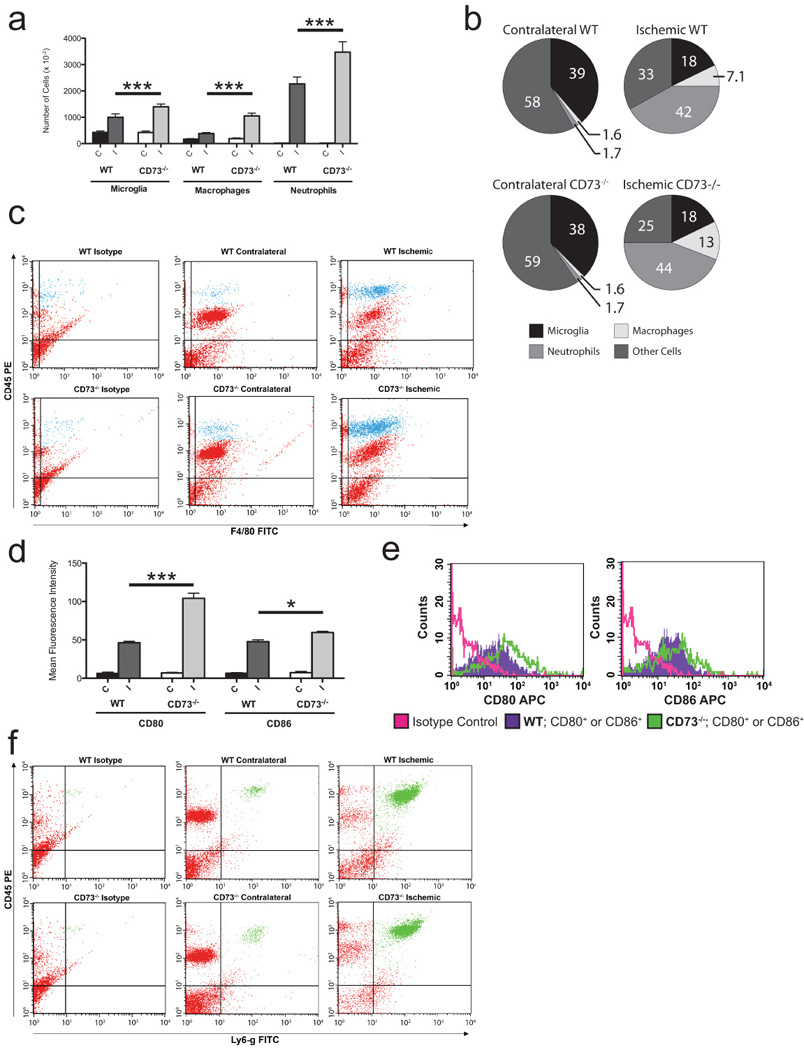
Role of CD73 in leukocyte sequestration in the ischemic brain 48 hrs after MCA occlusion: A) Absolute number of leukocyte subpopulations (i.e., microglia, macrophages and neutrophils) in contralateral (C) and ischemic (I) hemispheres in WT and CD73 null mice (n=6 per group; ***P<0.001 B) Relative contribution of microglia, macrophages and neutrophils in contralateral (C) and ischemic (I) hemispheres in WT and CD73 null mice 48 hrs after induction of brain ischemia (n=6 per group). C) Representative dot-plot scatter analysis of leukocytes isolated from contralateral (C) and ischemic (I) hemispheres within ischemic brains of WT and CD73 null mice; double staining for CD45 and F4/80 allowed the identification of 2 different populations: CD45low F4/80+ (microglia), CD45hi F4/80+ (macrophages). D) Mean fluorescent intensity of macrophages expressing CD80 and CD86 molecules isolated from contralateral (C) and ischemic (I) hemispheres of CD73−/− and WT mice (n=4 per group). E) Overlay histograms illustrate the difference in mean fluorescent intensity of macrophages expressing CD80 and CD86 molecules, between ischemic hemispheres of CD73−/− and WT mice. F) Representative scattergrams of CD45 and LY-6G stained leukocytes isolated from contralateral and ischemic hemispheres of WT and CD73 null mice. Strong positivity for both markers indicates infiltrating neutrophil population.
We further hypothesized that infiltrating macrophages exposed to the inflammatory environment present in the ischemic hemispheres of CD73−/− mice could become more activated compared with macrophages isolated from the less-inflamed ischemic hemispheres of wild-type controls. Moreover, CD45hiF4/80hi macrophages isolated from ischemic hemispheres of WT mice express higher levels of CD86 than CD80, while macrophages from ischemic hemispheres of CD73−/− mice express much higher levels of CD80 than CD86 with CD80 MFI of 46.2 ± 2.1 in WT vs. 103.7 ± 7.1 in CD73−/− and CD86 MFI of 46.04 ± 2.9 in WT vs. 59.4± 2.4 in CD73−/− (Figure 2d, e). Thus, the increased CD80:CD86 ratio in macrophages isolated from CD73−/− mice reflects the more pro-inflammatory status of those cells in comparison with their WT counterparts. Although the relative ratio of microglia and neutrophils did not change between wild type and CD73−/− ischemic hemispheres, absolute cell number of both populations were increased approximately 2-fold in the ischemic brains of CD73−/− mice vs. control animals, with 13.9x104 ± 1.1x104 microglia in CD73−/− vs. 10x104 ± 1.3x104 in WT mice and 34.7.4x104 ± 3.9x104 neutrophils in CD73−/− vs. 22.6x104 ± 2.6x104 in WT mice (Figure 2a, b, f). In addition to increased numbers of macrophages, microglia and neutrophils, a 42% relative increase in the total number of T cells in the ischemic hemisphere of CD73−/− mice vs. WT mice following MCA occlusion was observed (4.1x105 ± 8.0x104 vs. 1.6x105 ± 2.0x104, p=0.02; for CD73−/− vs. WT, respectively). When T cell subsets were examined, there was an increase in both CD4+ and CD8+ T cells infiltrating into the ischemic brain tissue. The ischemic hemisphere of CD73−/− mice contained significantly more CD4+ T cells (230x104 ± 6.1x104 vs. 6.2x104 ± 0.9x104, p=0.03) and CD8+ T cells (1.8x105 ± 2.9x104 vs. 1.0x105 ± 1.3x104, p=0.03) than their WT counterparts (Figure 3a, b).
Figure 3.

Infiltration of T cell subpopulations into the contralateral (C) and ipsilateral (I) hemispheres of wild-type and CD73−/− mice at forty-eight hours post stroke. A) Absolute numbers of CD45+/CD4+ (helper) T cell populations. B) Absolute numbers of CD45+/CD8+ (cytotoxic) T cell populations.
In addition to examining absolute levels of recruited effector lymphocyte and leukocyte populations, experiments were performed to determine local production of inflammatory cytokines and adhesion molecules that could drive leukosequestration in the ischemic zone. To confirm the pro-inflammatory phenotype of ischemic brains of CD73−/− mice, expression of pro-inflammatory cytokines and adhesion molecules was analyzed 48 hrs after induction of brain ischemia by quantitative RT-PCR of RNA isolated from the brains of wild-type or CD73−/− mice.
