

Abstract
Heterogeneity in the composition of neurotransmitter receptors is thought to provide functional diversity that may be important in patterning neural activity and shaping behavior (Dani and Bertrand, 2007; Sassoè-Pognetto, 2011). However, this idea has remained difficult to evaluate directly because of the complexity of neuronal connectivity patterns and uncertainty about the molecular composition of specific receptor types in vivo. Here we dissect how molecular diversity across receptor types contributes to the coordinated activity of excitatory and inhibitory motor neurons in the nematode Caenorhabditis elegans. We show that excitatory and inhibitory motor neurons express distinct populations of ionotropic acetylcholine receptors (iAChRs) requiring the ACR-12 subunit. The activity level of excitatory motor neurons is influenced through activation of nonsynaptic iAChRs (Jospin et al., 2009; Barbagallo et al., 2010). In contrast, synaptic coupling of excitatory and inhibitory motor neurons is achieved through a second population of iAChRs specifically localized at postsynaptic sites on inhibitory motor neurons. Loss of ACR-12 iAChRs from inhibitory motor neurons leads to reduced synaptic drive, decreased inhibitory neuromuscular signaling, and variability in the sinusoidal motor pattern. Our results provide new insights into mechanisms that establish appropriately balanced excitation and inhibition in the generation of a rhythmic motor behavior and reveal functionally diverse roles for iAChR-mediated signaling in this process.
Introduction
Nicotinic or ionotropic acetylcholine receptors (iAChRs) play diverse roles in nervous systems ranging from nematodes to mammals. A large variety of alternative iAChR subunit combinations, each with distinguishing features, participate in neural circuits throughout the CNS (Dani and Bertrand, 2007). In the mammalian brain, iAChRs are most commonly localized to synaptic terminals or extrasynaptic sites in which they function to modulate neurotransmitter release or regulate neuronal excitability, although considerable functional variability exists (Mulle et al., 1991; Grady et al., 2009; Mackey et al., 2012). Specific neuronal iAChR subtypes are preferentially targeted by addictive drugs, such as nicotine (Tapper et al., 2004; Fowler et al., 2011), and iAChRs are critically involved in the process of addiction as well as the pathophysiology of a variety of neurological disorders (Tuesta et al., 2011). Because of the diversity of molecules involved, the limited availability of pharmacological tools that target specific iAChR subtypes, and the complexity of neuronal connectivity patterns, it remains difficult to address how particular iAChR subunit combinations contribute to the function of specific neural circuits in the mammalian CNS.
Here we use genetic and electrophysiological approaches to investigate how specific iAChR subtypes contribute to synaptic function and circuit activity using an anatomically well-defined motor circuit of the nematode Caenorhabditis elegans. In C. elegans, excitatory cholinergic motor neurons (ACh MNs) make synaptic contacts onto both muscle cells and GABA MNs that, in turn, make inhibitory synaptic contacts onto opposing musculature (see Fig. 3A) (White et al., 1986). Proper function of this circuit produces temporally coordinated and balanced excitatory and inhibitory signals that pattern movement. Although the anatomical connectivity of this circuit has been well characterized, the signaling mechanisms that underlie coordinated MN activity are not well understood. Many of the 29 iAChR subunits encoded by the C. elegans genome are expressed in MNs (Cinar et al., 2005; Fox et al., 2005; Jones et al., 2007; Rand, 2007). In particular, ACh MNs express a class of heteromeric acetylcholine-gated ion channel complexes known as ACR-2R (Jospin et al., 2009; Barbagallo et al., 2010). ACR-2Rs are composed of five distinct subunits (ACR-2, ACR-3, UNC-38, UNC-63, and ACR-12), each of which is essential for function in heterologous expression studies. Loss of ACR-2R leads to relatively subtle changes in behavior; however, gain-of-function acr-2 mutations have profound consequences, including hyperactivation and, in extreme cases, death of ACh MNs (Jospin et al., 2009; Barbagallo et al., 2010). Loss-of-function and cell-specific expression experiments demonstrated that the effects of acr-2(gf) were dependent on expression of the partnering iAChR subunit acr-12 in ACh MNs.
Figure 3.

ACR-12 is differentially localized across MN populations. A, Schematic of C. elegans locomotory circuit. Red coloring indicates ACh MNs (VA, VB, DA, and DB) and blue indicates GABA MNs (DD and VD). Dark gray and light gray shading represent body wall muscles. In A and C, black triangles and boxes represent sites of cholinergic (excitatory) and GABAergic (inhibitory) innervation of muscles, respectively. Figure modified from WormAtlas (http://www.wormatlas.org/). B, Independent confocal images showing ACR-12–GFP localization in ventral and dorsal nerve cords of young adult animals as indicated (ufIs57). Positions of cell bodies are indicated by white arrowheads in B, D, and E. Scale bars, 10 μm. C, Diagram depicting subcellular localization of ACR-12–GFP and the presynaptic marker mCherry–RAB-3 in ACh and GABA MNs. Although ACh MNs form dyadic synapses onto both GABA MNs and muscles, only synaptic contacts between ACh and GABA MNs are depicted for clarity. ACR-12–GFP fluorescence (green shading) is diffuse in the processes of ACh VA and VB MNs. In contrast, ACR-12–GFP fluorescence appears punctate (green circles) in the processes of GABAergic VD and DD MNs and localizes opposite mCherry–RAB-3 fluorescent puncta (red circles) expressed in ACh MNs. D, Confocal image of Pacr-2::ACR-12–GFP (ufIs78) localization in ACh MNs of the posterior ventral nerve cord. E, Confocal images of Punc-47::ACR-12–GFP (ufIs92) localization in GABA MNs of the posterior ventral and dorsal nerve cords as indicated. F, Confocal image of the dorsal nerve cord showing apposition of mCherry–RAB-3 expressed in ACh MNs (Pacr-2, ufIs63) with ACR-12–GFP expressed in GABA MNs (Punc-47, ufIs92). Scale bar, 10 μm.
We show here that acr-12 is also expressed in GABA MNs, and a distinct population of iAChRs requiring ACR-12 appears specifically localized at postsynaptic sites on GABA MN processes. Loss of ACR-12 iAChRs from inhibitory MNs leads to reduced synaptic drive, decreased inhibitory neuromuscular signaling, and variability in the sinusoidal motor pattern characteristic of nematode movement. Together, our results suggest that ACR-12 iAChRs regulate motor circuit activity by contributing to the synaptic coupling of excitatory and inhibitory MNs.
