

Abstract
Pyrene-functionalized oligonucleotides (PFOs) are increasingly explored as tools in fundamental research, diagnostics and materials science. Their popularity is linked to the ability of pyrenes to function as polarity-sensitive and quenchable fluorophores, excimer-generating units, aromatic stacking moieties and nucleic acid duplex intercalators. These characteristics have motivated development of PFOs for detection of complementary DNA/RNA targets, single nucleotide polymorphisms (SNPs), and generation of π-arrays on nucleic acid scaffolds. This Review will highlight the physical properties and applications of PFOs that are likely to provide high degree of positional control of the chromophore in nucleic acid complexes. Particular emphasis will be placed on pyrene-functionalized Locked Nucleic Acids (LNAs) since these materials display distinctive properties such as large fluorescence quantum yields, efficient discrimination of SNPs, and recognition of mixed-sequence double stranded DNA.
1. Introduction
Development of pyrene-functionalized oligonucleotides (PFOs) is a research area that enjoys considerable attention. A simple Web of Science search using the terms “oligonucleotide and pyrene”, reveals that more than 230 original research articles have appeared on this topic since 2005. The motivation for this work ranges from a desire to obtain a deeper fundamental understanding of nucleic acid structures over to the prospect of developing enabling tools for wide-ranging applications in materials science, diagnostics and therapeutics. Representative applications include a) generation of self-assembled helical chromophore arrays with distinct photochemical and `communication' properties, b) specific detection of complementary DNA/RNA and single nucleotide polymorphisms (SNPs), and c) targeting of mixed-sequence double stranded DNA.
This Review will highlight PFOs that are likely to offer a high degree of positional control of the chromophore in nucleic acid complexes as these materials display properties which are difficult to mimic with more flexible analogs. Considerable emphasis will be placed on pyrene-functionalized LNAs (Locked Nucleic Acids), i.e., oligodeoxyribonucleotides (ONs) modified with monomers where pyrene moieties are attached via short linkers to LNA or analogs hereof, since higher positional control of the label is expected. The properties of oligonucleotides composed of non-nucleosidic pyrene-functionalized monomers are not covered herein and the reader is directed to recent reviews instead.1–3
The Review is structured to provide: a) a physicochemical background to assist interpretation of key experimental observations with PFOs, b) an overview of pyrene-functionalized nucleotide monomers and their hybridization properties, and c) an introduction to LNA and overview of pyrene-functionalized LNA monomers. This then sets the stage for a survey of recent PFO-based applications.
2. Characteristics of pyrene moieties in nucleic acid contexts
The surface area of the aromatic pyrene moiety (Fig. 1) is comparable to that of Watson-Crick base pairs (stacking area: pyrene ~184 Å2, A:T base pair ~223 Å2).4 As a result, pyrene moieties have a tendency to π-stack with aromatic units (nucleobases and/or other pyrene moieties) and, accordingly, to intercalate into DNA homoduplexes. The latter results in local unwinding of the duplex by ~3.4 Å, bathochromic pyrene absorption shifts, increased probability of fluorescence quenching due to increased interactions with nucleobases (vide infra), and increased duplex thermostability (i.e., increased thermal denaturation temperature Tm).5 Pyrene moieties exhibit a lower tendency for intercalation into A-type duplexes than into B-typew compressed and have less efficient nucleobase overlap.6
Fig. 1.
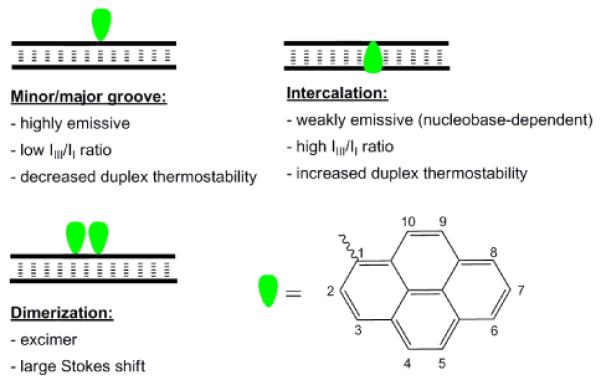
Position-dependent pyrene characteristics in nucleic acid duplexes.
The absorption spectrum of pyrene exhibits maxima at 275, 320 and 337 nm in deoxygenated CH2Cl2. The corresponding fluorescence emission spectrum exhibits five maxima (372, 378, 383, 393 and 413 nm); however, only vibronic bands I and III are typically observed due to signal broadening.7 The relative ratio of vibronic bands I and III is sensitive to solvent polarity, e.g., higher IIII/II-ratios are observed in apolar environments.8 This sensitivity can indicate the position of pyrene labels in nucleic acid duplexes due to differential polarities of the minor/major groove (more polar) and duplex core (more apolar). Nearby nucleobase moieties can quench pyrene fluorescence via photoinduced electron transfer (PET). Guanine moieties are regarded the strongest quenchers (via oxidative electron transfer from guanine); cytosine, and to a lesser degree, thymine are considered moderately strong quenchers via reductive electron transfer to nucleobase, while adenine is a weak quencher.9–11 Thus, higher fluorescence emission quantum yields ΦF (i.e., ratio of photons emitted as fluorescence relative to absorbed photons) are expected when a pyrene moiety is located in the polar grooves, than when intercalated in a GC-rich duplex core (Fig. 1).
Pyrene chromophores may display excimer fluorescence when a pyrene in an electronically excited state forms a π-stacking dimer with a pyrene in its ground state.12 Excimer emission is broad, featureless, and markedly red-shifted (λem ~490 nm) relative to pyrene monomer fluorescence. Formation of pyrene excimers can be exploited diagnostically and yield valuable structural information as the co-planar pyrenes must be separated by ~3.4 Å for efficient excimer formation (Fig. 1).
Development of modified pyrene fluorophores with red-shifted absorption and fluorescence emission maxima is desirable to minimize auto-fluorescence of biomolecules during in vivo applications. The photophysical properties of pyrene are easily modulated by direct electronic coupling to a nucleobase moiety or by extending π-conjugation. Electronic coupling to a nucleobase is most easily achieved by direct attachment of a pyren-1-yl moiety to the 5-position of 2'-deoxyuridine (2, Fig. 2). Excitation of the pyrene results in charge-transfer to the nucleobase giving a broad, red-shifted and solvent dependent emission band (λem ~495 nm).13 While 8-(pyren-1-yl)-2'-deoxyguanosine monomer 9 also displays electronic coupling (λem ~460 nm),14 attachment of pyren-1-yl to the C5-position of 2'-deoxycytidine15 or of pyren-2-yl to the C5-position of 2'-deoxyuridine does not.16 Electronic coupling to a nucleobase can also occur through unsaturated linkers; 5-(1-ethynylpyrenyl)-2'-deoxyuridine and 8-(1-ethynylpyrenyl)-2'-deoxyadenosine monomers 3 and 11 display bathochromic shifts in UV absorption and unstructured fluorescence emission (λem ~450 nm) due to exciplex formation between the pyrene and nucleobase moieties (Fig. 2). Interestingly, 5-(1-ethynylpyrenyl)-2'-deoxycytidine or 8-(1-ethynylpyrenyl)-2'-deoxyguanosine 12 do not result in electronic coupling.17
Fig. 2.
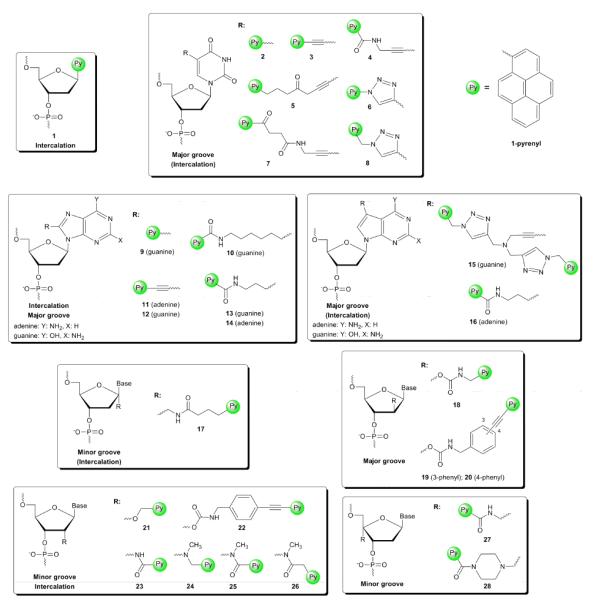
Pyrene-functionalized monomers and anticipated location of the pyrene moiety in nucleic acid duplexes.
Extension of π-conjugation through alkyne substitution is another approach to modulate the photophysical properties of pyrene moieties.18–19 For example, attachment of two phenylethynyl moieties at different positions on pyrene results in marked bathochromic shifts in absorption (up to 40 nm) and fluorescence emission (up to 80 nm) relative to mono-substituted pyrene derivatives.19
3. Pyrene-functionalized nucleotide monomers
Pyrene moieties can be linked to nucleosides: a) by replacing the nucleobase moiety, b) through attachment to the nucleobase moiety, and c) through attachment to the sugar moiety. The attachment point and linker length influence the position and characteristics of the pyrene label within a nucleic acid duplex. An overview of major PFO classes is provided in the following subsections along with a brief discussion of their hybridization properties and likely pyrene binding modes. The Reader should note that direct comparison of relative effects on duplex thermostability upon incorporation of different pyrene-functionalized monomers is not possible due to sequence-dependent effects and the lack of a consensus set of model targets. Moreover, the discussion of binding modes predominantly relies on indirect structural data (e.g., fluorescence emission spectra and molecular modeling) as direct structural data (e.g., NMR structures) only has been reported in few cases.
3.1 Monomers with nucleobase surrogates
Substitution of the nucleobase with a pyrene, positions the aromatic moiety into the base stack of nucleic acid duplexes. DNA duplexes that are singly modified with monomer 1 are destabilized relative to unmodified duplexes due to local disruption of Watson-Crick base pairing (ΔTm = −7 to −5 °C), and display universal base behavior, i.e., almost identical Tm-values regardless of the nucleotide opposite to the modification.20 The latter, most likely, reflects the ability of the pyrene moiety to provide a π-stacking surface comparable to a Watson-Crick base pair and to partially flip out the opposing nucleobase from the duplex core (vide infra).
3.2 Nucleobase-modified monomers
Numerous building blocks have been developed where pyrene moieties are attached to the C5-position of pyrimidines or C8-position of purines (Fig. 2). The synthesis of these monomers is relatively straightforward and involves palladium-catalyzed cross couplings (i.e., Sonogashira, Heck, Suzuki-Miyaura) on C5-halogenated pyrimidines or C8-halogenated purines as the key step. Pyrene moieties can, alternatively, be introduced via Cu (I) catalyzed azide alkyne Huisgen 1,3-dipolar cycloadditions on C5-ethynyl-modified pyrimidines to give triazole-linked pyrene-functionalized nucleotides.21 Small C5-substituents such as propyn-1-yl and 3-aminopropyn-1-yl, point toward the major groove of duplexes leading to considerable increases in thermal denaturation temperature relative to unmodified ONs (ΔTm/modification ~4 °C).22,23 Larger, more hydrophobic C5-substituents such as pyrene moieties that are attached via short rigid linkers as in 2–4, 6 and 8 (Fig. 2), typically result in markedly decreased duplex thermostability.21,24–26 Molecular modeling studies and fluorescence data (vide infra) suggest that the nucleobase of 4 adopts an anti conformation (Watson-Crick base pairing orientation) to position the pyrene in the major groove upon hybridization with complementary DNA, while adopting a syn conformation upon hybridization to mismatched DNA, which putatively results in pyrene intercalation.26 This binding mode is also likely when long flexible linkers are used as in monomers 5 and 7 (Fig. 2), which results in markedly increased duplex thermostability (ΔTm/mod +7 to +10 °C).24,27
C8-pyrene-functionalized purine monomers with short linkers between the pyrene and nucleobase moieties greatly destabilize DNA duplexes presumably since syn-conformations are adopted (e.g., 9, Fig. 2).14 The use of a more flexible linker (e.g. 14, Fig. 2) stabilizes DNA duplexes although the pyrene binding mode is not clear.28
C7-functionalized 7-deazapurines are well-tolerated in duplexes when carrying small substituents, leading to the hypothesis that these monomers predominantly adopt anti-conformations to place C7-substituents in the major groove.29 It is therefore surprising that only few C7-pyrene-functionalized 7-deazapurines have been reported in literature (e.g. 15 and 16, Fig. 2).30–31 Incorporation of monomer 16 into a 13-mer DNA sequence results in increased duplex thermostability (ΔTm/mod = +3.0 °C).30
3.3 Sugar-modified monomers
The furanose ring has been widely explored as an attachment site for pyrene moieties. Incorporation of C1'-pyrene-functionalized monomer 17 into ONs, obtained through post-synthetic conjugation of HBTU/HOBT-activated pyrene butyric acid with 1'-aminomethylthymidine under microwave conditions, increases DNA duplex thermostability (ΔTm/mod up to +3.5 °C/mod). Molecular modeling studies suggest that the substituent points toward the minor groove although intercalation cannot be excluded due to the flexible linker.32
Monomers 2733 and 2834 are two prominent examples of C4'-pyrene-functionalized monomers, which are obtained by post-synthetic conjugation of activated pyrene-1-carboxylic acid with 4'-amino methylthymidine and 4'-piperazinomethylthymidine, respectively (Fig. 2). Incorporation of monomer 27 into ONs only has a minor effect on DNA duplex thermostability, presumably because the pyrene moiety is directed toward the minor groove. Hybridization of DNA strands with mismatched nucleotides opposite of monomer 27 only leads to moderately decreased duplex thermostability suggesting an intercalative binding mode of the pyrene moiety, which is substantiated by fluorescence experiments (vide infra).33 Incorporation of the piperazino analog 28 into ONs results in markedly increased thermal affinity toward DNA (ΔTm ~ +7 °C) and decreased thermal affinity toward RNA complements (ΔTm ~ −8 °C).34 The pronounced DNA-selectivity suggests an intercalative binding mode of the pyrene moiety, while the excellent mismatch discrimination and fluorescence data indicate a minor groove binding mode.