As shown in Figure 3a-d, the amounts of mRNAs encoding the cytokines IL-1β, IL-6, TNF-α, and chemokine (C-X-C motif) ligand 1 (KC) were significantly greater in the ischemic hemispheres of CD73−/− animals compared with the amounts seen in WT mice. Although amounts of vascular cell adhesion molecule 1 (VCAM-1) mRNA were slightly increased in the non-ischemic hemispheres of CD73−/− mice, significantly greater amounts of VCAM-1 mRNA was present in the ischemic hemispheres of CD73−/− mice following middle cerebral artery occlusion (Figure 4e). These experiments went further to examine the induction of anti-inflammatory cytokine, IL-10, which is known to suppress TNF-α, IL-1 and IL-12, thereby contributing to both the limitation and resolution of inflammation (13). Though not statistically significantly different, there was a trend towards diminished IL-10 mRNA in ischemic brains of CD73−/− mice (Figure 4f). The upregulation of pro-inflammatory and downregulation of anti-inflammatory cytokines as well as leukocyte recruitment demonstrated the enhanced inflammation in the ischemic cerebrum of CD73−/− mice.
Figure 4.
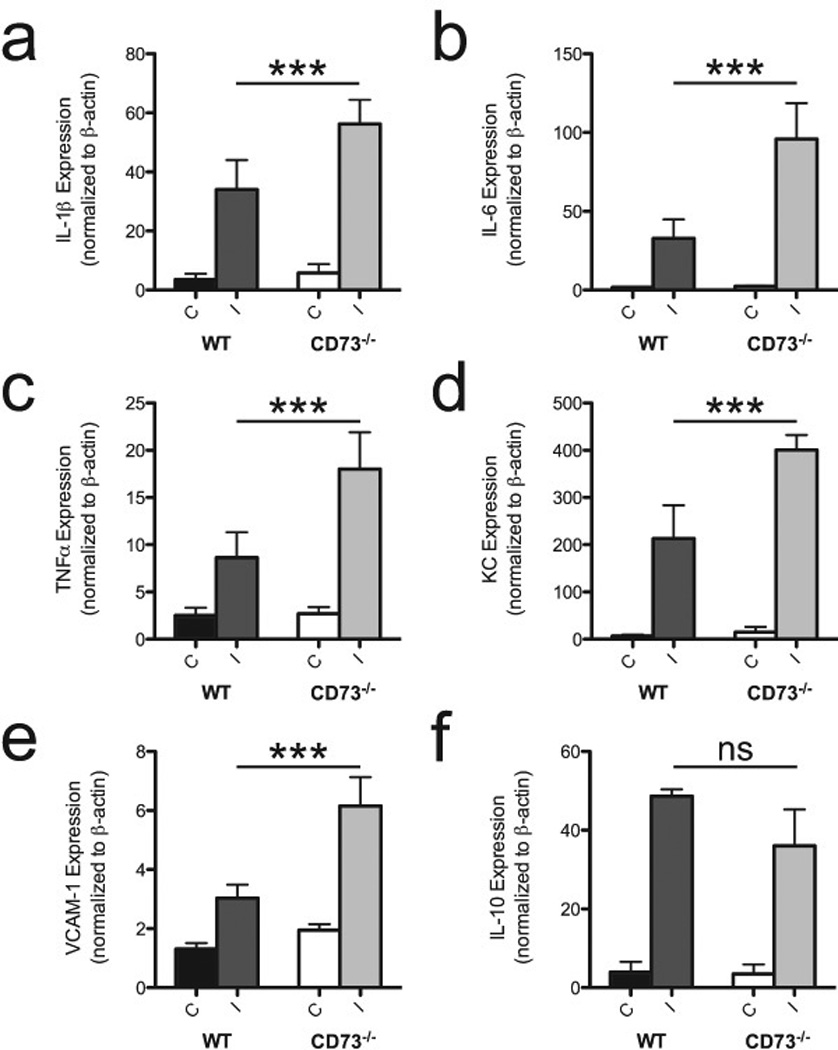
Role of CD73 on cytokine and adhesion molecule expression. mRNA levels were estimated using semiquantitative (RT)-PCR and normalized against β-actin mRNA. Shown are plots of expression of IL-1β (A); IL-6 (B); TNF-α (C); KC (D); VCAM-1 (E) and IL-10 (F) mRNAs in contralateral (C) and ischemic (I) hemispheres of WT and CD73−/− mice (n=4 per group). ***P<0.001, ns=not significant.
Rescue of CD73 genotype null mice from stroke sequelae with soluble 5’-nucleotidase
In order to fulfill Koch’s postulates regarding a causal role for a pathway in disease, we not only performed experiments in which the pathway was deleted, but also experiments in which the deleted pathway was reconstituted. To prove the assertion that CD73 plays an important role in regulation of leukocyte trafficking in brain ischemia, CD73−/− and wild-type mice were each reconstituted with 7U of soluble 5’-nucleotidase (5’-NT) purified from Crotalus Atrox venom, given 30 minutes before induction of brain ischemia. As in the earlier experiments, operators blinded to the identity of the experimental animals evaluated infarct volumes and neurological scores. The mice were then euthanized and ischemic and non-ischemic hemispheres were separated for flow cytometric analyses of infiltrating leukocyte populations. As shown in Figure 5a, 5’-NT treatment of CD73−/− mice was associated with the complete reconstitution of a wild type phenotype (cerebral infarct volumes in saline-treated CD73−/− mice were 68 ± 2.6 mm3 vs. 30.4 ± 2.9 mm3 in CD73−/− treated with 5’-NT). For comparison, 5’-NT was also able to reduce infarct volumes in WT animals (infarct volumes in wild-type mice treated with saline were 34.8 ± 0.75 mm3 vs. 22.7 ± 2.1 mm3 in wild-type animals treated with 5'-NT). The reduction of infarct volumes in CD73−/− mice treated with 5’-NT corresponded with improved neurological score after MCA occlusions (Figure 5b).
Figure 5.

In order to assess the therapeutic potential of soluble 5’ nucleotidase (CD73 analog) in preventing cerebral infarction, experiments were performed in a different cohort of mice. A) Average cerebral infarct volume was calculated 48 h after induction of ischemia in WT and CD73 null mice treated with soluble 5’ nucleotidase (5'NT) or vehicle (PBS) (n=6 per group), and B) Neurological deficit was measured using a 5-tired grading system in the same animals. Cells from contralateral (C) and ischemic (I) hemispheres of these mice were subjected to flow cytometric analysis to determine the absolute number of macrophages (C), microglia (F), and neutrophils (G) (n=6 per group). Similarly, mean fluorescent intensity was measured in macrophages isolated from ischemic (I) hemispheres of WT and CD73 null mice treated with soluble 5’nucleotidase or vehicle and labeled with antibodies against CD80 (D) and CD86 (E) (n=4 per group). *P<0.05; **P<0.01; ***P<0.001.
Leukocyte populations (neutrophils, microglia, and mononuclear fraction) were identified using the same combinations of antibodies as before. We had previously observed (Figure 2b) that CD73-deficiency primarily affects the mononuclear fraction of infiltrating cells (CD45hiF4/80hi macrophages) 48 h after induction of brain ischemia. In this next set of experiments, 5’-NT was administered 30 min prior to the ischemic episode. 5’-NT not only suppressed macrophage recruitment in ischemic wild-type mice (Figure 5c), but did so even more in ischemic mice lacking native CD73. 5’-NT reduced total numbers of infiltrating macrophages by 48% in WT animals and by 57% in CD73−/− mice. As a percentage of total infiltrating leukocytes, 5’-NT caused the macrophage population of ischemic CD73−/− mice to decrease to the same level as that seen in vehicle-treated wild-type controls (9.94% ± 0.5% CD73−/− treated with 5’-NT versus 9.33 ± 0. 3% in saline treated WT mice, data not shown).