Materials and Methods
C. elegans strains.
C. elegans strains were grown under standard laboratory conditions at 22°C. All strains are derivatives of the N2 Bristol strain (wild type). Transgenic strains were obtained by microinjection to achieve germ-line transformation. Multiple independent extragenic lines were obtained for each transgenic strain, and data presented are from a single representative transgenic line unless noted otherwise. In all cases, lin-15(n765ts) mutants were injected with the lin-15 rescuing plasmid (pL15Ek; 30 ng/μl) and one or more of the following plasmids: pPRB5 [Punc-47::mCherry], pPRB6 [Pacr-2::mCherry], pPRB47 [Pacr-2::mCherry-RAB-3], pPRB53 [ACR-12–GFPICL], pPRB77 [Pacr-2::ACR-12–GFPICL], pHP7 [Punc-47::ACR-12–GFPICL], and pAG21 [ACR-12–GFPC-term]. Stably integrated lines were generated by x-ray integration and outcrossed at least four times to wild type. The following strains were used in this study: IZ914: acr-12(ok367)X; IZ853: acr-12(ok367)X;ufIs57[ACR-12–GFPICL]; IZ984: acr-12(ok367);ufIs78[Pacr-2::ACR-12::GFPICL]; IZ556: acr-12(ok367);ufIs92[Punc-47::ACR-12–GFPICL]; IZ941: unc-49(e382)III;acr-12(ok367)X; IZ33: unc-29(x29);acr-16(ok789); IZ514: unc-29(x29);acr-16(ok789);acr-12(ok367); IZ629: ufIs38[ACR-12–GFPC-term];ufIs34[Punc-47::mCherry]; IZ651: ufIs38[ACR-12–GFPC-term];ufIs43[Pacr-2::mCherry]; IZ632: ufIs26[Punc-4::mCherry];ufIs38[ACR-12–GFPC-term]; IZ557: ufIs63[Pacr-2::mCherry-RAB-3];ufIs92[Punc-47::ACR-12–GFPICL]; CB382: unc-49(e382)III; IZ712: acr-12(uf77)X; RB1559: acr-2(ok1887)X.
Molecular biology.
The ACR-12–GFPICL transgene (pPRB53) was generated by cloning the green fluorescent protein (GFP) coding sequence in-frame into the sequence of an acr-12 genomic fragment (−1514 to +4799 bp relative to the translational start site) encoding the intracellular loop (ICL) between transmembrane domains TM3 and TM4. As is the case for other iAChR subunits, incorporation of GFP at this position yielded a functional construct (Nashmi et al., 2003; Francis et al., 2005), as demonstrated by ACR-12–GFPICL localization in neuronal processes and rescue of acr-12 mutants. pAG21 (ACR-12–GFPC-term) was provided by Alexander Gottschalk (Goethe University, Frankfurt, Germany) and was used in cell identifications (see Fig. 2). ACR-12–GFPC-term fluorescence was primarily confined to cell bodies. Punc-47::ACR-12–GFPICL (pHP7) and Punc-47::ACR-12–GFPICL (pPRB77) were generated by subcloning a 4.9 kb NruI/BglI fragment containing GFP from pPRB53 into constructs encoding the acr-12 cDNA control of the appropriate promoters (pBB25 and pHP3, respectively). Punc-47::mCherry (pPRB5) was generated by subcloning mCherry coding sequence downstream of a 1.3 kb promoter for the unc-47 gene. Pacr-2::mCherry (pPRB6) was generated by subcloning a 3.3 kb promoter for the acr-2 gene upstream of the mCherry coding sequence.
Figure 2.
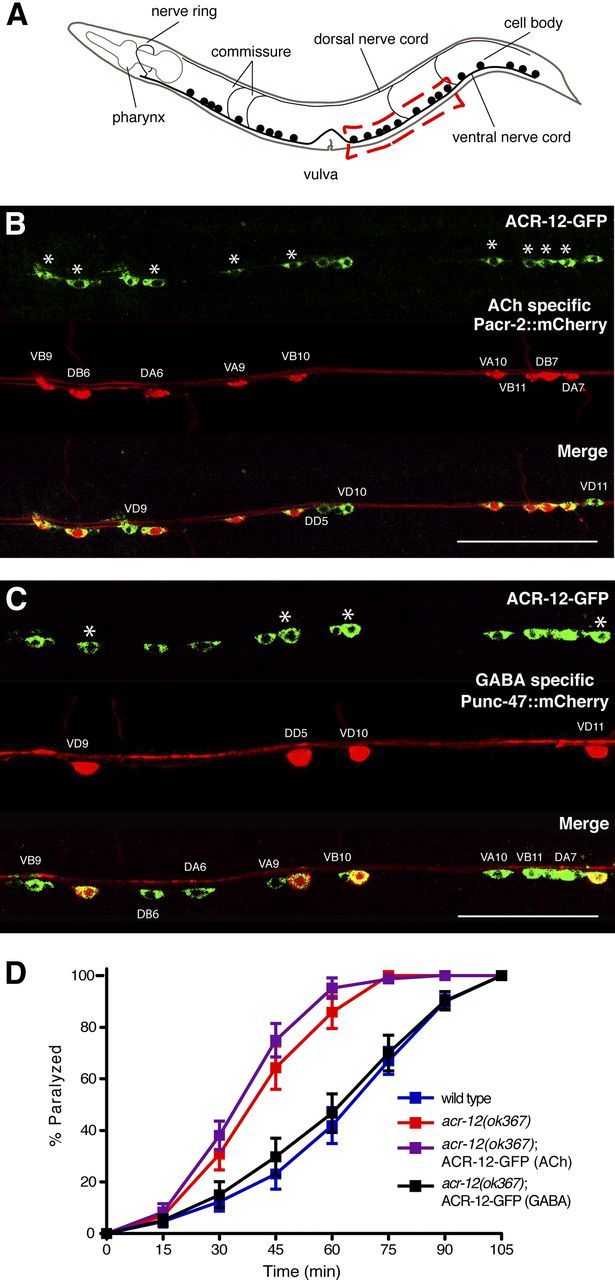
ACR-12 iAChRs regulate GABA MN activity. A, Diagram of C. elegans showing ventral nerve cord. Red box indicates approximate region imaged in B and C. B, Confocal image showing coexpression of ACR-12–GFP (ufIs38) and a ACh MN-specific marker (Pacr-2::mCherry, ufIs43) in the posterior ventral nerve cord. For B and C, asterisks indicate MN cell bodies coexpressing both reporters. Scale bars, 50 μm. C, Confocal image showing coexpression of ACR-12–GFP (ufIs38) and a GABA neuron-specific marker (Punc-47::mCherry, ufIs34) in the posterior ventral nerve cord. D, Time course of paralysis in the presence of aldicarb (1 mm) for wild-type (blue) (n = 14), acr-12(ok367) mutants (red) (n = 14), ACh-specific (Pacr-2) ACR-12–GFP rescue (purple) (n = 10), and GABA-specific (Punc-47) ACR-12–GFP rescue (black) (n = 10).