The 2'-position on the sugar ring has been a rigorously studied site for attachment of pyrene moieties. Single incorporation of 2'-O-(pyren-1-yl)methyluridine monomer 21 into DNA strands results in stabilization of DNA:DNA duplexes, the extent of which greatly depends on whether a pyrimidine (ΔTm = ~2 °C) or purine (ΔTm = 7–13 °C) is flanking the modification on the 3'-side (Fig. 2).35 Modified DNA:RNA duplexes are strongly destabilized and display similar sequence dependence (ΔTm = −10 °C to −1 °C).35 Incorporation of a single monomer 21 into RNA strands leads to minor stabilization of RNA:DNA duplexes (ΔTm = 0 to +3 °C) and destabilization of RNA:RNA duplexes (ΔTm = −3 to −4 °C).36 NMR studies demonstrate that the pyrene moiety predominantly points into the minor groove in RNA:RNA duplexes, while it is intercalated toward the 3'-side in DNA:DNA duplexes indicating that the abovementioned sequence-dependent hybridization properties are linked to the π-stacking surface size of the 3'-flanking nucleotide.37 These characteristics of 21 have been used to develop hybdridization probes for detection of specific RNA targets (vide infra).36
Korshun and coworkers studied a series of pyrene-functionalized 2'-carbamate monomers, which are obtained from the free nucleoside via a robust six step reaction sequence featuring a carbonyldiimidazole-activated carbamate formation as the key step.38–39 The effects upon incorporation of ribo-configured monomer 22 (Fig. 2) into DNA strands range from moderately increased to moderately decreased thermal affinity toward complementary DNA/RNA. The pyrene moiety is presumably located in the minor groove but structural evidence has not been obtained.38–39 Incorporation of the corresponding arabino-configured monomers 18–20 (Fig. 2) into DNA strands leads to markedly reduced thermostability of duplexes with DNA complements (ΔTm = −2 to −8 °C). Molecular modeling indicates that the pyrene moiety is located in the major groove.40–41
N2'-functionalization of 2'-amino-DNA analogs has been another popular approach toward introduction of pyrene moieties. The key synthetic step involves chemoselective N-alkylation or N-acylation of 2'-amino-DNA intermediates. Single incorporation of pyrene-functionalized monomers 2342, 2443, 2544 or 2644 into 5'-d(GTG ABA TGC) leads to pronounced increases in DNA:DNA duplex thermostability (vs DNA: ΔTm = +3, +15, +3, and +6 °C, respectively). The strong DNA-selectivity of these monomers (vs RNA: ΔTm = −6, +2, −8, and +1 °C, respectively), along with data from fluorescence spectroscopy, indicates that the pyrene binding mode is similar as for the previously discussed 2'-O-(pyren-1-yl)methyluridines, i.e., intercalation upon hybridization with DNA targets and minor groove positioning with RNA targets.
4. Pyrene-functionalized LNA
4.1 Biophysical characteristics of LNA
Locked nucleic acid (LNA) was independently developed in the end of the 1990's by the Wengel45 and Imanishi46 laboratories (Fig. 3). The oxymethylene bridge spanning the C2′- and C4′-positions efficiently locks the furanose ring in a C3′-endo conformation similar to the conformations of ribonucleotides in RNA duplexes.47 Moreover, the biyclic LNA skeleton restricts the glycosidic torsion angle due to a steric clash between H6(pyrimidine)/H8(purine) and the pseudoaxial H3', which is exacerbated by the large puckering amplitude of LNA nucleotides (Fig. 3).48 Incorporation of LNA monomers into ONs results in pre-organization of neighboring nucleotides toward increased N-type sugar conformation character, both in single stranded ONs47,49 and corresponding duplexes with complementary DNA/RNA.47,50 The limited internal flexibility of LNA nucleotides restricts conformational interconversion of adjacent nucleotide residues as they are mechanically coupled, whereby duplex dynamics are damped.51 As a result, LNA-modified ONs display very high thermal affinity toward single stranded DNA/RNA targets (ΔTm/mod up to +10 °C) and high target specificity (i.e., mismatch discrimination). LNA has, accordingly, been extensively explored as a fundamental research tool for modulation of gene expression via the antisense approach and evaluated as a RNA-targeting drug candidate against diseases of genetic origin.52
Fig. 3.
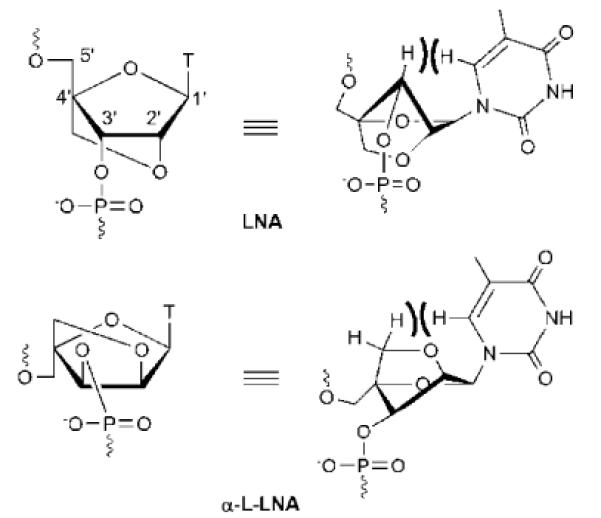
Structure of LNA-T and α-L-LNA-T monomers. Numbering of sugar ring carbons is shown for LNA.
One of the LNA diastereoisomers, i.e., α-L-LNA (Fig. 3) shares many of the desirable hybridization properties of LNA despite its `unnatural' configuration at the C2'-, C3'- and C4'-positions.53 Unlike LNA, which is considered an RNA mimic, α-L-LNA is considered a DNA mimic since duplexes between DNA/α-L-LNA-mixmers and DNA or RNA complements adopt geometries that globally resemble unmodified duplexes.54,55 It follows that α-L-LNA also is as an interesting antisense building block.56 For additional background on the LNA technology, we refer to the following reviews.57–58
In light of the beneficial hybridization properties and conformational rigidity of LNA, α-L-LNA and the corresponding 2'-amino analogs,59–60 the Wengel and Hrdlicka laboratories set out to develop pyrene-functionalized LNAs to study the fundamental properties of duplexes composed of PFOs that offer a high degree of positional control of the pyrene moiety (Fig. 4).
Fig. 4.
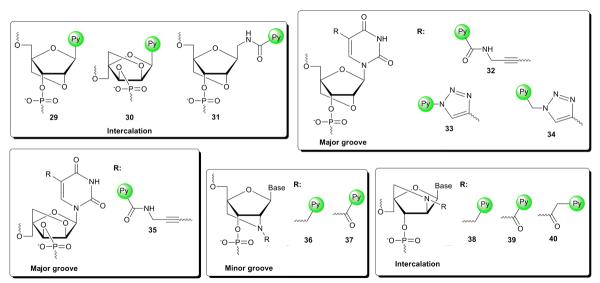
Pyrene-functionalized LNAs and anticipated location of the pyrene moiety in nucleic acid duplexes.
4.2 LNA monomers with nucleobase surrogates
Wengel and coworkers have developed three different LNA monomers where a pyrene has replaced the nucleobase moiety, i.e., LNA C-glycoside 29,61 α-L-LNA C-glycoside 30,62 and LNA aminomethyl C-glycoside monomer 31 (Fig. 4).63 Interestingly, the affinity-enhancing effect of the LNA skeletons is fully compromised as evidenced by the extensive destabilization of singly modified DNA duplexes (ΔTm down to −11 °C). These probes display similar universal base behavior as the structurally simpler monomer 1 (Fig. 2).
4.3 Nucleobase-modified LNA monomers
Hrdlicka and coworkers developed a series of C5-functionalized LNA and α-L-LNA pyrimidines based on the hypothesis that the conformationally restricted sugar ring and restricted glycosidic torsion angle would reduce the available conformational space for C5-substituents in the major groove relative to DNA analogs. C5-functionalized LNA carrying non-aromatic moieties are tolerated extraordinarily well in nucleic acid duplexes (ΔTm/mod up to + 13 ° C).64 Interestingly, incorporation of 5-[3-(1-pyrenecarboxamido)propynyl]-functionalized LNA and α-L-LNA monomers 32 and 35 into DNA duplexes (Fig. 4) results in similar decreases in DNA duplex thermostability (ΔTm/mod = −5 to −2 °C) as the corresponding 2'-deoxyuridine analog 4 (Fig. 2).11 Similar trends have been observed for C5-triazole-linked pyrene-functionalized LNA monomers 3365 and 3465 (Fig. 4) relative to their corresponding 2'-deoxyuridine analogs (for 6: ΔTm/mod = −14 to −4 °C, for 8: ΔTm/mod = −7 to +2 °C).21 Although the stabilizing effects of the parent LNA and α-L-LNA skeletons are largely compromised by these C5-substituents, fluorescence emission spectra suggest that high positional control of the fluorophore is realized (vide infra).11
4.4 N2'-Functionalized 2'-amino-LNA & 2'-amino-α-L-LNA
N2'-functionalized 2'-amino-LNA66 and 2'-amino-α-L-LNA67 were developed by Wengel, Hrdlicka and coworkers based on the hypothesis that a N2'-substituent attached via a short linker would be positioned more precisely within duplexes than with more flexible monomers. Most N2'-functionalized 2'-amino-LNA display similar thermal affinities toward complementary DNA/RNA targets as the corresponding parent LNA.66 ONs modifed with 2'-N-(pyren-1-yl)carbonyl-2'-amino-LNA monomer 37 also follow this trend, while ONs modified with the slightly more flexible 2'-N-(pyren-1-yl)methyl-2'amino-LNA monomer 36 display more modest thermal affinities toward complementary DNA/RNA (Fig. 4). Fluorescence emission spectra (vide infra) and molecular modeling studies suggest that the N2'-substituent predominantly is located in the minor groove (Fig. 5).
Fig. 5.
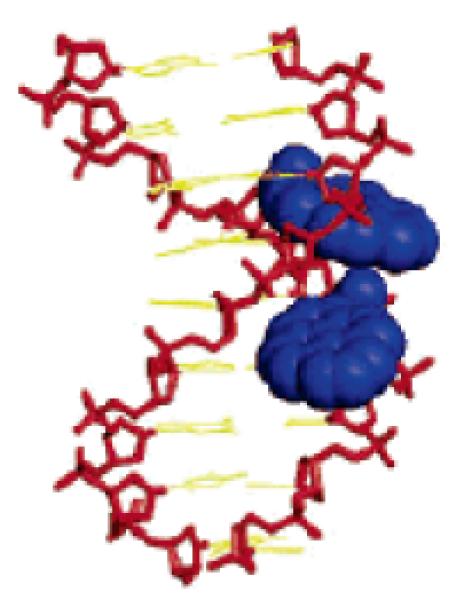
Placement of pyrene moieties in the minor groove using N2'-functionalized 2'-amino-LNA. Molecular modelling structure of duplex between 5'-d(GCA 37A37 CAC) and 3'-d(CGT ATA GTG). Monomer 37: 2'-N-(pyren-1-yl)carbonyl-2'-amino-LNA-T. (Reproduced with permission from ref. 90. Copyright 2005 American Chemical Society).
2'-Amino-α-L-LNA carrying non-aromatic moieties at the N2'-position display dramatically decreased thermal affinity toward DNA/RNA targets (e.g., ΔTm/mod down to −16.5 °C for a N2'-acetyl group).67 In contrast, exceptional increases in duplex thermostability are observed for ONs modified with pyrene-functionalized monomers 38, 39 and 40 (Fig. 4). DNA:DNA duplexes are significantly more stabilized than DNA:RNA duplexes (vs DNA: ΔTm/mod = 6–15, 10–19 and 10–16 °C, respectively; vs RNA: ΔTm/mod = −1 to +7, 4–11 and 6–12 °C, respectively).67 The extensive DNA selectivity, along with hybridization-induced bathochromic shifts of pyrene absorption maxima, increased circular dichroism signal intensity in the pyrene region, and molecular modeling studies, suggest pyrene intercalation in the duplex core. Closer scrutiny of the molecular structure reveals that the 2-oxo-5-azabicyclo[2.2.1]heptane skeleton of monomers 38–40 is locking the attachment points of the nucleobase and the pyrene moiety (i.e., C1' and N2') relative to each other. This, along with the short linkers connecting the pyrene units to the bicyclic skeleton, most likely, preorganizes the pyrene moieties to intercalate in the 3'-direction (Fig. 6).
Fig. 6.
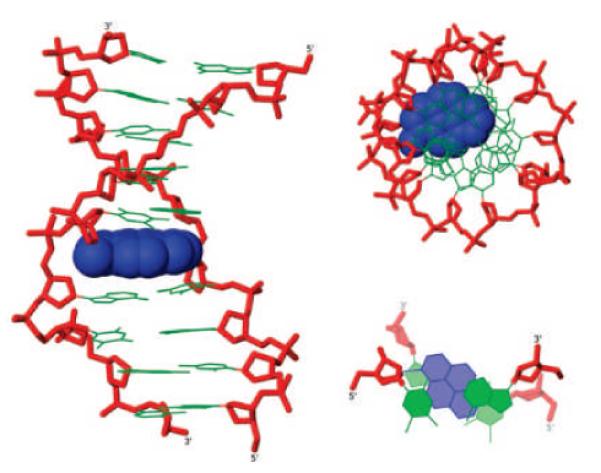
Placement of pyrene moieties in the duplex core using N2'-functionalized 2'-amino-α-L-LNA. Different representations of molecular modelling structure of duplex between 5'-d(GTG A38A TGC) and 3'-d(CAC TAT ACG). Monomer 38: 2'-N-(pyren-1-yl)methyl-2'-amino-α-L-LNA-T. (Reproduced with permission from ref. 67. Copyright 2009 American Chemical Society).