One other important facet of recruited leukocytes is related to their activation state, which can affect their immune functions. Treatment with soluble 5’-NT in both CD73−/− and wild-type mice not only resulted in a markedly reduced absolute number of infiltrating macrophages, but those infiltrating macrophages exhibited a striking reduction in activation phenotype 48 h after induction of brain ischemia (Figure 5d, 5e). As in previous experiments, these data were obtained by measuring expression of CD80 and CD86 co-stimulatory molecules. Although CD86 is constitutively expressed on a variety of antigen presenting cells (APCs), the expression of both CD80 and CD86 is elevated upon activation (14). B7-1 positive infiltrating macrophages (CD45hiF4/80hi CD80+) isolated from ischemic hemispheres of CD73−/− mice treated with 5’-NT were shown to express lower levels of B7-1 antigen (39%) compared with macrophages isolated from ischemic hemispheres of saline-treated CD73−/− mice (Figure 5d). Note that CD73-deficient macrophages treated with saline demonstrated far greater activation then wild-type macrophages with respect to CD80 expression (CD73−/− MFI 100.4 ± 8.9 vs. 48.6 ± 0.5 in WT, Figure 5d). However, in CD73−/− mice, treatment with soluble 5’-NT downregulated CD86 expression only 25%, resulting in a substantial decrease in the CD80:CD86 ratio compared to saline-treated mice (Figure 6e). Treatment of wild-type mice with soluble 5’-NT resulted in an additional reduction of both co-stimulatory molecules. (Figure 5d, e).
Figure 6.
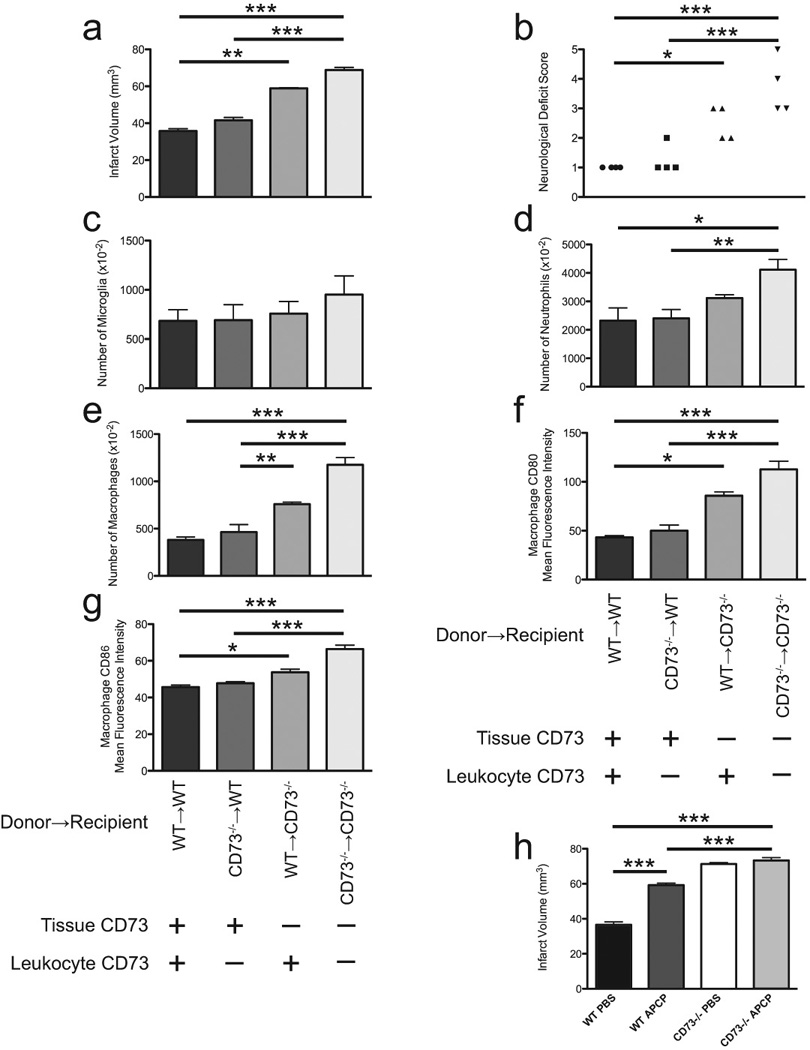
Selective inactivation of CD73 molecule on tissue only attenuates MCAO induced brain ischemia. A) Quantitative analyses of infarct volumes in marrow-reconstituted mice (n=4 per group). B) Locomotor activity determined by neurological deficit score shown for individual animals across the genotype 48 hrs after induction of brain injury. (n=4 per group). The contribution of CD73 on brain resident tissue and leukocytes to leukosequestration of microglia (C), neutrophils (D) and macrophages (E) was examined using bone-marrow reconstitution studies. (n=4 per group). Panels (F) and (G) show mean fluorescent intensity of macrophages expressing CD80 and CD86 molecules isolated from ischemic hemispheres of bone-marrow reconstituted mice (n=4 per group).
Thirty minutes prior to induction of brain ischemia, a different cohort of WT and CD73−/− mice were treated intraperitoneally with vehicle alone or the CD73 inhibitor AOPCP, (panel H) and 48 h later infarct volumes were calculated. (n=5 per group except n=8 for WT AOPCP). *P<0.05; **P<0.01; ***P<0.001.
Absolute numbers of both microglia and neutrophils were markedly reduced after treatment with soluble 5’-NT, whether this was administered to WT mice or to CD73−/− mice. When WT mice were examined in the setting of stroke, the administration of 5’-NT caused a 28% reduction in microglial numbers (7.3x104 ± 0.7x104 in 5’-NT-treated vs. 10.1x104 ± 0.85x104 in saline-treated mice, P<0.01, Figure 5f). When CD73−/− were similarly treated with 5’-NT, there was a 43% reduction in microglia detected in the ischemic hemisphere (7.91x104 ± 0.7x104 vs 14.6x104 ± 0.7x104, P< 0.001, Figure 5f).
Similar data were observed when neutrophil infiltration in the ischemic brain was examined. Treatment of wild-type animals with soluble 5’-NT resulted in 27% reduction of neutrophil infiltration when compared to wild-type saline-treated mice (16.7x104 ± 1.3x104 neutrophils per ischemic hemisphere for 5’-NT treated WT vs. 23x104 ± 1.4x104 for WT saline-treated controls, P<0.01). An even greater absolute reduction in infiltrating neutrophils was observed in CD73−/− mice treated with 5’-NT (19.6x104 ± 2.4x104 for 5’NT-treated CD73−/− mice vs. 3.7x105 ± 1.7x104 for CD73−/− saline-treated mice, P<0.001, Figure 5g). Here again, 5-NT reconstituted the CD73−/− mice to a WT level of neutrophil-infiltration. These data show for the first time that transient rescue from CD73 deficiency can be accomplished through administration of soluble CD73 in the setting of brain ischemia.