Microscopy.
Confocal microscopy was performed using a Carl Zeiss Axioskop 2 microscope system and LSM Pascal 5 imaging software (Carl Zeiss). All images used staged animals 24 h after the L4 stage and were processed using NIH ImageJ software. For all synapse quantification, a region of the dorsal cord directly across from the vulva was imaged. Synapses were quantified within a 50 μm region of interest by thresholding fluorescence intensity and using the analyze particles module in NIH ImageJ software. The percentage of overlap with presynaptic RAB-3 was determined by quantifying the number of mCherry puncta that were positioned either directly apposing or overlapping with ACR-12::GFP fluorescent signal.
Behavioral assays.
All behavioral analyses were performed using staged populations of young adult animals (24 h after L4) at room temperature (22–24°C). Strains were scored in parallel, with the researcher blinded to genotype. For aldicarb assays, staged populations of adult animals (≥10) were transferred to nematode growth medium plates containing 1 mm aldicarb (ChemService), and movement was assessed every 15 min for 2 h. Movies and still images for behavioral analyses were obtained using an Olympus SZ61 upright microscope equipped with a FireWire camera (Imaging Source). For gross movement, individual worms were transferred to large unseeded plates and assayed manually. Body bend amplitudes were determined using NIH ImageJ software. The distance between the deepest point of the bend and a line tangent to the tip of the head and the body was measured. This measurement was then normalized to the length of each animal and averaged to generate a value for average body bend amplitude.
Electrophysiology.
Endogenous postsynaptic currents (PSCs) were recorded from body wall muscles as described previously (Francis et al., 2005). The extracellular solution consisted of the following (in mm):150 NaCl, 5 KCl, 4 MgCl2, 1 CaCl2, 15 HEPES, and 10 glucose, pH 7.4, osmolarity adjusted with 20 sucrose. The intracellular fluid consisted of the following (in mm):115 K-gluconate, 25 KCl, 0.1 CaCl2, 50 HEPES, 5 Mg-ATP, 0.5 Na-GTP, 0.5 cGMP, 0.5 cAMP, and 1 BAPTA, pH 7.4, osmolarity adjusted with 10 sucrose. For some experiments measuring GABA-mediated currents, the intracellular solution contained 115 mm KCl and 25 mm K-gluconate, enabling measurement of inward inhibitory current responses (chloride efflux). At least 60–90 s of continuous data were used in the analysis. Data analysis was performed using Igor Pro (Wavemetrics) and Mini Analysis (Synaptosoft) software. Statistical comparisons were made by Student's t test using GraphPad Prism.
Results
Loss of ACR-12 leads to heightened excitability
acr-12 encodes a 573 aa iAChR α subunit with significant homology to the ACR-8-like group of C. elegans subunits and more distant homology to mammalian neuronal heteromeric α subunits such as α6 (Fig. 1) (Jones et al., 2007). To investigate the contribution of ACR-12 receptor complexes to the excitability of neurons in the motor circuit, we evaluated independent strains carrying putative loss-of-function alleles of acr-12 during acute exposure to the cholinesterase inhibitor aldicarb. Treatment of C. elegans with aldicarb leads to elevated levels of synaptic ACh and causes paralysis over time attributable to prolonged muscle contraction (Nguyen et al., 1995). The contractile state of muscles reflects the summed activity of excitatory ACh and inhibitory GABA synaptic inputs; genetic mutations that alter this balance will shift the time course over which aldicarb leads to paralysis. For example, manipulations that reduce or eliminate inhibitory GABA signaling cause enhanced muscle activation and more rapid paralysis (Loria et al., 2004; Vashlishan et al., 2008). acr-12(uf77) was isolated as a suppressor of acr-2(gf) and is a missense mutation affecting a splice acceptor site that results in premature termination before transmembrane domain 4 (amino acids 387) (Barbagallo et al., 2010). acr-12(ok367) is a deletion mutation that eliminates 1368 bp of chromosomal DNA, including sequence encoding transmembrane domains 1–3 (Fig. 1A,B). Strains carrying either of these alleles were viable, and gross connectivity of the nervous system was normal (data not shown), indicating that ACR-12 is not required for normal development of the nervous system. We found the acr-12(uf77) and acr-12(ok367) mutations both accelerated the time course of aldicarb-induced paralysis (Fig. 1C), suggesting that loss of ACR-12 led to enhanced excitability. In contrast, previous work showed that removal of ACR-2Rs slightly delayed paralysis in response to aldicarb treatment (Jospin et al., 2009; Barbagallo et al., 2010; Fig. 1C). The differential effects of aldicarb across acr-2 and acr-12 mutants suggested that ACR-12 has additional roles in the nervous system independent of ACR-2. To better define potential functions for ACR-12 signaling, we constructed double mutants lacking both acr-12 and unc-49. The unc-49 gene encodes an essential subunit of ionotropic GABAA-like receptors at inhibitory neuromuscular synapses, and mutations in unc-49 cause aldicarb hypersensitivity (Fig. 1C) (Bamber et al., 1999). unc-49;acr-12 double mutants exhibited no additional hypersensitivity beyond that of unc-49 single mutants, suggesting that acr-12 and unc-49 may act in the same pathway.
Figure 1.