5. Generation of pyrene arrays on nucleic acids
Nucleic acids have received considerable attention due to their potential as building blocks in bottom-up nanotechnology due to: a) their predictable and programmable self-assembly that is governed by Watson-Crick base pairing rules; b) their well-defined and naturally nanometer-sized dimensions; c) their straightforward synthesis and; d) the availability of experimental techniques for additional structural manipulation of nucleic acids (e.g., ligation, digestion, PCR amplification). A remarkable range of DNA-based structures such as nanowires, lattices and octahedrons have been reported.68 Periodic organization of matter (e.g., nanoparticles and proteins) on two-dimensional DNA arrays is one of many promising applications of bottom-up DNA nanotechnology. The use of nucleic acids as supramolecular scaffolds for arrangement of chromophores has lately received considerable interest due to the prospect of developing DNA-based light harvesting complexes.3,69,70 Pyrene arrays have been studied as a first step toward this end.
5.1 Pyrene arrays in the minor groove
Nakamura, Yamana and coworkers hybridized 2'-O-methyl RNA strands that are sequentially modified with 2'-O-(pyren-1-yl)methyluridine 21 monomers to RNA complements (Fig. 7).71 The intense excimer emission along with data from UV-vis spectroscopy, circular dichroism and molecular modeling studies, provides indirect evidence for formation of pyrene arrays in the minor groove (Fig. 7).71
Fig. 7.
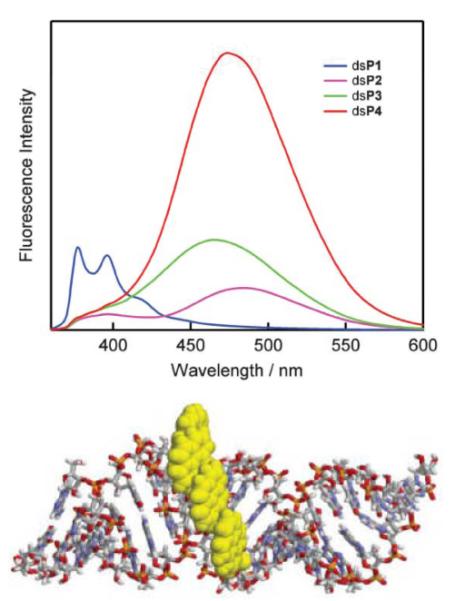
Formation of pyrene arrays in the minor groove using 2'-O-(pyren-1-yl)methyluridine 21 monomers. Upper: fluorescence emission spectra of duplexes between 19-mer 2'-O-methyluridine strands centrally modified with 1–4 sequential 21 monomers and complementary RNA (dsP1–dsP4, λex = 350 nm). (Reproduced with permission from ref. 144 Copyright 2005 RSC) Lower: molecular modelling structure of dsP4. (Reproduced with permission from ref. 71. Copyright 2007 RSC Publishing).
Wengel, Hrdlicka and coworkers used two complementary ONs modified with 2'-N-(pyren-1-yl)methyl-2'amino-LNA monomer 36 to form DNA duplexes featuring interstrand pyrene arrays in the minor groove (Fig. 8).72 Thus, positioning of monomers 36 i n “−1 interstrand zipper arrangements” results in extensive duplex stabilization and intense excimer formation signifying strong stacking arrangements of pyrene moieties. The increased duplex thermostability is particularly noteworthy considering that isolated 36 monomers only have minor effect on thermostability (Fig. 8).72 More brightly fluorescent analogs of this excimer-forming communication system, based on 2'-amino-LNA monomers that are N2-functionalized with 1-, 2- and 4-(phenylethynyl)pyrenes, have been developed recently.73 Incorporation of these excimer-forming switches at strategic positions has been utilized to signal stepwise self-assembly of higher order nucleic acid structures (Fig. 8).74
Fig. 8.
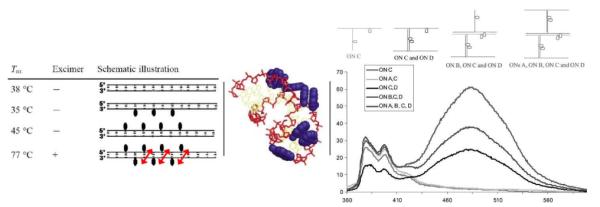
Formation of pyrene interstrand zipper arrays using 2'-N-(pyren-1-yl)methyl-2'-amino-LNA monomer 36. Left: Tm-values and fluorescence characteristics of DNA duplexes modified with monomer 36. Arrows indicate −1 zipper orientation.63Middle: molecular modelling structure of duplex with three −1 interstrand arrangements of monomer 36 (Tm = 77 °C). (Adapted from ref. 72. Copyright 2004 RSC Publishing). Right: fluorescence emission spectra illustrating stepwise self-assembly of higher order nucleic acid structures (using monomer 36). (Adapted with permission from ref. 74. Copyright 2005 Taylor & Francis).
Nakamura, Yamana and coworkers subsequently generated interstrand pyrene-zipper arrays in the minor groove using RNA duplexes modified with 2'-O-(pyren-1-yl)methyl uridine/adenosine pairs (i.e., 0-interstrand zippers) positioned as next-nearest neighbors.75 Array formation is supported by intense excimer emission and molecular modeling studies. While formation of the pyrene array counteracts the negative impact on RNA duplex thermostability by 2'-O-(pyren-1-yl)methyl nucleotides, the resulting duplexes are still less stable than unmodified RNA duplexes.75 The extraordinary duplex thermostability, directional preference and robustness of the 2'-N-(pyren-1-yl)methyl-2'amino-LNA communication system, underscores the potential advantages of pyrenefunctionalized LNA.72
5.2 Pyrene arrays in the major groove
Positioning of pyrenes in the major groove of nucleic acid duplexes was first suggested by Korshun and coworkers who utilized DNA duplexes modified with `+2 interstrand zipper arrangements' of arabino-configured 2'-carbamate monomer 18 (Fig. 2), resulting in the formation of excimers.40
Wagenknecht and coworkers observed a) strong exciton coupling of pyrene signals in the circular dichroism spectra and b) strongly increased and blue-shifted fluorescence emission (λem ~ 445 nm) upon hybridization of ONs containing five sequential incorporations of 5-(pyren-1-yl)-2'-deoxyuridine monomer 2 (Fig. 2) with complementary DNA, which indicates formation of an electronically coupled pyrene array in the major groove.76 Interestingly, hybridization with DNA strands containing two or more mismatched nucleotides opposite of the modified region, results in less intense fluorescence emission, suggesting that the presence of mismatched base pairs perturbs the electronically coupled organization of pyrenes.76 Similar observations have been made with the corresponding 5-(1-ethynylpyrenyl)-2'-deoxyuridine monomer 3 (Fig. 2).77
5.3 Pyrene arrays in the duplex core
The nucleic acid duplex core can be engineered to generate metal ion arrays.78 Similarly, pyrene arrays can be formed inside duplex cores due to the comparable π-stacking area of pyrene moieties and Watson-Crick base pairs. Kool and coworkers demonstrated that DNA duplexes containing `base pairs' between C-glycoside 1 and abasic sites Φ display similar thermostability as reference duplexes with native A:T pairs (Fig. 2).20 However, duplex integrity is lost if more than two out of ten regular base pairs are replaced by 1:Φ pairs.20 Similar results have been observed using LNA aminomethyl C-glycoside monomer 31.63
Hrdlicka, Wengel and coworkers studied hybridization properties of ONs modified with 5'-(YΦ)-units, i.e., where a 2′-N-(pyren-1-yl)acetyl-2′-amino-α-L-LNA monomer 40 (= Y) is flanked by an abasic site Φ on the 3′-side.79 These ONs display extraordinary thermal affinity toward DNA strands containing abasic sites in +1 interstrand positions (Fig. 9). Thus, a 13-mer DNA duplex containing four of these 5'-(YΦ):3'-(AΦ)-units separated by one base pair, exhibits a thermal denaturation temperature of ~60 °C. In contrast, the corresponding 13-mer reference duplex where 5'-(YΦ):3'-(AΦ)-units are replaced by 5'-(TA):3'-(AT) base pairs has a Tm of 35 °C. Molecular modeling studies strongly suggest that the pyrene moiety of N2'-functionalized 2'-amino-α-L-LNA monomer 40 is located in the duplex core void formed by the Φ:Φ pair and partakes in efficient π-π stacking with neighboring base pairs (Fig. 9).79
Fig. 9.
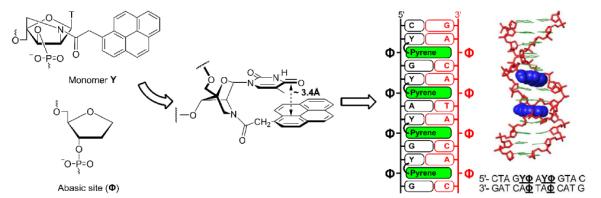
Left: illustration of pyrene array formation in the duplex core using 5'-(YΦ):3'-(AΦ)-units where Y denotes 2'-N-(pyren-1-yl)acetyl-2'-amino-α-L-LNA-T monomer 40 and monomer Φ is an abasic site analog. Right: molecular modelling structure of a 13-mer DNA duplex containing two separated 5'-(YΦ): 3'-(AΦ)-units. (Adapted with permission from ref. 79. Copyright 2008 American Chemical Society).
Häner and coworkers have recently developed so-called oligopyrenotides, i.e.,. `duplexes' composed of achiral phosphodiester-linked pyrene building blocks and a single chiral 1,2-diaminocyclohexane, which feature a continuous pyrene array in the core. This highlights the possibility of creating purely artificial self-assembling structures and demonstrates the potential of pyrene moieties as building blocks in nanotechnology.80
The use of nucleic acids as supramolecular scaffolds to arrange fluorophores in arrays can be used to force two or more fluorophores to interact in the excited state to form red-shifted excimers (between identical fluorophores) or exciplexes (between different fluorophores).81 Kool and coworkers studied all 4096 combinations (84 = 4096) of a tetramer that is modified at every position with any of eight building blocks (seven different C-glycosides or an abasic site). This resulted in the isolation of tetramers with very different emission characteristics (λmax = 376–633 nm) despite using identical excitation conditions (λem = 340–380 nm).82
6. Diagnostic applications of PFOs
Reliable detection of specific nucleic acid sequences is an integral component of a wide range of applications including quantification of DNA/RNA in real-time PCR,83 detection of acceptor sites for antisense oligonucleotides,84 visualization of RNA trafficking in cells,85 detection of SNPs,86 and pathogen detection.87 Development of fluorophore-modified ONs,88 and PFOs in particular, has received major attention over the past decade. Pyrene-functionalized monomers are used as components of a) hybridization probes, b) base discriminating fluorescent probes, c) excimer-forming probes, and d) quencher-free molecular beacons.
6.1 Hybridization probes
Hybridization probes are designed to display a) low levels of fluorescence emission in the single-stranded state, typically through quenching interactions between pyrene and nucleobase moieties,9 and b) high levels of fluorescence emission in the double-stranded state by placing the pyrene moiety in a non-quenching environment (Fig. 10).
Fig. 10.
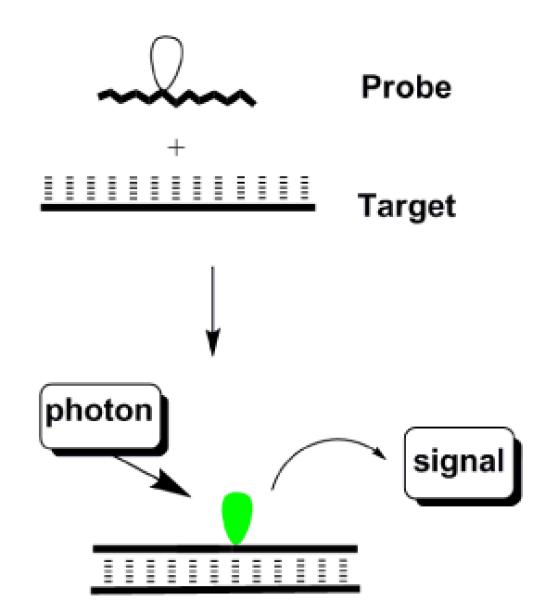
Principle of hybridization probes. White and green droplets represent silent and emissive fluorophore.
Yamana, Murakami and coworkers discovered that hybridization between RNA probes modified with 2'-O-(pyren-1-yl)methyluridine monomer 21 (Fig. 2) and RNA targets results in pronounced increases in fluorescence intensity (10–30-fold) to give duplexes with emission quantum yields up to 24%.36 Much smaller increases are observed upon hybridization with DNA targets,36 or if corresponding DNA probes are hybridized to DNA/RNA targets.35 This reflects the fact that intercalation of the pyrene moiety into the quenching base stack is a major binding mode in DNA:DNA and DNA:RNA, but not RNA:RNA duplexes.37 These data demonstrate how functional control is closely linked with positional control of the pyrene moiety. 2'-O-methyl-RNA probes modified with monomer 21 display extraordinary increases in fluorescence intensity upon hybridization with RNA (up to ~300-fold) but only if flanked by a 3'-cytosine or guanine moiety, which limits the generality of these probes. These neighboring bases are most likely needed to ensure sufficient quenching of the single stranded probe.84 A hexitol nucleic acid (HNA) analog of monomer 21, i.e., monomer 41 (Fig. 11), has recently been incorporated into HNA backbones. However, only very modest changes in fluorescence intensity were observed upon hybridization with RNA targets.89
Fig. 11.