Stroke sequelae in CD73 chimeric mice
In an attempt to differentiate the contribution of CD73 on brain resident cells from that of CD73 on circulating leukocytes, a series of CD73 chimeric mice were generated. Four groups of chimeras were made by myeloablation and bone marrow reconstitution according to the following schema (Donor → Recipient); WT marrow into WT recipient; CD73−/− marrow into WT recipient; WT marrow into CD73−/− recipient; and CD73−/− marrow into CD73−/− recipient. The first and last chimeras (KO → KO and WT → WT) served as transplantation controls. KO → WT chimeras served as an experimental condition in which endothelium and other resident cells express CD73, but circulating leukocytes do not. WT → KO chimeras served as an experimental condition in which CD73 is expressed on circulating leukocytes, but it is absent from resident vascular cells. All experiments were performed eight to ten weeks after reconstitution to allow for full bone marrow reconstitution.
Forty-eight hours after photothrombotic occlusion of the MCA, cerebral MRI scans were obtained to quantify infarction, neurological deficit was scored by an operator blinded to experimental conditions, and leukocyte trafficking was assessed by flow cytometry. Cerebral infarct volumes in the KO → KO group of mice was markedly larger (48%) then those in the WT → WT group (68.8 ± 0.7 mm3 vs 35.8 ± 1.2 mm3 respectively, P<0.001, Figure 5a). We next examined the effect of selective CD73 rescue by myeloablating naïve mice and reconstituting them with marrow cells possessing or lacking the CD73 gene, after which MCAO was performed. Infarct volumetric analysis demonstrated that expression of CD73 only on brain resident tissue (i.e., KO marrow → WT recipient) provided some protection to mice from stroke when compared to mice with global deficiency of CD73 (the KO → KO group). Quantitatively, this protection was measurable as a 40% reduction in infarct volumes (41.6 ± 0.7 mm3 vs. 68.8 ± 0.7 mm3 in KO → WT vs. KO → KO groups, respectively, P<0.001). These data show that there is a considerable contribution from tissue-resident CD73 in cerebroprotection after ischemic brain injury. We further examined the effect of selective expression of CD73 on bone-marrow derived cells, using a strategy of WT marrow implanted into CD73−/− recipients (WT → KO). This expression of CD73 on bone marrow cells only provided limited protection from cerebral ischemia (58.9 ± 0.2 mm3), which represent a 14% decrease in infarct volume in comparison with global lack of the CD73 molecule (KO → KO; P=NS). Consonant with these data, locomotor activity in the KO → WT group was substantially better than that in the KO → KO group, but there was no difference in basal locomotor activity between WT → WT and KO → WT mice (Figure 6b).
We next evaluated the effect of site-selective CD73 expression on the trafficking of leukocytes to the ischemic brain. Total numbers of nucleated cells infiltrating ischemic hemispheres paralleled infarct size as well as neurological deficit scores in each of the four groups of myeloablated and marrow-reconstituted mice under study (data not shown). By using a dual staining technique with anti-CD45 and anti- F4/80 antibodies, an infiltrating mononuclear fraction could be easily identified, and distinguished from the resident microglial population. Levels of CD45 and F4/80 expression distinguish between microglial (CD45loF4/80+) and CNS-associated macrophage populations (CD45hiF4/80hi) (15). As in our previous experiments, the relative percentage of CD45loF4/80+ cells (microglia) among all leukocytes and also the relative percentage of CD45hiLY6-Ghi cells (neutrophils) did not change across genotypes. However, the total numbers of infiltrating cells of either population were significantly higher in KO → KO mice when compared to control (WT → WT) mice, or chimeric animals that have tissue-resident CD73 (KO → WT; Figure 6c). Similarly, the presence of CD73 either on circulating or resident cells reduced the accumulation of neutrophils in the ischemic brain (Figure 6d).
After the induction of unilateral brain ischemia, the total number of infiltrating cells in the contralateral (non-ischemic) hemisphere did not vary with respect to CD73 genotype or chimerism (data not shown). In myeloablated and reconstituted mice completely devoid of CD73, there was more than a 50% increase in the relative ratio of CD45hiF4/80hi infiltrating macrophages compared to myeloablated and reconstituted control mice (WT → WT). In chimeric animals in which CD73 was present in brain resident tissue (KO → WT), macrophage infiltration was similar to control chimeras (WT → WT). In contrast, WT → KO mice, macrophage infiltration was increased significantly (by 37%) in comparison with WT → WT strokes (data not shown). Similarly, the total number of infiltrating macrophages was significantly increased (up to 2.5 fold) in KO → KO and WT → KO mice (Figure 6e). When CD73 was present on brain tissue but absent from leukocytes, there is little effect on leukocyte trafficking compared with WT → WT chimeras (Figure 6e). These data together indicate that CD73 has an important native role which suppresses leukocyte accumulation in an ischemic zone especially when that CD73 is expressed on brain resident tissue.
As in our previous experiments, in order to define the contribution of activated leukocytes following injury, the next set of experiments showed that macrophages isolated from completely CD73−/− chimeric mice (KO → KO) express ~ 60% more CD80 and ~ 26% more CD86 on their surface when compared with WT → WT controls. The presence of CD73 on brain resident tissue alone (KO → WT) significantly lowered expression of CD80 relative to CD86, thereby decreasing the CD80:CD86 ratio, reflecting attenuation of macrophage infiltration as well as overall inflammation in those mice when compared to animals with global lack of CD73 (CD80 MFI 50 ± 5.7 in KO → WT mice vs. 113 ± 8.3 in KO → KO mice; CD86 MFI 48 ± 1 in KO → WT vs. 66.4 ± 2.2 in KO → KO mice, Figure 6f, g). On the other hand, macrophages isolated from ischemic hemispheres of mice which have CD73 on leukocytes only (WT → KO) express significantly higher levels of CD80 when compared with macrophages isolated from control chimeric mice (CD80 MFI 85.7 ± 3.8 in WT → KO vs. 43 ± 2 in WT → WT), and when compared with macrophages isolated from the chimeras when CD73 is present on the brain tissue only (KO→WT CD80 MFI 49.9 ± 5.7, Figure 6f, g). These data imply that CD73 from bone marrow cells provides less neuroprotection than CD73 derived from the brain-resident tissue. Immunohistochemical experiments staining wild-type and CD73 null mice for CD73 or vWF revealed CD73 expression on cells of the choroid plexus and an absence on endothelial cells of the brain (Figure 7).
Figure 7.