ACR-12 gene sequence. A, ACR-12 amino acid sequence. cys-loop (*), vicinal cysteines present in α subunits (∼), GFP insertion site (green lettering), transmembrane domains (gray shading), ok367 deletion (blue lettering), and site of uf77 mutation (yellow shading) are indicated. B, Schematic of ACR-12 receptor subunit. Approximate locations of cys-loop (*), vicinal cysteines (∧∧), GFP insertion site (green box), transmembrane domains (M1–M4), ok367 deletion (red shading), and uf77 mutation (yellow star) are indicated. C, Time course of paralysis in the presence of aldicarb (1 mm) for wild-type (blue) (n = 16), acr-12(ok367) mutants (red) (n = 14), acr-12(uf77) mutants (gray) (n = 8), unc-49(e382) mutants (brown) (n = 11), unc-49;acr-12(ok367) double mutants (green) (n = 10), ACR-12–GFP rescue using the native promoter (purple) (n = 12), and acr-2(ok1887) mutants (black) (n = 4) are shown. Data represent mean ± SEM.
acr-12 is reported to have broad expression in the nervous system (Gottschalk et al., 2005), but a precise description of the MN classes that express acr-12 has remained unclear. To address this issue, we generated transgenic strains expressing ACR-12 tagged with GFP at the C terminus together with various red fluorescent reporters labeling defined classes of ventral cord (VC) MNs and examined the cellular distribution of ACR-12–GFP. ACR-12–GFP fluorescence was broadly visible in MN cell bodies and overlapped with a reporter that specifically labeled ACh MNs (Pacr-2::mCherry) (Fig. 2B). We also examined the coexpression of ACR-12–GFP with a red fluorescent protein reporter (Punc-4::mCherry) that labels a subset of ACh MNs (A class) and the VC neurons that are primarily involved in egg laying (Lickteig et al., 2001; Bany et al., 2003). We observed partial overlap of ACR-12–GFP with Punc-4::mCherry and noted that ACR-12–GFP fluorescence was absent from VC MNs (data not shown). Additional GFP fluorescence was clearly visible in neuronal cell bodies along the VC that were not labeled by either Pacr-2::mCherry or Punc-4::mCherry. The cell bodies of GABA MNs are also located in the ventral nerve cord. To test whether the additional neurons expressing ACR-12–GFP were GABA MNs, we imaged animals coexpressing ACR-12–GFP with a reporter labeling GABA neurons (Punc-47::mCherry) and observed overlapping fluorescent signals (Fig. 2C). Our results show that acr-12 is expressed in the major classes of ACh (DA, VA, DB, VB) and GABA (DD and VD) MNs that have primary functions in locomotory control. Because expression of some ACR-2R constituents (e.g., acr-2 and acr-3) is limited to ACh MNs (Jospin et al., 2009; Barbagallo et al., 2010), our findings strongly suggest that ACR-12 contributes to molecularly distinct iAChR populations that are differentially expressed across ACh and GABA MNs.
To dissect functional roles for ACR-12 across MN classes, we specifically expressed ACR-12 in either ACh or GABA MN of acr-12 mutants and tested the responses of these animals to aldicarb treatment (Fig. 2D). Specific expression of acr-12 in ACh MNs did not alter the aldicarb hypersensitivity of acr-12 mutants. In contrast, specific expression of acr-12 in GABA neurons restored wild-type sensitivity to aldicarb. Our results suggest that ACR-12 receptor complexes regulate GABA MN activity and inhibitory neuromuscular signaling under conditions when ACh levels are elevated.
iAChRs requiring ACR-12 have distinct patterns of localization and functional roles across MN classes
We examined the subcellular distribution of ACR-12 in MNs by expressing a rescuing ACR-12–GFP fusion protein under control of the native promoter (Fig. 3B). ACR-12–GFP expression was clearly visible in MN processes of both the ventral and dorsal nerve cords of adult animals and exhibited two contrasting patterns of fluorescence. In the ventral nerve cord, we observed regions of punctate and diffuse localization, whereas in the dorsal cord, we observed punctate fluorescence almost exclusively (Fig. 3B,C). In contrast, specific expression of ACR-12–GFP in ACh MNs produced only diffuse fluorescence in the ventral nerve cord and no detectable fluorescent signal in the dorsal nerve cord (Fig. 3D). Axons and dendrites of ventrally directed ACh MNs (ventral A and B classes) extend through the ventral nerve cord. ACh MNs innervating dorsal musculature (dorsal A and B classes) also have cell bodies in the ventral nerve cord but extend axons into the dorsal nerve cord, in which they make dyadic synaptic contacts with body wall musculature and dendrites of ventrally directed GABA MNs (ventral D class) (White et al., 1986) (Fig. 3A). We previously observed diffuse ACR-2–GFP fluorescence in the dendrites of ACh MNs without obvious postsynaptic localization (Barbagallo et al., 2010). Our current finding that ACR-12–GFP is also diffusely localized in ACh MN dendrites is consistent with the notion that ACR-2 and ACR-12 partner together in heteromeric complexes in ACh MNs. Specific expression of ACR-12–GFP in GABA MNs produced solely punctate fluorescence (Fig. 3E). Many of these ACR-12–GFP puncta were located immediately apposed to regions of ACh MN axons in which the synaptic vesicle marker mCherry–RAB-3 was concentrated (Fig. 3F) (Mahoney et al., 2006; Klassen and Shen, 2007), suggesting clustering opposite sites of neurotransmitter release. We quantified fluorescence in a 50 μm segment of the posterior dorsal nerve cord and found that a majority (87 ± 3%) of RAB-3 puncta overlapped with ACR-12–GFP signal. However, ACR-12 puncta were more numerous than RAB-3 (25 ± 2/50 μm and 15.9 ± 2/50 μm, respectively), and we noted a subset of ACR-12–GFP puncta without obvious RAB-3 apposition, perhaps because the acr-2 promoter does not drive RAB-3 expression in all of the neurons that are presynaptic to GABA MNs. Our results provide evidence that ACR-12 complexes in GABA MNs are clustered in receptor fields located opposite presynaptic specializations of ACh MNs.