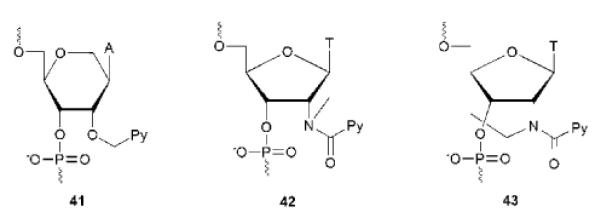
Pyrene-functionalized monomers utilized in hybridization probes. Py = pyren-1-yl.
Hrdlicka and coworkers have developed hybridization probes based on ONs modified with a single pyrene-functionalized triazole-linked 2'-deoxyuridine monomer 8 (Fig. 2). Hybridization with complementary DNA or RNA targets results in very large increases in fluorescence emission at λem = 382 nm (9- to 23-fold), except when monomer 8 is flanked by thymine moieties. Emission quantum yields up to 16% were observed.21
In a different study, Hrdlicka, Wengel and coworkers discovered that DNA strands modified with 2'-N-(pyren-1-yl)carbonyl-2'-amino-LNA monomer 37 display large increases in fluorescence intensity upon hybridization with complementary DNA and RNA (between 2.5- and 69-fold).90 To achieve such increases, two or more non-sequential 37 monomers must be incorporated into ONs since a) this lowers the fluorescence intensity of single stranded probes by facilitating quenching interactions between pyrene and/or nucleobase moieties, and b) each incorporation results in near-additive increases in fluorescence intensity.90 Duplexes between appropriately designed probes and DNA/RNA targets are highly thermostable and brightly fluorescent with emission quantum yields between 28–99% (Table 1). Equivalent RNA or 2'-O-methyl-RNA backbone-based probes lead to even larger hybridization-induced increases in fluorescence intensity due to more efficient fluorescence quenching of the single stranded probes and higher duplex quantum yields.91 Phosphorothioate DNA (PS-DNA) based probes display similar characteristics as the parent DNA probes, whereas single stranded LNA backbone-based probes display high quantum yields even when they are doubly modified. This presumably reflects pre-organization of the single-stranded LNA in a duplex-like conformation. The observed duplex quantum yields with these `Glowing LNA' are two- to four-fold higher than for state-of-the-art PFOs composed of 2'-O-(pyren-1-yl)methyluridine 2136 or C4-pyrene-functionalized monomer 2733 (Fig. 2), which is an unlocked variant of monomer 37. Moreover, Glowing LNA are a) much less sensitive to the nature of flanking nucleotides than other PFO-based hybridization probes,90,92 and b) equally suitable for detection of DNA and RNA targets.90–92 These desirable optical characteristics are hypothesized to be a consequence of the conformationally locked bicyclic skeleton and the short amide linkage of monomer 37 working in concert to firmly position the pyrene moiety in the non-quenching minor groove (Fig. 5).90
Table 1.
Thermal denaturation temperatures (Tm) and fluorescence quantum yields (ΦF) of singly and doubly modified Glowing LNA probes in different backbone contexts and their duplexes with complementary DNA/RNA (1X, 5'-GCA T(U)A37 CAC; 2X, 5'-GCA 37A37 CAC). (Reproduced with permission from ref. 91. Copyright 2010 American Chemical Society).
|
Tm (△Tm/mod)/°C |
quantum yield (ΦF) |
||||
|---|---|---|---|---|---|
| probe | +DNA | +RNA | SSP | +DNA | +RNA |
| DNA 1X | 32.5 (+3.0) | 33.0 (+6.0) | 0.50 | 0.79 | 0.91 |
| DNA 2X | 40.5 (+5.5) | 41.5 (+7.5) | 0.27 | 0.81 | 0.70 |
| PS-DNA 1X | 24.0 (+3.5) | 26.0 (+7.5) | 0.50 | 0.64 | 0.66 |
| PS-DNA 2X | 32.0 (+6.0) | 35.0 (+8.5) | 0.20 | 0.61 | 0.62 |
| RNA 1X | 31.5 (+4.5) | 42.5 (+4.5) | 0.84 | 0.89 | 0.93 |
| RNA 2X | 38.0 (+5.5) | 46.5 (+4.0) | 0.16 | 0.85 | 0.89 |
| O2'-Me 1X | 31.0 (+5.0) | 45.0 (+4.5) | 1.00 | 0.97 | 0.98 |
| O2'-Me 2X | 20.0 (−3.0) | 46.0 (+3.0) | 0.14 | 0.68 | 0.86 |
| LNA 1X | 54.5 (−3.5) | 64.0 (−5.0) | 0.95 | 0.90 | 0.89 |
| LNA 2X | 49.0 (−4.5) | 60.5 (−4.5) | 0.72 | 0.92 | 0.84 |
Several Glowing LNA analogs have been subsequently studied. Wengel and coworkers changed the fluorophore from (pyren-1-yl)carbonyl to various (phenylethynyl)pyrene moieties which generally results in red-shifted absorption and fluorescence emission. High quantum yields are observed for some of these analogs but it is currently unclear if they follow similar design guidelines and are as general as the parent Glowing LNA.19,73 Chattopadhyaya and coworkers attached the (pyren-1-yl)carbonyl moiety on two other conformationally restricted nucleotides, i.e., azetidine and aza-ENA (ethylene nucleic acid) thymine monomers 42 and 43, respectively (Fig. 11).93 DNA strands that are singly modified with monomer 42 display strongly reduced thermal affinity toward RNA targets and duplex quantum yields that vary considerably with the sequence context (ΦF = 0.06–0.73, lower with 3'-flanking G). Hybridization to complementary RNA only results in modestly increased quantum yields; however, it is possible to observe large hybridization-induced increases at λex = 378 nm since hybridization is accompanied with a marked blue-shift in fluorescence emission due to destacking of the pyrene and nucleobase moieties. ONs that are singly modified with monomer 43 seem to display smaller hybridization-induced increases in fluorescence intensity and duplex quantum yields (9–47%) than the parent Glowing LNA but direct comparison is complicated by the use of different sequence contexts.93 Even the conservative change from a (pyren-1-yl)carbonyl moiety to a slightly more flexible (pyren-1-yl)methyl moiety (i.e., monomer 36, Fig. 4) results in markedly less intense fluorescence emission.72 Collectively these examples indicate the importance of precise positioning of the pyrene moiety to achieve the desired function.
6.2 Base discriminating fluorescent probes
ONs modified with base discriminating fluorescent (BDF) monomers generate different levels of fluorescence emission based on the nature of the nucleotide opposite of the modification.94–95 Most BDF probes that are based on pyrene-functionalized monomers rely on a simple principle - the pyrene moiety is a) positioned in a non-quenching groove upon hybridization with complementary targets while, b) being intercalated up on hybridization with non-complementary targets, leading to quenching of fluorescence by flanking nucleobases (typically cytosine or guanine, Fig. 12). Unlike hybridization probes, BDF probes can discriminate between complementary and mismatched targets at non-stringent conditions where mismatched duplexes are formed. Accordingly, BDF probes are well-suited to discriminate SNPs, which are the most frequently occurring genetic variation in the human genome (>9 million SNPs) and important biomedical markers.96
Fig. 12.
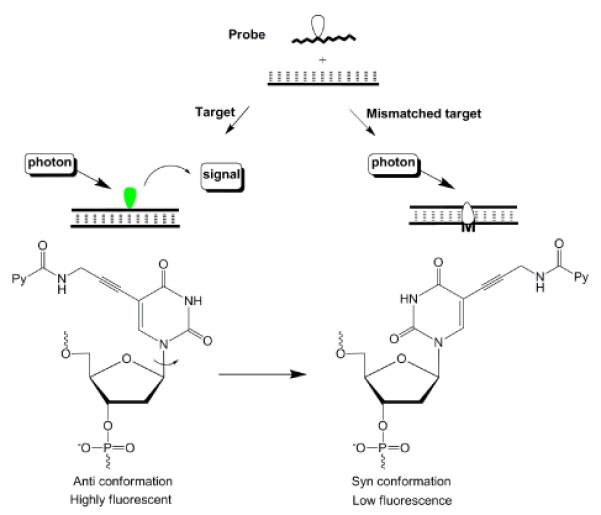
Principle of BDF probes.
The use of ONs modified with C5-pyrene-functionalized pyrimidine monomers has proven to be a robust approach toward successful BDF probe design. Among the numerous linker variants that have been studied,21,26,97–99 the C5-[3-(1-pyrenecarboxamido)propynyl]-functionalized 2'-deoxyuridine monomer 4 emerges as a particularly interesting BDF monomer (Fig. 2), which has been used to discriminate SNP sites in human breast cancer 1 (BRCA 1) gene.26,100 As discussed earlier, molecular modeling studies and fluorescence emission spectra suggest that the nucleobase of monomer 4 adopts an anti conformation (Watson-Crick base pairing orientation) upon hybridization with complementary DNA to position the pyrene in the major groove, while adopting a syn conformation with mismatched DNA leading to pyrene intercalation (Fig. 12).26
Hrdlicka and coworkers recently developed the corresponding LNA and α-L-LNA analogs 32 and 35 (Fig. 4) based on the hypothesis that indirect restriction of the glycosidic torsion angle (see section 4.1 and Fig. 3) would result in higher positional control of the polarity-sensitive C5-fluorophore and potentially enhance SNP discrimination.11 A systematic comparison of the DNA, LNA and α-L-LNA monomers revealed the following. First, ONs modified with DNA monomer 4 (i.e., parent probes) require flanking C/G-moieties for efficient fluorescent mismatch discrimination. Second, ONs modified with LNA analog 32 display higher quantum yields (ΦF = 0.44–0.67) and pyrene extinction coefficients upon hybridization with complementary DNA than parent probes; probes where 32 is flanked by C/G-moieties display more efficient fluorescent discrimination of mismatched targets (Fig. 13). Third, ONs modified with α-L-LNA analog 35 also display higher quantum yields (ΦF = 0.50–0.80) upon hybridization with complementary DNA; probes where 35 is flanked by A/T-moieties display improved fluorescent discrimination of mismatched targets compared to the parent probes.11 The higher duplex quantum yields observed with ONs modified with monomers 32 and 35, most likely, reflect higher positional control of the fluorophore in the major groove.
Fig. 13.
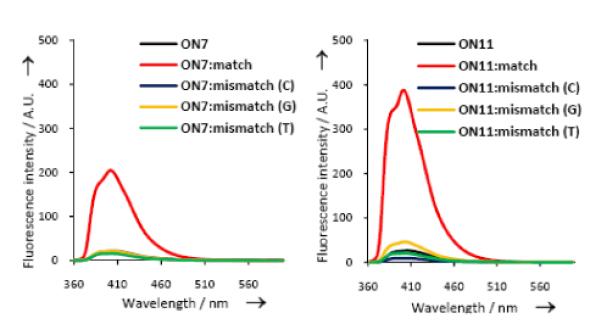
SNP discrimination using C5-[3-(1-pyrenecarboxamido)propynyl] DNA/LNA monomers. Fluorescence emission spectra of 5'-d(CG CAA GBG AAC GC-3'), where B = DNA monomer 4 (ON7) or LNA monomer 32 (ON11), against complementary or singly mismatched DNA. (Reproduced with permission from ref. 11. Copyright 2011 Wiley).
C7-deaza- and C8-pyrene-functionalized 2'-deoxyadenosines 16 and 14 (Fig. 2) also display BDF characteristics. ONs modified with monomer 16 display low emission upon hybridization with complementary DNA and higher emission with singly mismatched targets.30 Intriguingly, the opposite trends are observed for equivalent probes modified with monomer 14.28 The mechanism and binding modes of these flexible probes are not yet understood. C4-pyrene-functionalized monomer 2733 is a rare example of a BDF-monomer where the pyrene moiety is linked to the sugar moiety. The pyrene moiety is presumably located in the minor groove upon hybridization with complementary DNA (high fluorescence), while intercalating upon hybridization with mismatched DNA targets (low fluorescence).33
In an interesting extension of the BDF probe concept, C5-[3-(1-pyrenecarboxamido)propynyl]-functionalized 2'-deoxyuridine monomer 4 was incorporated into a DNA strand and positioned 2–5 nt from a flexible 5'-terminal fluorescein moiety. Duplexes with complementary DNA display Fluorescence Resonance Energy Transfer (FRET) from pyrene to fluorescein (λex= 340 nm, λem= 520 nm). In contrast, less efficient FRET is observed upon hybridization to singly mismatched DNA targets, presumably due to intercalation and quenching of the pyrene fluorophore.101
6.3 Excimer-forming probes
The large Stoke's shift (~120 nm) of pyrene-pyrene excimers render them attractive for nucleic acid diagnostics as this may reduce background fluorescence from unbound probes. As mentioned previously, duplexes between RNA targets and 2'-O-methyl RNA probes modified with two sequential 2'-O-(pyren-1-yl)methyluridine 21 monomers display excimer emission due to pyrene-pyrene interactions in the minor groove (Fig. 7). Duplexes with mismatched base pairs proximal to the modified region only display very low levels of excimer emission.102 These probes do not allow discrimination of mismatched DNA targets since the intercalative binding mode of the pyrene moiety in matched 2'-O-methyl-RNA:DNA heteroduplexes prevents excimer formation.93 ONs with sequential modifications of 2'-N-(pyren-1-ylmethyl)-2'-amino-LNA monomer 36 address this point presumably since the pyrene moieties are forced into the minor groove (vide infra).103
Dual probes104 based on end-labelled PFOs are another class of excimer-forming probes.105 Two short probes are assembled into a ternary complex upon binding with a complementary target, which generates an excimer signal. Monomer fluorescence is predominantly observed in absence of target (Fig. 14).104–105 Wengel and coworkers evaluated extensively LNA-modified ONs end-labelled with 2'-N-(pyren-1-ylmethyl)-2'-amino-LNA monomer 36 in SNP dual probe assays (Fig. 14).103 As expected, binding with complementary 17-mer DNA or RNA targets results in excimer emission due to productive pyrene-pyrene interactions in the ternary complex. In contrast, addition of singly mismatched DNA or RNA targets leads to significantly lower excimer emission (Fig. 14). Thermal denaturation temperatures of matched/mismatched complexes suggest that the decreased excimer intensity is a result of a) mismatch-induced local structural perturbation of the ternary complex whereby pyrene moieties adopt unsuitable conformations for excimer formation (positions 8–11, Fig. 14), or b) lack of ternary complex formation due to thermal mismatch discrimination, which is particularly efficient due to the short probe length and high LNA content (positions 2–7, Fig. 14).103 The LNA-based dual probe platform appears to result in more pronounced SNP-discrimination than other dual probes based on more flexible monomers, but direct comparison is complicated by the use of different nucleic acid targets.105–106 The covalently linked 17-mer analog of the 8- and 9-mer dual probes also displays excimer emission upon hybridization with complementary DNA and decreased excimer emission with mismatched DNA targets, but optical discrimination is only efficient in a narrower region (positions 7–10) through local structural pertubation.103 This underlines the advantage of the dual probe approach, i.e., the combination of local structural pertubation and thermal mismatch discrimination to achieve optical discrimination of SNP-containing targets.