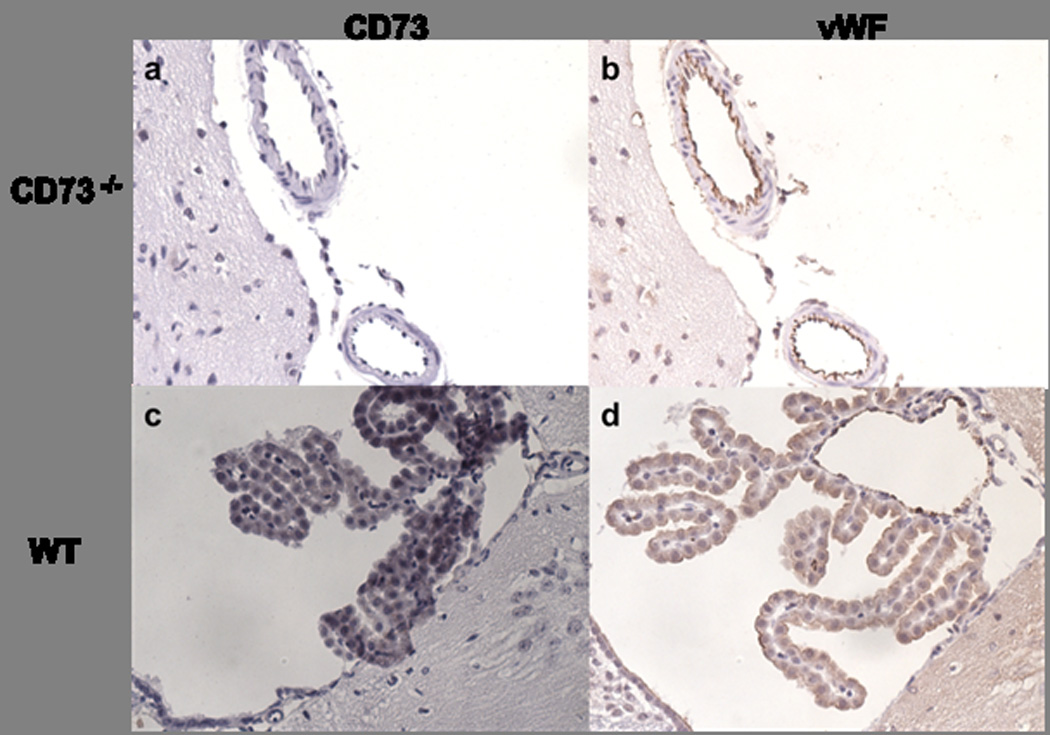
CD73 and von Willebrand factor (vWF) expression in the brain. CD73−/− mice (upper row) and wild-type mice (lower row) stained for CD73 in the first column and vWF in the second column. A) A lack of CD73 staining is shown in the knockout mice. B) vWF staining defines the vessel wall and together with the CD73 staining confirms the absence of CD73 in the vasculature. C) CD73 expression in the wild-type mouse can be observed in the choroid plexus. D) vWF staining of choroid plexus reveals surrounding vessels and the absence of CD73 expression.
Effect of CD73 inhibition on stroke outcome
We next pursued the functional contribution of CD73 to cerebroprotection by treating mice with an intraperitoneal injection of 20 mg/kg of the specific CD73-inhibitor, AOPCP, or vehicle control 30 minutes before induction of permanent brain ischemia. As shown in Figure 6h, treatment of WT animals with AOPCP increases infarct volume 38% in comparison with saline-treated WT mice. Infarct volume observed in CD73−/− mice after the treatment with AOPCP was not statistically different from infarct volume in CD73−/− animals treated with saline, but both are still larger (around 19%) than infarct volumes of WT mice treated with AOPCP. This observation suggested to us that treatment with AOPCP partially blocked the CD73 activity, at least in the concentration used in this study.
DISCUSSION
The ecto-5’-nucleotidase CD73, which catalyzes the hydrolysis of AMP to adenosine, is consider the rate-limiting step in the generation of extracellular adenosine (16). In this work, we hypothesized that the generation of extracellular adenosine by CD73 is required to maintain the balance between pro- and anti-inflammatory signaling during cerebral ischemia. We used both genetic and pharmacologic approaches to test this hypothesis, and found that CD73-null mice were more susceptible to stroke injury, contained more infiltrating macrophages and associated pro-inflammatory cytokines in ischemic brain tissues, and that these macrophages had a small increase in CD86 expression and a huge increase in CD80 expression. In addition, we found that injection of soluble 5’-NT attenuated these effects in CD73−/− mice, effectively rescuing them from ischemic brain injury. We furthermore used bone marrow transplantation to demonstrate the relative importance of brain tissue-derived CD73 compared to the CD73 on circulating cells. Data from these experiments showed that CD73 on circulating cells is less critical than CD73 on tissue resident cells in conferring ischemic cerebral protection. These results support our hypothesis and lead to the novel conclusion that brain tissue-derived CD73 attenuates stroke injury by reducing the infiltration and activation of macrophages.
Effect of CD73 on stroke
Our studies revealed that MCAO resulted in significant increases in infarct volume (~50%), neurological deficit (~56%), and brain edema (~30%) in CD73−/− mice compared to wild-type controls. Rat brain MCAO has been shown to increase CD73 expression in infarcted tissue (4), and this and other evidence suggests an increase in the ability of brain tissue to hydrolyze extracellular nucleotides released as a consequence of severe tissue damage (4, 17). With regard to this latter point, it was recently shown that hypoxia drives transcriptional increase of both surface ecto-nucleotidases, CD39 and CD73 (18). Consequently, this amplifies the extracellular production of adenosine from adenine nucleotide precursors. In fact, our previous work showed that, following challenge by cerebral ischemia, CD39-deficient animals developed worse clinical outcomes than their corresponding controls (6, 19). Hypoxia can also induce the expression of CD73 via binding of hypoxia-inducible factor (HIF)1-α to the CD73 promoter region (9, 20). On the other hand, activation of the A2A adenosine receptor by adenosine has been shown to suppress immune responses by inhibiting activated immune cells (21). These observations suggested that dephosphorylation of AMP by CD73 could represent a major pathway of extracellular adenosine formation and inflammatory suppression during the oxygen deficit of brain ischemia. In addition, administration of exogenous AMP in rats was shown to increase infarct volume, cause hypotension, hyperglycemia, hypocalcemia and consequent metabolic acidosis (22). This suggests that CD73 deficiency could increase stroke injury by both suppressing anti-inflammatory adenosine production and increasing extracellular AMP levels. The deleterious effects of AMP during ischemic stroke may be mediated by activation of AMP-activated protein kinase in the vascular endothelial cells, leading to increased nitric oxide production and vessel dilation and then to additional cerebral hypoperfusion and brain damage (23).
CD73 control of leukocyte trafficking
The extent of neuronal damage correlates with the degree of the innate immune response, with numerous studies demonstrating the critical role of cellular and humoral immune activity after ischemic brain injury (24, 25). Forty-eight hours after induction of permanent brain ischemia, we found a near doubling of the total number of infiltrating neutrophils and microglial cells in the ischemic hemispheres of CD73−/− mice, a far larger increase that seen in wild type controls. Consistent with the present findings, studies of hypoxia-associated lung inflammation and lipopolysaccharide-induced acute lung injury confirmed an anti-inflammatory role for both CD73 and CD39 in these models (26). Lennon et al., examining the interaction of leukocytes, particularly neutrophils, at cell-cell junctions, showed that inhibition of CD73 using either AOPCP or the anti-CD73 monoclonal antibody IE9 inhibited the resealing of endothelial and epithelial barriers by 85% (27), suggesting the necessity for purine nucleotide metabolism in this pathway. This observation fits nicely with our data demonstrating a loss of barrier function in the ischemic brains of CD73−/− mice. In contrast, a recent study has shown that CD73−/− mice are protected from experimental autoimmune encephalomyelitis, suggesting that CD73-dependent adenosine production and signaling through the A2A adenosine receptor are required for the efficient entry of lymphocytes into the central nervous system (28). These disparate findings may be explained by differences in the mechanism governing inflammatory cell trafficking in the central nervous system versus the lungs, or differences between the subtypes of inflammatory cells (neutrophils vs. lymphocytes), or differences in leukocyte egress mechanisms. It is also possible that the absence of CD73 in other brain tissues could be due to the interaction of leukocytes and the endothelium. Adenosine, in our case generated by CD73, has been shown to decrease leukocyte trafficking through the inhibition of cytokine release from endothelial cells (29). Additionally, catalytic activity of CD73 on the cell surface is inhibited by the adhesion of lymphoid cells (30). While CD73 may not be present on the endothelial cells of the brain, it is observed on the leukocytes that accumulate at the vasculature interfaces and infiltrate into the ischemic tissue. The adenosine generated by CD73 has been shown to be more important than CD73 itself in regulating lymphocyte entry into the CNS in the case of experimental autoimmune encephalomyelitis (28).