ACR-12 expression in GABA MNs is required for normal inhibitory signaling
To directly test the requirement for ACR-12 in regulating MN activity, we used standard electrophysiology recording techniques to measure the frequency of synaptic events at the neuromuscular junction (NMJ) in vivo (Francis and Maricq, 2006). We used two independent strategies to distinguish between GABA and ACh PSCs: (1) recordings were made under ionic conditions in which GABA- and ACh-mediated events were separable based on the directionality of the currents (for details, see Materials and Methods); and (2) we recorded from unc-29;acr-16 double mutants that are devoid of functional iAChRs on body wall muscles and lack excitatory neurotransmission at the NMJ (Francis et al., 2005; Touroutine et al., 2005). Consistent with previous findings for acr-2 mutants (Jospin et al., 2009), deletion of acr-12 significantly reduced the rate of endogenous EPSCs (37 ± 14% decrease, p = 0.01) (Fig. 4) but did not affect EPSC amplitude (wild-type, 28.7 ± 1.3 pA; acr-12, 25.5 ± 1.2 pA, p = 0.09). The reduction in EPSC frequency was rescued by expression of an ACR-12 rescuing transgene under control of the native promoter. When recording outward GABA-mediated IPSCs (25 mm intracellular Cl−), we also observed a significant reduction in the rate of inhibitory events in mutants lacking ACR-12 (73 ± 24% decrease, p < 0.01; Fig. 5). We did not observe a significant difference in the amplitude of endogenous IPSCs (wild-type, 26 ± 1.8 pA; acr-12, 23.4 ± 2 pA, p > 0.05; Fig. 5A,B). When recording inward GABA-mediated IPSCs from unc-29;acr-16 double mutants (115 mm intracellular Cl−), we observed a lower basal rate of IPSCs compared with wild-type animals. This may reflect the differences in recording conditions used in each set of experiments or secondary effects attributable to the absence of excitatory transmission at the NMJ. Deletion of acr-12 significantly reduced the frequency of IPSCs in unc-29;acr-16 double mutants (52 ± 13% decrease, p < 0.01; Fig. 5C,D). To distinguish whether the reduction in IPSC frequency arose as a consequence of loss of ACR-12 from GABA MNs or arose as a result of reduced excitation levels in presynaptic ACh MNs, we recorded from animals that expressed acr-12 specifically in GABA MNs. The reduced IPSC rate was rescued by specific expression of acr-12 in GABA MNs but not ACh MNs, consistent with cell-autonomous action of ACR-12 in GABA neurons in determining IPSC rate. Finally, we recorded endogenous IPSCs from acr-2 mutants and, consistent with a previous report (Jospin et al., 2009), found no significant reduction in IPSC rate (Fig. 5A,B). Thus, the reduction in endogenous IPSC frequency in acr-12(ok367) mutants cannot be explained by decreased excitation of presynaptic ACh MNs. Our results provide evidence that independent populations of ACR-12 receptors act cell autonomously in ACh and GABA MNs to regulate their activity.
Figure 4.

EPSC frequency is reduced in acr-12 mutants. A, Representative recordings of endogenous EPSCs recorded at −60 mV for the indicated genotypes. Rescue refers to transgenic expression of ACR-12 in acr-12 mutants using the native promoter. B, Average (mean ± SEM) EPSC frequency for the genotypes indicated. **p < 0.01 from wild type. Wild type, n = 11; acr-12, n = 13; rescue, n = 6.
Figure 5.

Loss of ACR-12 receptors from GABA MNs reduces inhibitory signaling. A, Representative recordings of endogenous IPSCs recorded at 0 mV for the indicated genotypes. Rescue refers to transgenic expression of ACR-12 in acr-12 mutants under control of either the native, unc-47 (GABA) or acr-2 (ACh) promoters. B, Average (mean ± SEM) endogenous IPSC frequency for the genotypes indicated. Wild type, n = 7; acr-12, n = 9; acr-12 rescue (native), n = 4; acr-12 rescue (GABA), n = 5, acr-12 rescue (ACh), n = 4; acr-2, n = 4. **p < 0.01 from wild type. C, Representative recordings of endogenous IPSCs recorded at −60 mV for the indicated genotypes. Recordings were made using 115 mm KCl in the pipette. D, Average (mean ± SEM) endogenous IPSC frequency for the genotypes indicated. unc-29;acr-16, n = 12; unc-29;acr-16;acr-12, n = 11. **p < 0.01 from unc-29;acr-16.
ACR-12-mediated signaling onto MNs regulates locomotion
To determine how ACR-12-mediated signaling onto MNs contributes to normal locomotory behavior, we tracked animals during exploratory movements on agar plates. We found that deletion of acr-12 produced a reduction in movement velocity (Fig. 6). This movement deficit could be rescued completely by expression of an ACR-12 rescuing transgene under control of the native promoter. Additionally, specific expression of ACR-12 in ACh or GABA MNs partially rescued the decreased movement, suggesting that normal levels of iAChR activity in both neuron classes are important for establishing the velocity of movement. Consistent with this idea, acr-12 mutants displayed a more dramatic reduction in velocity than acr-2 mutants. We also noted that the sinusoidal locomotory wave appeared more erratic in acr-12 mutants (Fig. 7A). To examine this in more detail, we monitored consecutive body bends during extended periods of uninterrupted forward movement. Wild-type animals displayed remarkable consistency in their movement with only a few irregularities in their sinusoidal tracks. In the absence of ACR-12 receptors (ok367 and uf77), the motor pattern was less stable, showing significantly increased variability from one body bend to the next (Fig. 7B, p < 0.01). This variability also led to a modest reduction in the average amplitude of body bends (∼14% for both uf77 and ok367, p < 0.01; Fig. 7C). In contrast, deletion of the acr-2 gene did not cause an irregular waveform or a reduction in body bend amplitude, indicating that loss of ACR-2/ACR-12 heteromeric iAChRs from ACh MNs could not account for the changes in waveform. Both of these phenotypes were completely reversed by ACR-12 expression using the native promoter or partially by specific expression of ACR-12 in GABA MNs, implicating involvement of ACR-12 iAChRs expressed by GABA MNs. Additionally, specific expression of acr-12 in ACh MNs partially rescued the variable waveform but not the reduced amplitude, suggesting that elevated acr-12 levels in ACh MNs can partially offset the behavioral effects of loss of ACR-12 iAChRs from inhibitory MNs. Together, our results suggest that two classes of iAChRs containing the ACR-12 subunit act in a distributed manner to coordinate MN activity during movement, leading to consistent locomotory output.
Figure 6.

ACR-12 iAChRs regulate the velocity of movement. Average (mean ± SEM) number of body bends per minute for the genotypes indicated. Wild type, n = 36; acr-12(ok367), n = 29; ACR-12–GFP rescue (native promoter), n = 10; GABA-specific (Punc-47) ACR-12–GFP rescue, n = 10; ACh-specific (Pacr-2) ACR-12–GFP rescue, n = 10; and acr-2(ok1887), n = 17. *p < 0.05 from wild type; **p < 0.01 from wild type.
Figure 7.