Fig. 14.
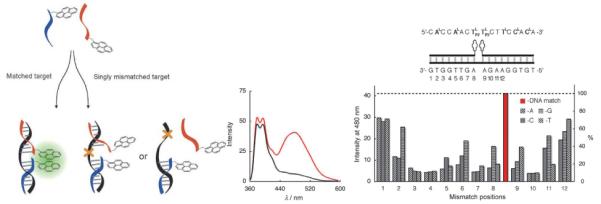
Left: principle of SNP-discrimination by excimer-forming dual probes. Middle: fluorescence emission spectra of LNA-based dual probes end-functionalized with monomer 36 and complementary (red) or singly mismatched DNA target (black). Right: probe/target sequences; discrimination of SNPs in positions 1–12 (λem = 480 nm). (Adapted with permission from ref. 103. Copyright 2007 Wiley).
Two recent developments within the dual probe technology merit special mention. First, excimer-forming PFO-based dual probes have been coupled with time-resolved fluorescence spectroscopy to discriminate between short-lived background fluorescence and pyrene excimers with long fluorescence lifetimes (> 40 ns); this facilitated SNP-analysis of biological samples.107 Secondly, dual probes based on hybridization-induced formation of exciplexes between a pyrene- and a N,N'-dialkylnapthalene-labelled probe have been developed.106 The main advantage of this approach is the potential for multiplexed DNA detection as different exciplex partners with unique emission characteristics can be designed. A potential limitation of this detection platform, however, is that trifluoroethanol is a necessary additive since water quenches exciplex fluorescence emission.108
Another approach that employs pyrene-pyrene excimer fluorescence and which does not require stringent control of experimental temperatures for SNP-detection, is based on ONs with two incorporations of 2′-N-(pyren-1-yl)acetyl-2′-amino-α-L-LNA monomer 40 as next-nearest neighbors (Fig. 15).109 Duplexes with complementary DNA/RNA targets predominantly display pyrene monomer fluorescence, whereas DNA/RNA duplexes with mismatched base pairs proximal to the modified region (positions 4–7, Fig. 15) display significantly increased excimer emission (2- to 11-fold increase at λem = 480 nm). As previously discussed, the pyrene moiety of monomer 40 is forced into the core of matched duplexes resulting in extensive duplex stabilization and monomer fluorescence. Mismatched duplexes appear to induce a more flexible geometry, which allows the pyrene moieties to adopt extrahelical orientations suitable for formation of pyrene-pyrene excimers.109 A related platform has been developed based on non-nucleosidic pyrenefunctionalized “Intercalating Nucleic Acid” (INA) monomers (Fig. 19). SNP discrimination appears to be less efficient but comparative studies have not been performed.110
Fig. 15.
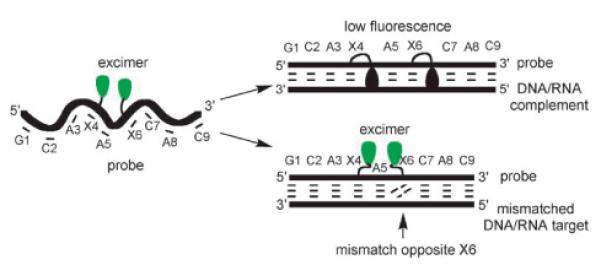
Principle of mismatch detection using doubly modified 2'-N-(pyren-1-yl)acetyl-2'-amino-α-L-LNA 40. (Reproduced with permission from ref. 109. Copyright 2007 Wiley).
Fig. 19.
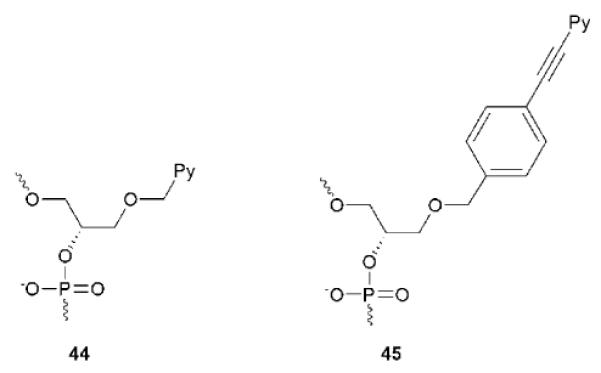
Non-nucleosidic monomers used for dsDNA-targeting.
6.4 Quencher-free molecular beacons
In the original molecular beacon (MB) design, single stranded probes adopting a stem-loop structure are end-functionalized with a fluorophore-quencher pair.111 In the absence of targets, the fluorophore and quencher are in close proximity of each other leading to low fluorescence emission levels. Binding to a single stranded target that is complementary to the loop region results in MB opening, spatial separation of the fluorophore-quencher pair, and increased fluorescence emission (Fig. 16A). A key advantage of MBs over linear probes is that they display improved thermal discrimination of mismatched targets, which reduces the risk of false positives.111 Numerous quencher-free MBs, including several that are based on PFOs, have recently been developed based on prospects of simpler design, lower costs, and potential for secondary modification via end-functionalization.112
Fig. 16.
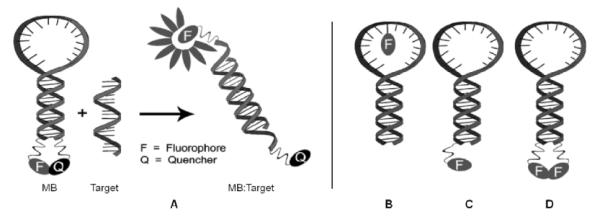
A: General principle of a conventional MB. B–D: Different quencher-free MB designs. (Adapted with permission from ref. 112. Copyright 2008 RSC Publishing).
Hrdlicka and co-workers designed quencher-free MBs based on 29-mer DNA or 2'-O-methyl RNA strands, which are modified with four separated incorporations of Glowing LNA monomer 37 in the 15-mer loop region (Fig. 16B, shown for singly modified MB).91 Hybridization with complementary DNA/RNA targets results in 3- to 9-fold increases in fluorescence intensity and formation of brightly fluorescent duplexes (ΦF = 0.45–0.85). These changes arise, in all probability, due to translocation of the pyrene moieties from a single-stranded environment where quenching interactions with nucleobases occur to the non-quenching minor groove. Glowing LNA beacons display excellent thermal discrimination of singly mismatched DNA/RNA targets and high biostability (> 48h), which enabled their use for detection of specific mRNA in murine 3T3-L1 cell cultures.91
In a different approach, Kim and coworkers incorporated 8-(1-ethynylpyrenyl)-2'-deoxyadenosine monomer 11 into MBs as a 5'-dangling residue which results in a) efficient π–stacking with the terminal base pair, and b) quenching of fluorescence through photoinduced electron transfer which depends on the nature of the 5'-flanking nucleotide in the following manner: C>G>T>A (Fig. 16C).10 In related approaches,113–114 Saito and coworkers, e.g., introduced a pyrene-labeled pyrrolocytidine monomer in the middle of a MB stem leading to quenching of fluorescence through interactions with the pairing guanine moiety.114 Binding of these MBs to complementary DNA results in a) MB opening, b) localization of the pyrene moiety in a single stranded region, and c) dequenching due to reduced nucleobase interactions.10,114
In yet another approach, incorporation of two flexible non-nucleosidic pyrene units at the termini of 29-mer DNA-based MBs leads to efficient pyrene-pyrene-stacking and formation of excimer fluorescence (Fig. 16D).115 Similarly, incorporation of two 8-(1-ethynylpyrenyl)-2'-deoxyadenosine 11 monomers opposite of each other within the stem of a 27-mer DNA-based MB, leads to excimer emission in the closed state presumably since nucleobases adopt syn conformations.116 Hybridization with complementary DNA results in MB opening, separation of the pyrene moieties, and a marked increase in the monomer/excimer intensity ratio (Fig. 17). Virtually no change in the monomer/excimer intensity ratio is observed upon hybridization with singly mismatched DNA targets, which underlines the high thermal mismatch discrimination of MBs.115–116
Fig. 17.
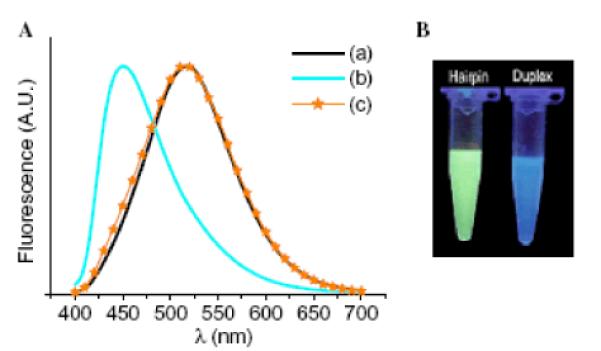
Excimer-forming MBs. Left: Fluorescence emission spectra (λex = 386 nm) of (a) MB alone, (b) MB + complementary DNA and, (c) MB + singly mismatched DNA. Right: Optical detection of complementary DNA. MB sequence: 5'-d(GC11 GAG AAG TTA GAA CCT ATG CTC 11GC). (Reproduced with permission from ref. 116. Copyright 2006 Elsevier Ltd.).
The opposite principle has been used in MBs with two proximal incorporations of flexible C8-pyrene functionalized monomer 10 in one stem arm. In the absence of target strands, the pyrene moieties predominantly display monomer fluorescence. Upon binding with complementary targets, the beacon opens up, the stem becomes single-stranded, and the pyrene moieties interact with each other to generate excimer fluorescence.117 Similar observations were made with MBs modified with 1–4 flexible pyrene moieties at the 5'-end and a 3'-DABCYL quencher.118
Wagenknecht and coworkers incorporated 5-(1-ethynylpyrenyl)-2'-deoxyuridine monomer 3 into the stem of MBs in close proximity of a 2'-deoxyuridine modified at the C5-position with a `Nile Red' fluorophore. In the closed state, efficient FRET from pyrene to Nile Red results in fluorescence emission at 435 nm and 660 nm (pyrene and Nile Red, respectively). Opening of the MB upon DNA target binding, results in separation of the FRET donor-acceptor pair and an increase in the I435/I660 ratio enabling ratiometric DNA detection.119
6.5 Miscellaneous diagnostic applications of PFOs
Pyrene-functionalized monomers have been used to study formation of alternative nucleic acid structures.98,120–121 Silverman and coworkers systematically modified the P4–P6 domain of Tetrahymena group I intron with N2'-pyrene-functionalized 2'-amino-DNA monomers to study RNA folding. Mg2+-induced formation of tertiary structures results in increases in fluorescence emission when long-linked monomers are used, presumably since the pyrene moiety is translocated from a stacking to a non-stacking environment.120
Kim and coworkers attached 8-(1-ethynylpyrenyl)-2'-deoxyadenosine monomer 11 as a dangling residue of G-rich ONs.121 In the single stranded state these ONs exhibit fluorescence emission at λem = 451 nm; addition of potassium ions results in a) G-quadruplex formation, b) stacking between pyrene and terminal guanosines, and c) exciplex formation. As a result, red-shifted fluorescence emission is observed (λem = 485 nm, Fig. 18).121 This is particularly interesting given the recent focus on G-quadruplexes as materials for bottom-up fabrication of nanomaterials.122
Fig. 18.
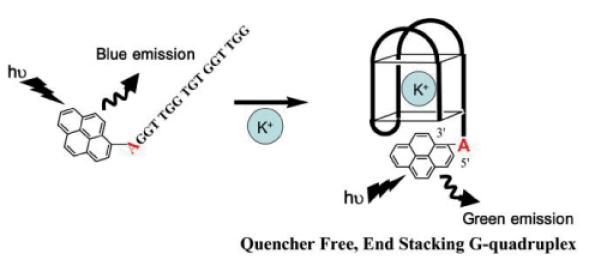
Fig. 18. Fluorescence signalling of quadruplex formation. (Reproduced with permission from ref. 121. Copyright 2007 RSC Publishing).