One of the most interesting observations in our study was an increase in the relative ratio (44%) and total number of infiltrating macrophages in ischemic hemispheres of CD73−/− mice compared to wild-type controls. The most important cellular response to post-ischemic inflammation is by cells of the innate immune system, predominantly resident microglia/brain macrophages and blood-derived monocytes/macrophages (31). Macrophages in the ischemic zone are more highly activated cells, and therefore could be primed to exacerbate local inflammation and promote incremental tissue damage (32). In the inflammatory milieu, macrophages have multiple functions, including acting as phagocytic cells to clear the area of cell debris to promote remodeling and repair of damaged tissue. Those functions are responsible for the dual role of macrophages in inflamed tissues - to promote the cell-mediated immunity and to promote resolution of inflammation (33). In addition, both CD4+ and CD8+ T cell numbers were increased in the ischemic brains of CD73−/− mice. CD25+Foxp3+ T regs and CD25-uncommitted primed precursor T helper cells have been shown to express CD73. Through the generation of adenosine by these cells, the inflammatory response seems to be diminished. It is possible the adenosine generation by CD73 present on the infiltrating T cells of the wild-type mice resulted in protection and smaller infarct volumes (34). IFN-β is known to increase CD73 expression on brain endothelial cells. The adenosine generated by CD73 may mediate the inhibitory effects of IFN-β on the transmigration of CD4+ T lymphocytes (35). This is in concordance with the increased numbers of T lymphocytes seen in CD73 null mice. The binding of various ligands to adenosine receptors on monocytes and macrophages strongly suppressed TLR4-mediated LPS induction of the pro-inflammatory cytokines IL-12 and TNF-α (36, 37), which may be one of the central mechanisms whereby adenosine receptor occupancy prevents inflammation-induced tissue injury. In addition, adenosine facilitates anti-inflammatory IL-10 production by stimulation of A2A and A2B receptors on murine peritoneal macrophages (13, 38), which may also contribute to the anti-inflammatory and immunosuppressive action of adenosine. A recent report showed that IL-1β and TNF-α were produced by a subset of microglia and macrophages after induction of permanent brain ischemia (39). These data are in agreement with our observation that the mRNA expression of the pro-inflammatory cytokines IL-6, KC, TNF-α, and IL-1β are increased in ischemic hemispheres of CD73−/− animals compared to wild-type controls, a finding consistent with the greater inflammation and macrophage infiltration we observed in CD73−/− mice. Previous work in ischemia/reperfusion has shown protective effects of IL-10 due to its ability to suppress macrophage activation, down regulate pro-inflammatory cytokine production and suppress leukocyte-endothelial cell interactions (40). Additionally, IL-10 has been shown as a central mediator of the T-reg cell cerebroprotective effects (41). The diminished amount of IL-10 mRNA in ischemic hemispheres of CD73 −/− mice could, therefore, be a contributing factor to increased cerebral expression of pro-inflammatory cytokines and increased cerebral infarct volumes.
Activation of innate immune mechanisms is often triggered or amplified by expression of a class of molecules on the surface of macrophages. These molecules, termed “activation/co-stimulatory” molecules, engage ligands external to the macrophages which lead to an internal activation signal. CD80 and CD86 are two classic co-stimulatory molecules implicated in innate immune functions. Our data showed that macrophages from ischemic hemispheres of CD73−/− mice express higher levels of CD80 relative to CD86 when compared with macrophages from wild-type mice. This observation is consistent with the similar observations in SJL mice immunized for induction of experimental autoimmune encephalomyelitis (42). Temporal up-regulation of CD80 surface expression relative to CD86 was found on splenic and CNS, but not on lymph node B cells, T cells and macrophages, demonstrating CD80 predominance on cells infiltrating the CNS of mice with active disease (43). In that work, the changes in the expression patterns in both spleen and CNS-infiltrating F4/80+ macrophages had important functional consequences, as CD80 becomes the predominant costimulatory ligand in T-cells proliferation assays. Srinivasan et al. showed that increased expression of CD80 on antigen-presenting cells, after induction of experimental autoimmune encephalomyelitis, correlated with greater inflammation and disease progression in the central nervous system (14). Increased infiltration of CD80-expressing macrophages was also observed in colonic inflamed mucosa in a murine model of inflammatory bowel disease (44). If CD80 and CD86 are able to direct and activate the adaptive immune response, the outcome will depend on the level of expression of the two molecules on antigen-presenting cells. On most antigen-presenting cells, CD86 is constitutively-expressed, whereas CD80 is induced after activation (45). Despite interacting with the same receptors (CD28/CD152), differences in the nature of expression (constitutive versus induced) and kinetics of interaction of CD80 and CD86 have been attributed to their different functions in disease processes. To our knowledge, this study demonstrates for the first time that CD73 presence in inflamed CNS milieu can regulate expression of co-stimulatory molecules such as CD80 (B7-1) and CD86 (B7-2) on infiltrating macrophages, thereby playing an important role in the regulation of the immune response in the setting of stroke.
Rescue of the CD73 null phenotype by 5’nucleotidase
We showed that intraperitoneal injection of soluble 5’-NT resulted in significant attenuation of infarct volume, improved functional outcome and reduced leukocyte infiltration 48 hrs after induction of brain ischemia. This supports our hypothesis that the conversion of extracellular AMP to adenosine by the 5'-NT, CD73, is a key control point for the regulation of vascular inflammation associated with brain ischemia. A significant increase in leukocyte adherence to the vascular endothelium has previously been shown in CD73−/− mice in a model of cremaster muscle ischemia-reperfusion injury (46). CD73 and its active metabolite adenosine dose-dependently inhibited VCAM-1 but not ICAM-1 expression. In a model of wire induced vascular injury, CD73 deficiency increased monocyte adhesion to ex vivo perfused carotid arteries that was mediated by VLA-4/VCAM-1 (46). These data are in agreement with our observation, as we found more VCAM-1 mRNA in ischemic hemispheres of CD73−/− mice compared to wild-type controls. Moreover, CD73-deficient mice had more severe vascular leakage and greater neutrophil infiltration during hypoxia than wild-type animals, which could be reversed by administration of soluble CD73 (9). These data suggest that CD73 can control inflammation by regulating the dynamics of leukocyte-endothelial interaction.