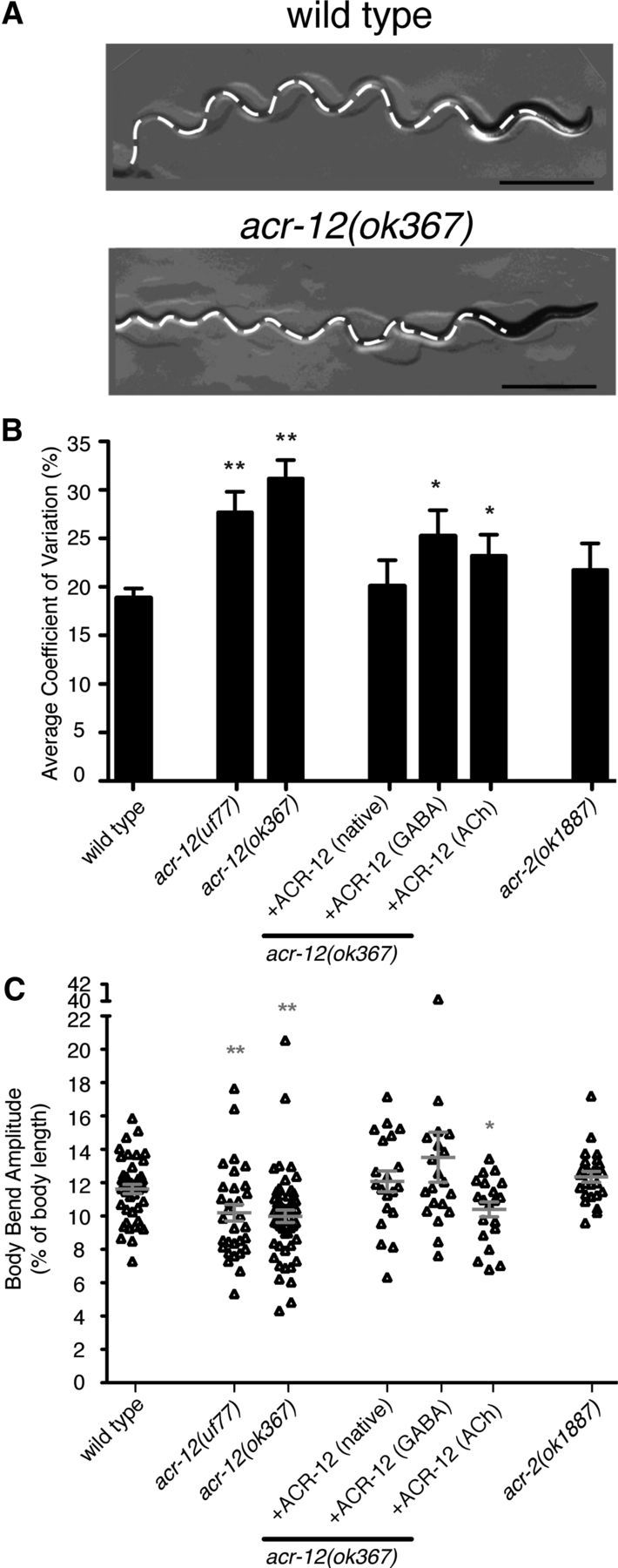
ACR-12 iAChRs are required for consistent motor pattern. A, Still images of locomotory paths for wild-type and acr-12(ok367) mutants as indicated. Tracks are indicated by white dashed lines. The image shows a more severe example of the variable waveform phenotype in acr-12 mutants. Scale bar, 2 mm. B, Coefficient of variance for body bend amplitude averaged over 10 consecutive body bends for genotypes indicated. Wild type, n = 23; acr-12(uf77), n = 11; acr-12(ok367), n = 52; rescue (native promoter), n = 11; rescue (GABA-specific), n = 34; rescue (ACh-specific), n = 14; acr-2(ok1887), n = 13. *p < 0.05 from wild type; **p < 0.01 from wild type. C, Scatter plot of body bend amplitudes for genotypes indicated. Wild type, n = 45; acr-12(uf77), n = 31; acr-12(ok367), n = 52; rescue (native), n = 20; rescue (GABA-specific), n = 20; rescue (ACh-specific), n = 20; acr-2(ok1887), n = 24. Each point represents an average of three consecutive body bends for an individual animal. Mean ± SEM is indicated by horizontal lines. *p < 0.05 from WT; **p < 0.01 from wild type.
Discussion
Although the anatomical connectivity of the C. elegans nervous system was described almost 30 years ago, we have only recently been able to reconcile this static picture of nervous system wiring with measures of functional connectivity. One of the barriers to progress in understanding the neural circuit mechanisms responsible for the generation of movement has been an inability to independently manipulate ACh and GABA MNs. For example, mutations that alter the activity of ACh MNs also impact synaptically coupled GABA MNs. To investigate the mechanisms by which functional connectivity among MN classes may be achieved, we analyzed the contribution of iAChRs to motor circuit activity. Our results show that at least two classes of iAChRs requiring ACR-12 participate in shaping motor output. Using genetic, behavioral, and electrophysiological analysis of acr-12 mutants, we have been able to investigate the contribution of each receptor class to motor circuit activity and behavior. Our data suggest that synaptic coupling of ACh and GABA MNs is achieved, at least in part, through ACR-12 iAChRs located on GABA MNs. Mutations in acr-12 caused increased sensitivity to drugs that enhance cholinergic activity, reduced inhibitory MN activity, and altered the locomotory pattern during movement. The behavioral requirement for ACR-12 iAChRs on GABA MNs was most apparent when levels of ACh MN activity were high, suggesting that a primary function of these receptors may be to limit hyperactive motor output.