Okamoto, Saito and coworkers used a CG-alternating ON modified with 8-(1-ethynylpyrenyl)-2'-deoxyguanosine monomer 12 and C5-[3-(1-pyrenecarboxamido)propynyl]-functionalized 2'-deoxycytosine (i.e., the C-analog of monomer 4, Fig. 2), to monitor salt-induced transitions from a right-handed B-type to left-handed Z-type DNA-duplex. Presumably, monomers 4 and 12 adopt anti- and syn-conformations, respectively, which positions the pyrene moieties far apart at low salt concentrations (B-type) while leading to π-stacking and excimer formation at high salt concentrations (Z-type).98
PFOs have also been utilized to study protein-nucleic acid interactions. Seitz, Weinhold and coworkers reported that DNA duplexes modified with pyrene glycoside 1 (Fig. 2) bind ~400 times more strongly to adenine-specific DNA methyltransferase (MTase) M·TaqI than corresponding unmodified DNA duplexes. The DNA methylations catalyzed by this enzyme commence by flipping the target nucleotide to an extrahelical position. The resulting void is more efficiently stabilized by the pyrene moiety of monomer 1 than by natural nucleobases.123 Stivers and coworkers used DNA duplexes modified with monomer 1 to study the mechanism of uracil DNA glycosylase, i.e., a DNA repair enzyme which also proceeds via base-flipping of a nucleotide.124
Kim and coworkers incorporated monomer 3 (Fig. 2) into the U25 bulge region of HIV TAR RNA which undergoes a substantial conformational change upon binding with Tat-protein mimics. Binding leads to reduced interactions between the pyrene moiety and adjacent base pairs and dequenching of fluorescence emission. Such probes may, accordingly, be useful to identify RNA-binding entities.125
7. Targeting of double stranded DNA using PFOs
Chemically modified ONs are widely utilized to modulate gene expression, which has enabled gene function studies and led to development of drug candidates against diseases of genetic origin. The vast majority of these efforts have focused on targeting of RNA.126 Development of probes for sequence-specific targeting of double stranded DNA (dsDNA) has received far less attention, despite the prospect of powerful tools for modulation of gene expression, gene manipulation, and detection of dsDNA in living cells.127–128 From a chemical perspective, dsDNA is a significantly more complex target than single stranded DNA/RNA since the Watson-Crick hydrogen-bonding faces are buried in the duplex core. This leaves few molecular `handles' for sequence-specific recognition of dsDNA. Probes targeting dsDNA in biological contexts face the additional challenge of compacted and protein-associated chromosomal DNA. Triplex forming oligonucleotides (TFOs), which bind in the major groove through relatively weak Hoogsteen base pairs, are the most extensively explored dsDNA-targeting technology. Limitations of TFOs include: a) requirement for long homopurine target regions (>15 nt) to ensure sufficient binding affinity/specificity; b) the need for slightly acidic pH to ensure cytosine protonation which is required for Hoogsteen base pairs between TC-motif TFOs and dsDNA; and c) undeisred formation of secondary structures by G-rich TFOs at physiological potassium ion concentrations, which affects dsDNA binding capabilities. For further discussion of the TFO-technology, the Reader is directed to the review by Arimondo and coworkers.129 The limitations of the TFO-technology have spurred a search for alternative dsDNA-targeting strategies including minor groove binding polyamides, peptide nucleic acids (PNAs), pseudo-complementary PNA (pc-PNA), and other approaches which have been recently reviewed by Concordet, Giovannangeli and coworkers and that are not further discussed herein.130
PFOs have been explored for dsDNA-targeting applications. Pedersen, Filichev and coworkers introduced non-nucleosidic “Twisted Intercalating Nucleic Acid” (TINA) monomer 45 as bulges in TC-motif TFOs (Fig. 19).131 Increases in triplex thermostability are observed that vary with TFO length, pH and the number, position, and distance between modifications (ΔTm/mod 0–20 °C).131–132 Molecular modeling studies suggest that the pyrene moieties are situated in the Watson-Crick-paired dsDNA part of the triplex, while the phenyl moiety stacks with the nucleobases of the TFO.133 The low triplex thermostability at physiological pH and TFO-based sequence limitations remain challenges that need to be addressed.
Hrdlicka and coworkers have proposed a radically different approach for dsDNA-targeting which utilizes Invader LNA probes (Fig. 20), i.e., DNA duplexes modified with one or more +1 interstrand zipper arrangements of 2'-N-(pyren-1-yl)methyl-2'-amino-α-L-LNA thymine monomer 38 (Fig. 4).134–135 Invader LNA display low probe duplex thermostability, which is related to the “nearest neighbor exclusion principle”5; the 2-oxo-5-azabicyclo[2.2.1]heptane skeleton presumably forces the pyrene moieties of +1 zipper-positioned 38 monomers to intercalate into the same region as demonstrated by excimer formation. Due to constraints in local helix expandability, this `energetic hotspot' leads to partial duplex unwinding and decreased thermostability.134–135 Other interstrand zipper arrangments of monomer 38 result in much higher duplex thermostability, which underlines the importance of positional control to achieve functional control.134–135 Both strands of the Invader LNA probes display very high thermal affinity toward complementary DNA (ΔTm/mod up to +15 °C).67 These large differences in duplex thermostability have been exploited to enable spontaneous recognition of isosequential, mixed-sequence dsDNA targets under non-denaturing conditions (Fig. 20).134–135 The recognition process is conveniently monitored in-real time by the decrease in excimer emission as the pyrene moieties are forced apart (Fig. 20), or by taking advantage of the different electrophoretic mobilities of Invader LNA, dsDNA targets and probe:target-duplexes.135 Thus, addition of a pre-annealed 13-mer Invader LNA to an equimolar quantity of the isosequential dsDNA target at 110 mM NaCl, pH 7 and low incubation temperatures, results in 50% recognition within ~40 min; shorter targets, higher incubation temperatures and/or the use of Invader LNA with more than one energetic hotspot, accelerate molecular recognition. It is particularly noteworthy that dsDNA-recognition even occurs in buffers of very high ionic strengh (710 mM NaCl, pH 7, t50% ~125 min). When Invader LNA are mixed with dsDNA where one base pair is exchanged relative to Invader LNA (i.e., non-isosequential dsDNA targets), much slower and less pronounced decay of the excimer signal is observed, underlining that the recognition process is sequence-specific. The recognition mechanism is not fully understood, but may involve partial melting of dsDNA targets and Invader LNA probes due to base pair breathing, which reveals nucleation sites for the formation of probe-target duplexes.135 A related hypothesis has been formulated by Ly and coworkers to explain binding of single stranded γ-PNA to mixed-sequence dsDNA at low ionic strengths.136 Subsequent studies have revealed that the free energy for molecular recognition of dsDNA by Invader LNA can be increased through changes in the linker chemistry between the pyrene moiety and the sugar skeleton of 2'-amino-α-L-LNA monomers.137 The demanding synthesis of Invader LNA monomers is a potential limitation of this mixed-sequence dsDNA-targeting technology.60,67 Gratifyingly, O2'-alkylated RNA and N2'-functionalized 2'-amino-DNA monomers have very recently been established as structural and functional analogs of Invader LNA monomers, which are synthetically more easily accessible.138 The potential of Invader LNA for targeting of biologically relevant mixed-sequence dsDNA under physiological conditions is underscored by their ability to reduce RNA-formation in in vitro transcription assays.139 Similar results have been observed with short “Invader LNA-like” probes modified with INA monomer 44 (Fig. 19) even though less free energy is available for dsDNA-targeting, presumably due to the high flexibility of 44.140
Fig. 20.
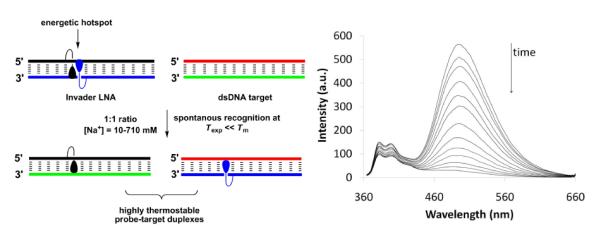
Left: illustration of the Invader LNA concept. Right: time course of steady-state fluorescence spectra upon addition of pre-annealed Invader LNA to pre-annealed isosequential dsDNA target (λex = 335 nm, 20 °C). Invader LNA: 5'-d(GGT A38A TAT AGG C):3'-d(CCA TA38 ATA TCC G) using monomer 38. Isosequential dsDNA target: as for Invader LNA except 38 is placed by T. (Adapted from ref. 135. Copyright 2010 RSC Publishing).
8. Summary and Perspective
Pyrene-functionalized ONs (PFOs) are valuable model systems for fundamental studies of nucleic acid structures due to their straightforward synthetic manipulation, microenvironment-sensitive fluorescence, π-stacking ability and propensity for intercalation into DNA duplexes. An large number of pyrene-functionalized nucleotide monomers has emerged over the past ~10 years leading to major advances towards a) generation of self-assembled helical chromophore arrays, b) sensitive detection of DNA/RNA targets and discrimination of single nucleotide polymorphisms (SNPs), and c) sequence-unrestricted targeting of dsDNA. The majority of these advances have been predicated by the development of pyrene-functionalized monomers with low inherrent flexibility and, putatively, high positional control of the pyrene moiety (e.g., short linkers; conformationally restricted sugar moieties). The following key examples underline this point: a) the extensive pyrene arrays obtained with duplexes between RNA strands and 2'-O-methyl RNA probes that are sequentially modified with 2'-O-(pyren-1-yl)methyluridine 21 monomers, rely on the preference of pyrene moieties to adopt non-intercalating binding modes in A-type duplexes (Fig. 7)71; b) the extraordinary high fluorescence emission quantum yields of duplexes between Glowing LNA and complementary DNA/RNA targets (ΦF up to 0.99) are a consequence of the conformationally locked bicyclic skeleton and the short amide linkage of monomer 37 working in concert to rigidly position the pyrene moiety in the non-quenching minor groove (Fig. 5, Table 1);90–91 and c) the ability of double-stranded Invader LNA to spontanously dissociate and bind to mixed-sequence dsDNA targets under physiological and non-denaturing conditions is related to the fact that intercalators linked to the N2'-position of 2'-amino-α-L-LNA are preorganized to point toward the duplex core (Fig. 6 and 20).134–135 Future studies on PFOs would greatly benefit from NMR or X-ray characterization in order to more confidently link the position of the chromophore with observed physical properties and to guide rational design of new monomers with enabling functions.
The proof-of-principle studies highlighted herein underline the many potential applications of PFOs within nucleic acid based diagnostics, therapeutics and materials science. Several challenges remain, e.g., development of highly sensitive and discriminating SNP-typing probes that can: i) be used against a wide range of targets regardless of sequence context, ii) be used for in vivo applications, and iii) compete with large-scale SNP-typing methodologies on cost, sample throughput, and/or sensitivity/specificity.141 Moreover, realization of sequence-unrestricted dsDNA targeting would not only provide a tool for modulation of gene expression and gene manipulation, but also pave the way toward detection of dsDNA target regions by fluorescence in situ hybridization (FISH) at non-denaturing conditions or even in living cells. An occasionally discussed drawback of pyrene is its blue-light excitation which can lead to high levels of cellular autofluorescence and damage to cells and assays. While development of chromophores with red-shifted emission characteristics certainly is desirable, it should be kept in mind that pyrene-functionalized probes displaying high fluorescence emission quantum yields can be coupled to milder and potentially less damaging excitation sources.
The highlighted developments of PFOs, in concert with the considerable advances in structural DNA nanobiotechnology68 and bioconjugation techniques,142 set the stage for a merger of these areas to generate self-assembled functional nanomaterials.143 For example, chemically modified DNA nanogrids lend themselves as supramolecular scaffolds for organization of chromophores for development of light harvesting complexes. PFOs will undoubtedly continue to play important roles as model systems and key components of such functional nanomaterials.
Acknowledgements
Nucleic acid oriented research in the Hrdlicka laboratory is supported by Award Number R01 GM088697 from the National Institute of General Medical Sciences, National Institutes of Health; Idaho NSF EPSCoR; University of Idaho Biological Applications of Nanotechnology (BANTech) Center; Institute of Translational Health Sciences (ITHS) (supported by grants UL1 RR025014, KL2 RR025015, and TL1 RR025016 from the NIH National Center for Research Resources); NIH Grant Number P20 RR016454 from the INBRE Program of the National Center for Research Resources; and the United States Department of Agriculture (award 2009-34479-19833).