Stroke sequelae in CD73 chimeric mice
Since CD73 is expressed both on brain-resident cells and on circulating leukocytes, a bone marrow transplantation model was used to create chimeric mice to distinguish between the relative contributions of resident and circulating CD73 in brain ischemia. We found that brain tissue-derived CD73 attenuated both macrophage influx and activation in the settings of stroke CD73, whereas CD73 on circulating cells inhibited these inflammatory parameters to a lesser degree. Our data cannot rule out the possibility that circulating cells may provide an additional source of adenosine available at the vascular endothelial interface.
In summary, our studies using CD73−/− mice demonstrated a significant protective role for CD73 during cerebral ischemia. This protection was primarily due to the attenuation of the number of infiltrating inflammatory cells as well as suppression of macrophage activation, and was associated with attenuated expression of pro-inflammatory cytokines IL-6, KC, TNF-α and IL-1β. These conclusions are supported by our finding that soluble 5’-nucleotidase mitigated the inflammatory phenotype of both CD73−/− and wild-type animals. Based on these observations, we propose the novel conclusion that CD73 derived from brain resident tissue has a critical role in controlling leukocyte extravasation and tissue injury after brain ischemia.
Acknowledgments
This work was supported by NIH grants HL086676 (D.J.P.), the J. Griswold Ruth MD & Margery Hopkins Ruth Professorship, and the A. Alfred Taubman Medical Research Institute at the University of Michigan.
The authors acknowledge the expert technical assistance of Dr. Sunitha Yanamadala and Dr. Richard Ransom and important discussions and provision of CD73 null mice by Dr. Linda F. Thompson.
Footnotes
DISCLOSURES
The authors have no financial conflict of interest.
Abbreviations used in this paper: WT, wild type; MCA, middle cerebral artery; MCAO, permanent occlusion of middle cerebral artery; AOPCP, adenosine 5’(α, β-methylene) diphosphate; MRI, magnetic resonance imaging; KC, chemokine (C-X-C motif) ligand 1; VCAM-1, vascular cell adhesion molecule 1; 5'-NT, 5’-nucleotidase; KO, CD73–/– knock-out.
REFERENCES
- 1.Lindsberg PJ, Grau AJ. Inflammation and infections as risk factors for ischemic stroke. Stroke. 2003;34:2518–2532. doi: 10.1161/01.STR.0000089015.51603.CC. [DOI] [PubMed] [Google Scholar]
- 2.Ross DT, Ebner FF. Thalamic retrograde degeneration following cortical injury: an excitotoxic process? Neuroscience. 1990;35:525–550. doi: 10.1016/0306-4522(90)90327-z. [DOI] [PubMed] [Google Scholar]
- 3.Huang J, Kim LJ, Mealey R, Marsh HC, Jr, Zhang Y, Tenner AJ, Connolly ES, Jr, Pinsky DJ. Neuronal protection in stroke by an sLex-glycosylated complement inhibitory protein. Science. 1999;285:595–599. doi: 10.1126/science.285.5427.595. [DOI] [PubMed] [Google Scholar]
- 4.Braun N, Lenz C, Gillardon F, Zimmermann M, Zimmermann H. Focal cerebral ischemia enhances glial expression of ecto-5'-nucleotidase. Brain Res. 1997;766:213–226. doi: 10.1016/s0006-8993(97)00559-3. [DOI] [PubMed] [Google Scholar]
- 5.Salmi M, Jalkanen S. Cell-surface enzymes in control of leukocyte trafficking. Nat Rev Immunol. 2005;5:760–771. doi: 10.1038/nri1705. [DOI] [PubMed] [Google Scholar]
- 6.Pinsky DJ, Broekman MJ, Peschon JJ, Stocking KL, Fujita T, Ramasamy R, Connolly ES, Jr, Huang J, Kiss S, Zhang Y, Choudhri TF, McTaggart RA, Liao H, Drosopoulos JH, Price VL, Marcus AJ, Maliszewski CR. Elucidation of the thromboregulatory role of CD39/ectoapyrase in the ischemic brain. J Clin Invest. 2002;109:1031–1040. doi: 10.1172/JCI10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Latini S, Pedata F. Adenosine in the central nervous system: release mechanisms and extracellular concentrations. J Neurochem. 2001;79:463–484. doi: 10.1046/j.1471-4159.2001.00607.x. [DOI] [PubMed] [Google Scholar]
- 8.Su EJ, Fredriksson L, Geyer M, Folestad E, Cale J, Andrae J, Gao Y, Pietras K, Mann K, Yepes M, Strickland DK, Betsholtz C, Eriksson U, Lawrence DA. Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat Med. 2008;14:731–737. doi: 10.1038/nm1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, Colgan SP. Crucial role for ecto-5'-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med. 2004;200:1395–1405. doi: 10.1084/jem.20040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hata R, Mies G, Wiessner C, Fritze K, Hesselbarth D, Brinker G, Hossmann KA. A reproducible model of middle cerebral artery occlusion in mice: hemodynamic, biochemical, and magnetic resonance imaging. J Cereb Blood Flow Metab. 1998;18:367–375. doi: 10.1097/00004647-199804000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Day SM, Reeve JL, Pedersen B, Farris DM, Myers DD, Im M, Wakefield TW, Mackman N, Fay WP. Macrovascular thrombosis is driven by tissue factor derived primarily from the blood vessel wall. Blood. 2005;105:192–198. doi: 10.1182/blood-2004-06-2225. [DOI] [PubMed] [Google Scholar]
- 12.Fux M, van Rooijen N, Owens T. Macrophage-independent T cell infiltration to the site of injury-induced brain inflammation. J Neuroimmunol. 2008;203:64–72. doi: 10.1016/j.jneuroim.2008.06.025. [DOI] [PubMed] [Google Scholar]
- 13.Nemeth ZH, Lutz CS, Csoka B, Deitch EA, Leibovich SJ, Gause WC, Tone M, Pacher P, Vizi ES, Hasko G. Adenosine augments IL-10 production by macrophages through an A2B receptor-mediated posttranscriptional mechanism. J Immunol. 2005;175:8260–8270. doi: 10.4049/jimmunol.175.12.8260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srinivasan M, Gienapp IE, Stuckman SS, Rogers CJ, Jewell SD, Kaumaya PT, Whitacre CC. Suppression of experimental autoimmune encephalomyelitis using peptide mimics of CD28. J Immunol. 2002;169:2180–2188. doi: 10.4049/jimmunol.169.4.2180. [DOI] [PubMed] [Google Scholar]
- 15.Xia W, Hilgenbrink AR, Matteson EL, Lockwood MB, Cheng JX, Low PS. A functional folate receptor is induced during macrophage activation and can be used to target drugs to activated macrophages. Blood. 2009;113:438–446. doi: 10.1182/blood-2008-04-150789. [DOI] [PubMed] [Google Scholar]