ACR-12 functions at synapses on GABA MNs
To address how specific iAChR classes contribute to motor circuit activity, we analyzed the expression, subcellular localization, and loss-of-function phenotype of acr-12 mutants. ACR-12::GFP fluorescence was diffusely distributed in ACh MNs, consistent with previous reports suggesting that heteromeric ACR-2Rs containing ACR-12 may act to maintain appropriate levels of activation through an extrasynaptic mechanism (Jospin et al., 2009; Barbagallo et al., 2010). In contrast, ACR-12::GFP fluorescence was punctate in GABA MNs and localized opposite putative synaptic release sites in ACh MNs, suggesting that ACR-12 iAChRs located on GABA neurons are localized to synapses. Interestingly, iAChRs play similarly diverse roles in the mammalian nervous system in which they are primarily postsynaptic at autonomic synapses and primarily presynaptic or extrasynaptic in the brain (Dani and Bertrand, 2007). The two localization patterns we observed might result from differences in subunit composition of the receptor or differential expression of localization factors unique to each MN subtype. Additional characterization of the receptor constituents in GABA MNs will be key to addressing this important question, as well as understanding potential effects of differing biophysical properties across receptor types. At least two subunits of iAChRs present on ACh MNs (ACR-2R) are not expressed in GABA MNs (Jospin et al., 2009), implying that distinct receptor subunit combinations are expressed by each MN class. However, a complete description of ACR-12 partners in GABA MNs is complicated by the fact that most iAChR subunits characterized to date have broad expression patterns in the nervous system, and many are also expressed in muscle.
Our analysis provided strong evidence ACR-12 receptors regulate GABA MN activity; however, several observations indicate that a residual level of GABA MN signaling persists in acr-12 mutants. First, acr-12 mutants exhibited variability in the locomotory wave and slowed gross movement but did not phenocopy mutants in which GABA signaling is absent. For example, acr-12 mutants did not display bilateral muscle contractions in response to tactile stimulation (shrinking) that are characteristic of GABA-deficient mutants (McIntire et al., 1993; Schuske et al., 2004). Second, acr-12 mutants carrying an additional mutation that eliminated neuromuscular GABA signaling (unc-49;acr-12 double mutants) showed enhanced sensitivity to aldicarb treatment compared with acr-12 single mutants. Finally, electrophysiological recordings from body wall muscles showed that GABA IPSCs continued at a reduced rate in acr-12 mutants but were not eliminated. These findings suggest the presence of additional mechanisms for regulating inhibitory activity, either intrinsic to GABA MNs or mediated through signaling pathways onto GABA MNs that function independently of ACR-12. Alternatively, it is possible that there is no absolute requirement for ACR-12 in the formation of functional heteromeric iAChR complexes on GABA neurons. However, because acr-12 encodes an α subunit and there is typically a stringent requirement for α subunits in iAChR function, this latter possibility appears less likely. Two non-identical α subunits (UNC-38 and UNC-63) are required for functional reconstitution of the levamisole-sensitive iAChR from C. elegans body wall muscles (Boulin et al., 2008), and three non-identical α subunits, including ACR-12, are each required for functional reconstitution of the ACR-2R from C. elegans ACh MNs (Jospin et al., 2009).
ACR-12 iAChRs in motor control
During nematode locomotion, waves of MN activity drive muscle contractions along the length of the body and propagate movement (Pierce-Shimomura et al., 2008; Kawano et al., 2011; Leifer et al., 2011). We found that mutations in acr-12 caused significant phenotypic changes during normal, exploratory movements. However, the most dramatic effects of ACR-12 loss occurred under conditions in which levels of cholinergic activity were elevated by pharmacological treatment (aldicarb). In many organisms, specific MN pools are recruited depending on the behavioral output (Goulding, 2009). The effects of the acr-12 mutation observed in our experiments may suggest a similar process occurs in C. elegans. Thus, under normal conditions, ACR-12-mediated synaptic activation of GABA MNs may be less stringently required, accounting for the relatively modest effects of acr-12 mutations during exploratory movements. In contrast, with high levels of ACh MN activity, an increased requirement for inhibitory signaling may be achieved through ACR-12 iAChR-mediated recruitment of GABA MNs. In this case, synaptic activation of GABA MNs may reinforce the sinusoidal motor pattern and prevent impaired movement attributable to hyperactive motor states. Therefore, a primary role for ACR-12-mediated signaling in the motor circuit may be to limit hyperactive motor output through activation of GABA MNs.
In summary, we have used genetic techniques and electrophysiology to provide a detailed analysis of neuron and circuit function in C. elegans. We have shown that ACR-12 contributes to at least two distinct receptor populations expressed by excitatory and inhibitory MNs. We propose that iAChRs requiring ACR-12 are located at synapses onto GABA MNs, and, through activation of these iAChRs, inhibitory MNs are recruited to pattern undulatory movements.
Footnotes
This work was made possible by support from National Institutes of Health (NIH) Grant R01NS064263 and Muscular Dystrophy Association (M.M.F.) and NIH Predoctoral National Research Service Award (B.B.). Some nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources. We thank members of the Francis laboratory for critical reading of this manuscript and helpful discussions, Mark Alkema and Claire Benard for helpful discussions, Patrick Boyle and David Madsen for technical assistance, and Alexander Gottschalk for reagents.
The authors declare no competing financial interests.