Footnotes
Part of a themed issue on the advances in DNA-based nanotechnology
Notes and references
- 1.Kashida H, Liang XG, Asanuma H. Curr. Org. Chem. 2009;13:1065–1084. [Google Scholar]
- 2.Filichev VV, Pedersen EB. Wiley Encyclopedia of Chemical Biology. 2009;1:493–524. [Google Scholar]
- 3.Malinovskii VL, Wenger D, Häner R. Chem. Soc. Rev. 2010;39:410–422. doi: 10.1039/b910030j. [DOI] [PubMed] [Google Scholar]
- 4.Guckian KM, Schweitzer BA, Ren RXF, Sheils CJ, Tahmassebi DC, Kool ET. J. Am. Chem. Soc. 2000;122:2213–2222. doi: 10.1021/ja9934854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dougherty G, Pilbrow JR. Int. J. Biochem. 1984;16:1179–1192. doi: 10.1016/0020-711x(84)90215-5. [DOI] [PubMed] [Google Scholar]
- 6.Lu XJ, Olson WK. Nucleic Acids Res. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yao CX, Kraatz HB, Steer RP. Photochem. Photobiol. Sci. 2005;4:191–199. doi: 10.1039/b414577c. [DOI] [PubMed] [Google Scholar]
- 8.Kalyanasundaram K, Thomas JK. J. Am. Chem. Soc. 1977;99:2039–2044. [Google Scholar]
- 9.Manoharan M, Tivel KL, Zhao M, Nafisi K, Netzel TL. J. Phys. Chem. 1995;99:17461–17472. [Google Scholar]
- 10.Seo YJ, Ryu JH, Kim BH. Org. Lett. 2005;7:4931–4933. doi: 10.1021/ol0518582. [DOI] [PubMed] [Google Scholar]
- 11.Østergaard ME, Kumar P, Baral B, Guenther DC, Anderson BA, Ytreberg FM, Deobald L, Paszczynski AJ, Sharma PK, Hrdlicka PJ. Chem. Eur. J. 2011;17:3157–3165. doi: 10.1002/chem.201002109. [DOI] [PubMed] [Google Scholar]
- 12.Winnik FM. Chem. Rev. 1993;93:587–614. [Google Scholar]
- 13.Amann N, Pandurski E, Fiebig T, Wagenknecht HA. Chem. Eur. J. 2002;8:4877–4883. doi: 10.1002/1521-3765(20021104)8:21<4877::AID-CHEM4877>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 14.Valis L, Mayer–Enthart E, Wagenknecht HA. Bioorg. Med. Chem. Lett. 2006;16:3184–3187. doi: 10.1016/j.bmcl.2006.03.063. [DOI] [PubMed] [Google Scholar]
- 15.Raytchev M, Mayer E, Amann N, Wagenknecht HA, Fiebig T. ChemPhysChem. 2004;5:706–712. doi: 10.1002/cphc.200301205. [DOI] [PubMed] [Google Scholar]
- 16.Wanninger-Weiss C, Wagenknecht HA. Eur. J. Org. Chem. 2008:64–69. [Google Scholar]
- 17.Mayer E, Valis L, Wagner C, Rist M, Amann N, Wagenknecht HA. ChemBioChem. 2004;5:865–868. doi: 10.1002/cbic.200300845. [DOI] [PubMed] [Google Scholar]
- 18.Maeda H, Maeda T, Mizuno K, Fujimoto K, Shimizu H, Inouye M. Chem. Eur. J. 2006;12:824–831. doi: 10.1002/chem.200500638. [DOI] [PubMed] [Google Scholar]
- 19.Astakhova IV, Korshun VA, Wengel J. Chem. Eur. J. 2008;14:11010–11026. doi: 10.1002/chem.200801077. [DOI] [PubMed] [Google Scholar]
- 20.Matray TJ, Kool ET. J. Am. Chem. Soc. 1998;120:6191–6192. doi: 10.1021/ja9803310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Østergaard ME, Guenther DC, Kumar P, Baral B, Deobald L, Paszczynski AJ, Sharma PK, Hrdlicka PJ. Chem. Commun. 2010:4929–4931. doi: 10.1039/c0cc01133a. [DOI] [PubMed] [Google Scholar]
- 22.Heystek LE, Zhou HQ, Dande P, Gold B. J. Am. Chem. Soc. 1998;120:12165–12166. [Google Scholar]
- 23.Znosko BM, Barnes TW, krugh TR, Turner DH. J. Am. Chem. Soc. 2003;125:6090–6097. doi: 10.1021/ja021285d. [DOI] [PubMed] [Google Scholar]
- 24.Kottysch T, Ahlborn C, Brotzel F, Richert C. Chem. Eur. J. 2004;10:4017–4028. doi: 10.1002/chem.200306044. [DOI] [PubMed] [Google Scholar]
- 25.Skorobogatyi MV, Malakhov AD, Pchelintseva AA, Turban AA, Bondarev SL, Korshun VA. ChemBioChem. 2006;7:810–816. doi: 10.1002/cbic.200600040. [DOI] [PubMed] [Google Scholar]
- 26.Okamoto A, Kanatani K, Saito I. J. Am. Chem. Soc. 2004;126:4820–4827. doi: 10.1021/ja039625y. [DOI] [PubMed] [Google Scholar]
- 27.Bag SS, Kundu R, Matsumoto K, Saito Y, Saito I. Bioorg. Med. Chem. Lett. 2010;20:3227–3230. doi: 10.1016/j.bmcl.2010.04.063. [DOI] [PubMed] [Google Scholar]
- 28.Saito Y, Hanawa K, Motegi K, Omoto K, Okamoto A, Saito I. Tetrahedron Lett. 2005;46:7605–7608. [Google Scholar]
- 29.Seela F, Zulauf M. Helv. Chim. Acta. 1999;82:1878–1898. [Google Scholar]
- 30.Saito Y, Miyauchi Y, Okamoto A, Saito I. Chem. Commun. 2004:1704–1705. doi: 10.1039/b405832a. [DOI] [PubMed] [Google Scholar]
- 31.Seela F, Ingale SA. J. Org. Chem. 2010;75:284–295. doi: 10.1021/jo902300e. [DOI] [PubMed] [Google Scholar]
- 32.Grünefeld P, Richert C. J. Org. Chem. 2004;69:7543–7551. doi: 10.1021/jo049062o. [DOI] [PubMed] [Google Scholar]
- 33.Dohno C, Saito I. ChemBioChem. 2005;6:1075–1081. doi: 10.1002/cbic.200400325. [DOI] [PubMed] [Google Scholar]
- 34.Bryld T, Højland T, Wengel J. Chem. Commun. 2004:1064–1065. doi: 10.1039/b402414a. [DOI] [PubMed] [Google Scholar]
- 35.Yamana K, Iwase R, Furutani S, Tsuchida H, Zako H, Yamaoka T, Murakami A. Nucleic Acids Res. 1999;27:2387–2392. doi: 10.1093/nar/27.11.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamana K, Zako H, Asazuma K, Iwase R, Nakano H, Murakami A. Angew. Chem. Int. Ed. 2001;40:1104–1106. doi: 10.1002/1521-3773(20010316)40:6<1104::aid-anie11040>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 37.Nakamura M, Fukunaga Y, Sasa K, Ohtoshi Y, Kanaori K, Hayashi H, Nakano H, Yamana K. Nucleic Acids Res. 2005;33:5887–5895. doi: 10.1093/nar/gki889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Korshun VA, Stetsenko DA, Gait MJ. J. Chem. Soc. Perkin. Trans. 2002;1:1092–1104. [Google Scholar]
- 39.Dioubankova NN, Malakhov AD, Shenkarev ZO, Korshun VA. Tetrahedron. 2004;60:4617–4626. [Google Scholar]
- 40.Dioubankova NN, Malakhov AD, Stetsenko DA, Gait MJ, Volynsky PE, Efremov RG, Korshun VA. ChemBioChem. 2003;4:841–847. doi: 10.1002/cbic.200300678. [DOI] [PubMed] [Google Scholar]
- 41.Astakhova IV, Malakhov AD, Stepanova IA, Ustinov AV, Bondarey SL, Paramonov AS, Korshun VA. Bioconjugate Chem. 2007;18:1972–1980. doi: 10.1021/bc700280h. [DOI] [PubMed] [Google Scholar]
- 42.Kalra N, Parlato MC, Parmar VS, Wengel J. Bioorg. Med. Chem. Lett. 2006;16:3166–3169. doi: 10.1016/j.bmcl.2006.03.072. [DOI] [PubMed] [Google Scholar]
- 43.Kalra N, Babu BR, Parmar VS, Wengel J. Org. Biomol. Chem. 2004;2:2885–2887. doi: 10.1039/B411626G. [DOI] [PubMed] [Google Scholar]
- 44.Ratje RL, Anderson BA, Hrdlicka PJ. unpublished results.
- 45.Koshkin AA, Singh SK, Nielsen P, Rajwanshi VK, Kumar R, Meldgaard M, Olsen CE, Wengel J. Tetrahedron. 1998;54:3607–3630. [Google Scholar]
- 46.Obika S, Nanbu D, Hari Y, Andoh J, Morio K, Doi T, Imanishi T. Tetrahedron Lett. 1998;39:5401–5404. [Google Scholar]
- 47.Petersen M, Nielsen CB, Nielsen KE, Jensen GA, Bondensgaard K, Singh SK, Rajwanshi VK, Koshkin AA, Dahl BM, Wengel J, Jacobsen JP. J. Mol. Recognit. 2000;13:44–53. doi: 10.1002/(SICI)1099-1352(200001/02)13:1<44::AID-JMR486>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 48.Nielsen JT, Arar K, Petersen M. Nucleic Acids Res. 2006;34:2006–2014. doi: 10.1093/nar/gkl144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Konorov SO, Schulze HG, Addison CJ, Haynes CA, Blades MW, Turner RFB. J. Raman Spectrosc. 2009;40:1162–1171. [Google Scholar]
- 50.Petersen M, Bondensgaard K, Wengel J, Jacobsen JP. J. Am. Chem. Soc. 2002;124:5974–5982. doi: 10.1021/ja012288d. [DOI] [PubMed] [Google Scholar]
- 51.Nielsen KE, Spielmann HP. J. Am. Chem. Soc. 2005;127:15273–15282. doi: 10.1021/ja051026z. [DOI] [PubMed] [Google Scholar]
- 52.Koch T, Ørum H. In: Antisense Drug Technology – Principles, Strategies, and Applications. 2nd ed Crooke ST, editor. CRC Press; Boca Raton: 2008. pp. 519–564. [Google Scholar]
- 53.Sørensen MD, Kværnø L, Bryld T, Håkansson AE, Verbeure B, Gaubert G, Herdewijn P, Wengel J. J. Am. Chem. Soc. 2002;124:2164–2176. doi: 10.1021/ja0168763. [DOI] [PubMed] [Google Scholar]
- 54.Nielsen KME, Petersen M, Håkansson AE, Wengel J, Jacobsen JP. Chem. Eur. J. 2002;8:3001–3009. doi: 10.1002/1521-3765(20020703)8:13<3001::AID-CHEM3001>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 55.Nielsen JT, Stein P, Petersen M. Nucleic Acids Res. 2003;31:5858–5867. doi: 10.1093/nar/gkg800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fluiter K, Frieden M, Vreijling J, Rosenbohm C, De Wissel MB, Christensen SM, Koch T, Ørum H, Baas F. ChemBioChem. 2005;6:1104–1109. doi: 10.1002/cbic.200400419. [DOI] [PubMed] [Google Scholar]
- 57.Petersen M, Wengel J. Trends Biotechnol. 2003;21:74–81. doi: 10.1016/S0167-7799(02)00038-0. [DOI] [PubMed] [Google Scholar]
- 58.Kaur H, Babu BR, Maiti S. Chem. Rev. 2007;107:4672–4697. doi: 10.1021/cr050266u. [DOI] [PubMed] [Google Scholar]
- 59.Singh SK, Kumar R, Wengel J. J. Org. Chem. 1998;63:10035–10039. doi: 10.1021/jo9806658. [DOI] [PubMed] [Google Scholar]
- 60.Kumar TS, Madsen AS, Wengel J, Hrdlicka PJ. J. Org. Chem. 2006;71:4188–4201. doi: 10.1021/jo060331f. [DOI] [PubMed] [Google Scholar]
- 61.Babu BR, Prasad AK, Trikha S, Thorup N, Parmar VS, Wengel J. J. Chem. Soc. Perkin Trans. 2002;1:2509–2519. [Google Scholar]
- 62.Raunak B. R. Babu, Sørensen MD, Parmar VS, Harrit NH, Wengel J. Org. Biomol. Chem. 2004;2:80–89. doi: 10.1039/b310719a. [DOI] [PubMed] [Google Scholar]
- 63.Verhagen C, Bryld T, Raunkær M, Vogel S, Buchalova K, Wengel J. Eur. J. Org. Chem. 2006:2538–2548. [Google Scholar]
- 64.Østergaard ME, Kumar P, Baral B, Raible DJ, Kumar TS, Anderson BA, Guenther DC, Deobald L, Paszczynski AJ, Sharma PK, Hrdlicka PJ. ChemBioChem. 2009;10:2740–2743. doi: 10.1002/cbic.200900500. [DOI] [PubMed] [Google Scholar]
- 65.Kumar P, hrdlicka PJ. unpublished results.