- 16.Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Physiological roles for ecto-5'-nucleotidase (CD73) Purinergic Signal. 2006;2:351–360. doi: 10.1007/s11302-005-5302-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X, Zhou T, Zhi X, Zhao F, Yin L, Zhou P. Effect of hypoxia/reoxygenation on CD73 (ecto-5'-nucleotidase) in mouse microvessel endothelial cell lines. Microvasc Res. 2006;72:48–53. doi: 10.1016/j.mvr.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 18.Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, Robson SC, Colgan SP. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J. Exp. Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hyman MC, Petrovic-Djergovic D, Visovatti SH, Liao H, Yanamadala S, Bouis D, Su EJ, Lawrence DA, Broekman MJ, Marcus AJ, Pinsky DJ. Self-regulation of inflammatory cell trafficking in mice by the leukocyte surface apyrase CD39. J. Clin. Invest. 2009;119:1136–1149. doi: 10.1172/JCI36433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. Ecto-5'-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sitkovsky MV. Use of the A(2A) adenosine receptor as a physiological immunosuppressor and to engineer inflammation in vivo. Biochem Pharmacol. 2003;65:493–501. doi: 10.1016/s0006-2952(02)01548-4. [DOI] [PubMed] [Google Scholar]
- 22.Zhang F, Wang S, Luo Y, Ji X, Nemoto EM, Chen J. When hypothermia meets hypotension and hyperglycemia: the diverse effects of adenosine 5'-monophosphate on cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 2009;29:1022–1034. doi: 10.1038/jcbfm.2009.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki K, Uchida K, Nakanishi N, Hattori Y. Cilostazol activates AMP-activated protein kinase and restores endothelial function in diabetes. Am. J. Hypertens. 2008;21:451–457. doi: 10.1038/ajh.2008.6. [DOI] [PubMed] [Google Scholar]
- 24.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 25.Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, Arumugam TV, Orthey E, Gerloff C, Tolosa E, Magnus T. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849–1857. doi: 10.1161/STROKEAHA.108.534503. [DOI] [PubMed] [Google Scholar]
- 26.Eltzschig HK, Thompson LF, Karhausen J, Cotta RJ, Ibla JC, Robson SC, Colgan SP. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 2004;104:3986–3992. doi: 10.1182/blood-2004-06-2066. [DOI] [PubMed] [Google Scholar]
- 27.Lennon PF, Taylor CT, Stahl GL, Colgan SP. Neutrophil-derived 5'-adenosine monophosphate promotes endothelial barrier function via CD73-mediated conversion to adenosine and endothelial A2B receptor activation. J Exp Med. 1998;188:1433–1443. doi: 10.1084/jem.188.8.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mills JH, Thompson LF, Mueller C, Waickman AT, Jalkanen S, Niemela J, Airas L, Bynoe MS. CD73 is required for efficient entry of lymphocytes into the central nervous system during experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2008;105:9325–9330. doi: 10.1073/pnas.0711175105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bourma M, van der Wildenberg FA, Buurman WA. Adenosine inhibits cytokine release and expression of adhesion molecules by activated human endothelial cells. Am J Physiol. 1996;270:C522–C529. doi: 10.1152/ajpcell.1996.270.2.C522. [DOI] [PubMed] [Google Scholar]
- 30.Henttinen T, Jalkanen S, Yegutkin GG. Adherent leukocytes prevent adenosine formation and impair endothelial barrier function by ecto-5’-nucleotidase/CD73-dependent mechanism. J Biol Chem. 2003;278:24888–24895. doi: 10.1074/jbc.M300779200. [DOI] [PubMed] [Google Scholar]
- 31.Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184:53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mordue DG, Sibley LD. A novel population of Gr-1+-activated macrophages induced during acute toxoplasmosis. J Leukoc Biol. 2003;74:1015–1025. doi: 10.1189/jlb.0403164. [DOI] [PubMed] [Google Scholar]
- 33.Hunter M, Wang Y, Eubank T, Baran C, Nana-Sinkam P, Marsh C. Survival of monocytes and macrophages and their role in health and disease. Front. Biosci. 2009;14:4079–4102. doi: 10.2741/3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deaglio S, Dwyer K, Gao W, Friedman D, Usheva A, Erat A, Chen J, Enjyoji K, Linden J, Oukka M, Kuchroo V, Strom T, Robson S. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niemelä J, Ifergan I, Yegutkin GG, Jalkanen S, Prat A, Airas L. IFN-β regulates CD73 and adenosine expression at the blood-brain barrier. Eur J Immunol. 2008;38:2718–2726. doi: 10.1002/eji.200838437. [DOI] [PubMed] [Google Scholar]
- 36.Kreckler LM, Wan TC, Ge ZD, Auchampach JA. Adenosine inhibits tumor necrosis factor-alpha release from mouse peritoneal macrophages via A2A and A2B but not the A3 adenosine receptor. J Pharmacol Exp Ther. 2006;317:172–180. doi: 10.1124/jpet.105.096016. [DOI] [PubMed] [Google Scholar]
- 37.Hasko G, Kuhel DG, Chen JF, Schwarzschild MA, Deitch EA, Mabley JG, Marton A, Szabo C. Adenosine inhibits IL-12 and TNF-[alpha] production via adenosine A2a receptor-dependent and independent mechanisms. Faseb J. 2000;14:2065–2074. doi: 10.1096/fj.99-0508com. [DOI] [PubMed] [Google Scholar]
- 38.Hasko G, Pacher P, Deitch EA, Vizi ES. Shaping of monocyte and macrophage function by adenosine receptors. Pharmacol Ther. 2007;113:264–275. doi: 10.1016/j.pharmthera.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clausen BH, Lambertsen KL, Babcock AA, Holm TH, Dagnaes-Hansen F, Finsen B. Interleukin-1beta and tumor necrosis factor-alpha are expressed by different subsets of microglia and macrophages after ischemic stroke in mice. J Neuroinflammation. 2008;5:46. doi: 10.1186/1742-2094-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujita T, Minamoto K, Liao H, Naka Y, Pinsky D. Potentiation of Endogenous Fibrinolysis and Rescue from Lung Ischemia/Reperfusion Injury in Interleukin (IL)-10-reconstituted IL-10 Null Mice. J. Biol. Chem. 2000;275:21468–21476. doi: 10.1074/jbc.M002682200. [DOI] [PubMed] [Google Scholar]
- 41.Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, Giese T, Veltkamp R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15:192–193. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- 42.Karandikar NJ, Vanderlugt CL, Eagar T, Tan L, Bluestone JA, Miller SD. Tissue-specific up-regulation of B7-1 expression and function during the course of murine relapsing experimental autoimmune encephalomyelitis. J. Immunol. 1998;161:192–199. [PubMed] [Google Scholar]
- 43.Miller SD, Vanderlugt CL, Lenschow DJ, Pope JG, Karandikar NJ, Dal Canto MC, Bluestone JA. Blockade of CD28/B7-1 interaction prevents epitope spreading and clinical relapses of murine EAE. Immunity. 1995;3:739–745. doi: 10.1016/1074-7613(95)90063-2. [DOI] [PubMed] [Google Scholar]
- 44.Eri R, Kodumudi KN, Summerlin DJ, Srinivasan M. Suppression of colon inflammation by CD80 blockade: evaluation in two murine models of inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:458–470. doi: 10.1002/ibd.20344. [DOI] [PubMed] [Google Scholar]
- 45.Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2:116–126. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- 46.Zernecke A, Bidzhekov K, Ozuyaman B, Fraemohs L, Liehn EA, Luscher-Firzlaff JM, Luscher B, Schrader J, Weber C. CD73/ecto-5'-nucleotidase protects against vascular inflammation and neointima formation. Circulation. 2006;113:2120–2127. doi: 10.1161/CIRCULATIONAHA.105.595249. [DOI] [PubMed] [Google Scholar]