- 66.Sørensen MD, Petersen M, Wengel J. Chem. Commun. 2003:2130–2131. doi: 10.1039/b307026c. [DOI] [PubMed] [Google Scholar]
- 67.Kumar TS, Madsen AS, Østergaard ME, Sau SP, Wengel J, Hrdlicka PJ. J. Org. Chem. 2009;74:1070–1081. doi: 10.1021/jo802037v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aldaye FA, Palmer AL, Sleiman HF. Science. 2008;321:1795–1799. doi: 10.1126/science.1154533. [DOI] [PubMed] [Google Scholar]
- 69.Varghese R, Wagenknecht HA. Chem. Commun. 2009:2615–2624. doi: 10.1039/b821728a. [DOI] [PubMed] [Google Scholar]
- 70.Nguyen TN, Brewer A, Stulz E. Angew. Chem. Int. Ed. 2009;48:1974–1977. doi: 10.1002/anie.200805657. [DOI] [PubMed] [Google Scholar]
- 71.Nakamura M, Shimomura Y, Ohtoshi Y, Sasa K, Hayashi H, Nakano H, Yamana K. Org. Biomol. Chem. 2007;5:1945–1951. doi: 10.1039/b705933g. [DOI] [PubMed] [Google Scholar]
- 72.Hrdlicka PJ, Babu BR, Sørensen MD, Wengel J. Chem. Commun. 2004:1478–1479. doi: 10.1039/b404446k. [DOI] [PubMed] [Google Scholar]
- 73.Astakhova IV, Lindegaard D, Korshun VA, Wengel J. Chem. Commun. 2010;46:8362–8364. doi: 10.1039/c0cc03026k. [DOI] [PubMed] [Google Scholar]
- 74.Lindegaard D, Babu BR, Wengel J. Nucleos. Nucleot. Nucleic Acids. 2005;24:679–681. doi: 10.1081/ncn-200060247. [DOI] [PubMed] [Google Scholar]
- 75.Nakamura M, Murakami Y, Sasa K, Hayashi H, Yamana K. J. Am. Chem. Soc. 2008;130:6904–6905. doi: 10.1021/ja801054t. [DOI] [PubMed] [Google Scholar]
- 76.Mayer-Enthart E, Wagenknecht HA. Angew. Chem. Int. Ed. 2006;45:3372–3375. doi: 10.1002/anie.200504210. [DOI] [PubMed] [Google Scholar]
- 77.Barbaric J, Wagenknecht HA. Org. Biomol. Chem. 2006;4:2088–2090. doi: 10.1039/b605251g. [DOI] [PubMed] [Google Scholar]
- 78.Tanaro K, Shionoya M. Coord. Chem. Rev. 2007;251:2732–2742. [Google Scholar]
- 79.Kumar TS, Madsen AS, Østergaard ME, Wengel J, Hrdlicka PJ. J. Org. Chem. 2008;73:7060–7066. doi: 10.1021/jo800551j. [DOI] [PubMed] [Google Scholar]
- 80.Häner R, Garo F, Wenger D, Malinovskii VL. J. Am. Chem. Soc. 2010;132:7466–7471. doi: 10.1021/ja102042p. [DOI] [PubMed] [Google Scholar]
- 81.Chiba J, Takeshima S, Mishima K, Maeda H, Nanai Y, Mizuno K, Inouye M. Chem. Eur. J. 2007;13:8124–8130. doi: 10.1002/chem.200700559. [DOI] [PubMed] [Google Scholar]
- 82.Teo YN, Wilson JN, Kool ET. J. Am. Chem. Soc. 2009;131:3923–3933. doi: 10.1021/ja805502k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kubista M, Andrade JM, Bengtsson M, Forootan A, Jonak J, Lind K, Sindelka R, Sjöback R, Sjögreen B, Strömbom L, Ståhlberg A, Zorix N. Mol. Asp. Med. 2006;27:95–125. doi: 10.1016/j.mam.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 84.Mahara A, Iwase R, Sakamoto T, Yamaoka T, Yamana K, Murakami A. Bioorg. Med. Chem. 2003;11:2783–2790. doi: 10.1016/s0968-0896(03)00227-x. [DOI] [PubMed] [Google Scholar]
- 85.Bratu DP, Cha BJ, Mhlanga MM, Kramer FR, Tyagi S. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13308–13313. doi: 10.1073/pnas.2233244100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sobrino B, Brion M, Carracedo A. Forensic Sci. Int. 2005;154:181–194. doi: 10.1016/j.forsciint.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 87.Alocilja EC, Radke SM. Biosens. Bioelectron. 2003;18:841–846. doi: 10.1016/s0956-5663(03)00009-5. [DOI] [PubMed] [Google Scholar]
- 88.Asseline U. Curr. Org. Chem. 2006;10:491–518. [Google Scholar]
- 89.Wang G, Bobkov GV, Mikhailov SN, Schepers G, Van Aerschot A, Rozenski J, Van der Auweraer M, Herdewijn P, De Feyter S. ChemBioChem. 2009;10:1175–1185. doi: 10.1002/cbic.200900031. [DOI] [PubMed] [Google Scholar]
- 90.Hrdlicka PJ, Babu BR, Sørensen MD, Harrit N, Wengel J. J. Am. Chem. Soc. 2005;127:13293–13299. doi: 10.1021/ja052887a. [DOI] [PubMed] [Google Scholar]
- 91.Østergaard ME, Cheguru P, Papasani MR, Hill RA, Hrdlicka PJ. J. Am. Chem. Soc. 2010;132:14221–14228. doi: 10.1021/ja1057295. [DOI] [PubMed] [Google Scholar]
- 92.Østergaard ME, Maity J, Babu BR, Wengel J, Hrdlicka PJ. Bioorg. Med. Chem. Lett. 2010;20:7265–7268. doi: 10.1016/j.bmcl.2010.10.089. [DOI] [PubMed] [Google Scholar]
- 93.Honcharenko D, Zho C, Chattopadhyaya J. J. Org. Chem. 2008;73:2829–2842. doi: 10.1021/jo702747w. [DOI] [PubMed] [Google Scholar]
- 94.Okamoto A, Saito Y, Saito I. J. Photochem. Photobiol. C. 2005;6:108–122. [Google Scholar]
- 95.Dodd DW, Hudson RHE. Mini–Rev. Org. Chem. 2009;6:378–391. [Google Scholar]
- 96.Kim S, Misra A. Annu. Rev. Biomed. Eng. 2007;9:289–320. doi: 10.1146/annurev.bioeng.9.060906.152037. [DOI] [PubMed] [Google Scholar]
- 97.Hwang GT, Seo YJ, Kim SJ, Kim BH. Tetrahedron Lett. 2004;45:3543–3546. [Google Scholar]
- 98.Okamoto A, Ochi Y, Saito I. Chem. Commun. 2005:1128–1130. doi: 10.1039/b416965d. [DOI] [PubMed] [Google Scholar]
- 99.Yoshida Y, Niwa K, Yamada K, Tokeshi M, Baba Y, Saito Y, Okamoto A, Saito I. Chem. Lett. 2010;39:116–117. [Google Scholar]
- 100.Okamoto A, Tainaka K, Ochi Y, Kanatani K, Saito I. Mol. Biosyst. 2006;2:122–126. doi: 10.1039/b515923g. [DOI] [PubMed] [Google Scholar]
- 101.Saito Y, Bag SS, Kusakabe Y, Nagai C, Matsumoto K, Mizuno E, Kodate S, Suzuka I, Saito I. Chem. Commun. 2007:2133–2135. doi: 10.1039/b618465k. [DOI] [PubMed] [Google Scholar]
- 102.Mahara A, Iwase R, Sakamoto T, Yamana K, Yamaoka T, Murakami A. Angew. Chem. Int. Ed. 2002;41:3648–3650. doi: 10.1002/1521-3773(20021004)41:19<3648::AID-ANIE3648>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 103.Umemoto T, Hrdlicka PJ, Babu BR, Wengel J. ChemBioChem. 2007;8:2240–2248. doi: 10.1002/cbic.200700408. [DOI] [PubMed] [Google Scholar]
- 104.Kolpashchikov DM. Chem. Rev. 2010;110:4709–4723. doi: 10.1021/cr900323b. [DOI] [PubMed] [Google Scholar]
- 105.Paris PL, Langenhan JM, Kool ET. Nucleic Acids Res. 1998;26:3789–3793. doi: 10.1093/nar/26.16.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bichenkova EV, Sardarian A, Savage HE, Rogert C, Douglas KT. Assay Drug Devel. Technol. 2005;3:39–46. doi: 10.1089/adt.2005.3.39. [DOI] [PubMed] [Google Scholar]
- 107.Marti AA, Li XX, Jockusch S, Li ZM, Raveendra B, Kalachikov S, Russo JJ, Morozova I, Puthanveettil SV, Ju JY, Turro NJ. Nucleic Acids Res. 2006;34:3161–3168. doi: 10.1093/nar/gkl406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bichenkova EV, Gbaj A, Walsh L, Savage HE, Rogert C, Sardarian AR, Etchells LL, Douglas KT. Org. Biomol. Chem. 2007;5:1039–1051. doi: 10.1039/b700293a. [DOI] [PubMed] [Google Scholar]
- 109.Kumar TS, Wengel J, Hrdlicka PJ. ChemBioChem. 2007;8:1122–1125. doi: 10.1002/cbic.200700144. [DOI] [PubMed] [Google Scholar]
- 110.Christensen UB, Pedersen EB. Helv. Chim. Acta. 2003;86:2090–2097. [Google Scholar]
- 111.Wang K, Tang Z, Yang CJ, Kim Y, Fang X, Li W, Wu Y, Medley CD, Cao Z, Li J, Colon P, Lin H, Tan W. Angew. Chem. Int. Ed. 2009;48:856–870. doi: 10.1002/anie.200800370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Venkatesan N, Seo YJ, Kim BH. Chem. Soc. Rev. 2008;37:648–663. doi: 10.1039/b705468h. [DOI] [PubMed] [Google Scholar]
- 113.Saito Y, Mizuno E, Bag SS, Suzuka I, Saito I. Chem. Commun. 2007:4492–4494. doi: 10.1039/b709715h. [DOI] [PubMed] [Google Scholar]
- 114.Saito Y, Shinohara Y, Bag SS, Takeuchi Y, Matsumoto K, Saito I. Tetrahedron. 2009;65:934–939. [Google Scholar]
- 115.Fujmoto K, Shimizu H, Inouye M. J. Org. Chem. 2004;69:3271–3275. doi: 10.1021/jo049824f. [DOI] [PubMed] [Google Scholar]
- 116.Seo YJ, Hwang GT, Kim BH. Tetrahedron Lett. 2006;47:4037–4039. [Google Scholar]
- 117.Matsumoto K, Shinohara Y, Bag SS, Takeuchi Y, Morii T, Saito Y, Saito I. Bioorg. Med. Chem. Lett. 2009;19:6392–6395. doi: 10.1016/j.bmcl.2009.09.060. [DOI] [PubMed] [Google Scholar]
- 118.Conlon P, Yang CJ, Wu Y, Chen Y, Martinez K, Kim Y, Stevens N, Marti AA, Jockusch S, Turro NJ, Tan W. J. Am. Chem. Soc. 2008;130:336–342. doi: 10.1021/ja076411y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Varghese R, Wagenknecht HA. Org. Biomol. Chem. 2010;8:526–528. doi: 10.1039/b918381g. [DOI] [PubMed] [Google Scholar]
- 120.Smalley MK, Silverman SK. Nucleic Acids Res. 2006;34:152–166. doi: 10.1093/nar/gkj420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Seo YJ, Lee IJ, Yi JW, Kim BH. Chem. Commun. 2007:2817–2819. doi: 10.1039/b707278c. [DOI] [PubMed] [Google Scholar]
- 122.Alberti P, Bourdoncle A, Sacca B, Lacroix L, Mergny JL. Org. Biomol. Chem. 2006;4:3383–3391. doi: 10.1039/b605739j. [DOI] [PubMed] [Google Scholar]
- 123.Beuck C, Singh I, Bhattacharya A, Heckler W, Parmar VS, Seitz O, Weinhold E. Angew. Chem. Int. Ed. 2003;42:3958–3960. doi: 10.1002/anie.200219972. [DOI] [PubMed] [Google Scholar]
- 124.Krosky DJ, Song F, Stivers JT. Biochemistry. 2005;44:5949–5959. doi: 10.1021/bi050084u. [DOI] [PubMed] [Google Scholar]
- 125.Jeong HS, Kang S, Lee JY, Kim BH. Org. Biomol. Chem. 2009;7:921–925. doi: 10.1039/b816768k. [DOI] [PubMed] [Google Scholar]
- 126.Crooke ST, Vickers T, Lima W, Wu H. In: Antisense Drug – Technology – Principles, Strategies and Applications. 2nd ed. Crooke ST, editor. CRC Press; Boca Raton: 2008. pp. 3–46. [Google Scholar]
- 127.Rogers FA, Lloyd JA, Glazer PM. Curr. Med. Chem. – Anti–Cancer Agents. 2005;5:319–326. doi: 10.2174/1568011054222300. [DOI] [PubMed] [Google Scholar]
- 128.Ghosh I, Stains CI, Ooib AT, Segal DJ. Mol. Biosyst. 2006;2:551–560. doi: 10.1039/b611169f. [DOI] [PubMed] [Google Scholar]
- 129.Duca M, Vekhoff P, Oussedik K, Halby L, Arimondo PB. Nucleic Acids Res. 2008;36:5123–5138. doi: 10.1093/nar/gkn493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Simon P, Cannata F, Concordet JP, Giovannangeli C. Biochimie. 2008;90:1109–1116. doi: 10.1016/j.biochi.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 131.Filichev VV, Pedersen EB. J. Am. Chem. Soc. 2005;127:14849–14858. doi: 10.1021/ja053645d. [DOI] [PubMed] [Google Scholar]
- 132.Schneider UV, Mikkelsen ND, Jøhnk N, Okkels LM, Westh H, Lisby G. Nucleic Acids Res. 2010;38:4394–4403. doi: 10.1093/nar/gkq188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Paramasivam M, Cogoi S, Filichev VV, Bomholt N, Pedersen EB, Xodo LE. Nucleic Acids Res. 2008;36:3494–3507. doi: 10.1093/nar/gkn242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hrdlicka PJ, Kumar TS, Wengel J. Chem. Commun. 2005:4279–4281. doi: 10.1039/b506986f. [DOI] [PubMed] [Google Scholar]
- 135.Sau SP, Kumar TS, Hrdlicka PJ. Org. Biomol. Chem. 2010;8:2028–2036. doi: 10.1039/b923465a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chenna V, Rapireddy S, Sahu B, Ausin C, Pedroso E, Ly DH. ChemBioChem. 2008;9:1–4. doi: 10.1002/cbic.200800441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sau SP, Hrdlicka PJ. unpublished results.
- 138.Hrdlicka PJ, et al. unpublished results.
- 139.Papasani MR, Hrdlicka PJ, Hill RA, et al. unpublished results.
- 140.Filichev VV, Vester B, Hansen LH, Pedersen EB. Nucleic Acids Res. 2005;33:7129–7137. doi: 10.1093/nar/gki1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.LaFramboise T. Nucleic Acids Res. 2009;37:4181–4193. doi: 10.1093/nar/gkp552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Weisbrod SH, Marx A. Chem. Commun. 2008:5675–5685. doi: 10.1039/b809528k. [DOI] [PubMed] [Google Scholar]
- 143.Wengel J. Org. Biomol. Chem. 2004;2:277–280. doi: 10.1039/b313986g. [DOI] [PubMed] [Google Scholar]
- 144.Nakamura M, Ohtoshi Y, Yamana K. Chem. Commun. 2005:5163–5165. doi: 10.1039/b507808c. [DOI] [PubMed] [Google Scholar]