References
- Bamber BA, Beg AA, Twyman RE, Jorgensen EM. The Caenorhabditis elegans unc-49 locus encodes multiple subunits of a heteromultimeric GABA receptor. J Neurosci. 1999;19:5348–5359. doi: 10.1523/JNEUROSCI.19-13-05348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bany IA, Dong MQ, Koelle MR. Genetic and cellular basis for acetylcholine inhibition of Caenorhabditis elegans egg-laying behavior. J Neurosci. 2003;23:8060–8069. doi: 10.1523/JNEUROSCI.23-22-08060.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbagallo B, Prescott HA, Boyle P, Climer J, Francis MM. A dominant mutation in a neuronal acetylcholine receptor subunit leads to motor neuron degeneration in Caenorhabditis elegans. J Neurosci. 2010;30:13932–13942. doi: 10.1523/JNEUROSCI.1515-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulin T, Gielen M, Richmond JE, Williams DC, Paoletti P, Bessereau JL. Eight genes are required for functional reconstitution of the Caenorhabditis elegans levamisole-sensitive acetylcholine receptor. Proc Natl Acad Sci U S A. 2008;105:18590–18595. doi: 10.1073/pnas.0806933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinar H, Keles S, Jin Y. Expression profiling of GABAergic motor neurons in Caenorhabditis elegans. Curr Biol. 2005;15:340–346. doi: 10.1016/j.cub.2005.02.025. [DOI] [PubMed] [Google Scholar]
- Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- Fowler CD, Lu Q, Johnson PM, Marks MJ, Kenny PJ. Habenular alpha5 nicotinic receptor subunit signalling controls nicotine intake. Nature. 2011;471:597–601. doi: 10.1038/nature09797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox RM, Von Stetina SE, Barlow SJ, Shaffer C, Olszewski KL, Moore JH, Dupuy D, Vidal M, Miller DM., 3rd A gene expression fingerprint of C. elegans embryonic motor neurons. BMC Genomics. 2005;6:42. doi: 10.1186/1471-2164-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis MM, Maricq AV. Electrophysiological analysis of neuronal and muscle function in C. elegans. Methods Mol Biol. 2006;351:175–192. doi: 10.1385/1-59745-151-7:175. [DOI] [PubMed] [Google Scholar]
- Francis MM, Evans SP, Jensen M, Madsen DM, Mancuso J, Norman KR, Maricq AV. The Ror receptor tyrosine kinase CAM-1 is required for ACR-16-mediated synaptic transmission at the C. elegans neuromuscular junction. Neuron. 2005;46:581–594. doi: 10.1016/j.neuron.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Gottschalk A, Almedom RB, Schedletzky T, Anderson SD, Yates JR, 3rd, Schafer WR. Identification and characterization of novel nicotinic receptor-associated proteins in Caenorhabditis elegans. EMBO J. 2005;24:2566–2578. doi: 10.1038/sj.emboj.7600741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulding M. Circuits controlling vertebrate locomotion: moving in a new direction. Nat Rev Neurosci. 2009;10:507–518. doi: 10.1038/nrn2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady SR, Moretti M, Zoli M, Marks MJ, Zanardi A, Pucci L, Clementi F, Gotti C. Rodent habenulo-interpeduncular pathway expresses a large variety of uncommon nAChR subtypes, but only the alpha3beta4* and alpha3beta3beta4* subtypes mediate acetylcholine release. J Neurosci. 2009;29:2272–2282. doi: 10.1523/JNEUROSCI.5121-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AK, Davis P, Hodgkin J, Sattelle DB. The nicotinic acetylcholine receptor gene family of the nematode Caenorhabditis elegans: an update on nomenclature. Invert Neurosci. 2007;7:129–131. doi: 10.1007/s10158-007-0049-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jospin M, Qi YB, Stawicki TM, Boulin T, Schuske KR, Horvitz HR, Bessereau JL, Jorgensen EM, Jin Y. A neuronal acetylcholine receptor regulates the balance of muscle excitation and inhibition in Caenorhabditis elegans. PLoS Biol. 2009;7:e1000265. doi: 10.1371/journal.pbio.1000265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano T, Po MD, Gao S, Leung G, Ryu WS, Zhen M. An imbalancing act: gap junctions reduce the backward motor circuit activity to bias C. elegans for forward locomotion. Neuron. 2011;72:572–586. doi: 10.1016/j.neuron.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Klassen MP, Shen K. Wnt signaling positions neuromuscular connectivity by inhibiting synapse formation in C. elegans. Cell. 2007;130:704–716. doi: 10.1016/j.cell.2007.06.046. [DOI] [PubMed] [Google Scholar]
- Leifer AM, Fang-Yen C, Gershow M, Alkema MJ, Samuel AD. Optogenetic manipulation of neural activity in freely moving Caenorhabditis elegans. Nat Methods. 2011;8:147–152. doi: 10.1038/nmeth.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lickteig KM, Duerr JS, Frisby DL, Hall DH, Rand JB, Miller DM., 3rd Regulation of neurotransmitter vesicles by the homeodomain protein UNC-4 and its transcriptional corepressor UNC-37/groucho in Caenorhabditis elegans cholinergic motor neurons. J Neurosci. 2001;21:2001–2014. doi: 10.1523/JNEUROSCI.21-06-02001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria PM, Hodgkin J, Hobert O. A conserved postsynaptic transmembrane protein affecting neuromuscular signaling in Caenorhabditis elegans. J Neurosci. 2004;24:2191–2201. doi: 10.1523/JNEUROSCI.5462-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey ED, Engle SE, Kim MR, O'Neill HC, Wageman CR, Patzlaff NE, Wang Y, Grady SR, McIntosh JM, Marks MJ, Lester HA, Drenan RM. alpha6* nicotinic acetylcholine receptor expression and function in a visual salience circuit. J Neurosci. 2012;32:10226–10237. doi: 10.1523/JNEUROSCI.0007-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney TR, Liu Q, Itoh T, Luo S, Hadwiger G, Vincent R, Wang ZW, Fukuda M, Nonet ML. Regulation of synaptic transmission by RAB-3 and RAB-27 in Caenorhabditis elegans. Mol Biol Cell. 2006;17:2617–2625. doi: 10.1091/mbc.E05-12-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntire SL, Jorgensen E, Horvitz HR. Genes required for GABA function in Caenorhabditis elegans. Nature. 1993;364:334–337. doi: 10.1038/364334a0. [DOI] [PubMed] [Google Scholar]
- Mulle C, Vidal C, Benoit P, Changeux JP. Existence of different subtypes of nicotinic acetylcholine receptors in the rat habenulo-interpeduncular system. J Neurosci. 1991;11:2588–2597. doi: 10.1523/JNEUROSCI.11-08-02588.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashmi R, Dickinson ME, McKinney S, Jareb M, Labarca C, Fraser SE, Lester HA. Assembly of alpha4beta2 nicotinic acetylcholine receptors assessed with functional fluorescently labeled subunits: effects of localization, trafficking, and nicotine-induced upregulation in clonal mammalian cells and in cultured midbrain neurons. J Neurosci. 2003;23:11554–11567. doi: 10.1523/JNEUROSCI.23-37-11554.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M, Alfonso A, Johnson CD, Rand JB. Caenorhabditis elegans mutants resistant to inhibitors of acetylcholinesterase. Genetics. 1995;140:527–535. doi: 10.1093/genetics/140.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce-Shimomura JT, Chen BL, Mun JJ, Ho R, Sarkis R, McIntire SL. Genetic analysis of crawling and swimming locomotory patterns in C. elegans. Proc Natl Acad Sci U S A. 2008;105:20982–20987. doi: 10.1073/pnas.0810359105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand JB. Acetylcholine. WormBook. 2007:1–21. doi: 10.1895/wormbook.1.131.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassoè-Pognetto M. Molecular and functional heterogeneity of neural circuits: an example from the olfactory bulb. Brain Res Rev. 2011;66:35–42. doi: 10.1016/j.brainresrev.2010.06.003. [DOI] [PubMed] [Google Scholar]
- Schuske K, Beg AA, Jorgensen EM. The GABA nervous system in C. elegans. Trends Neurosci. 2004;27:407–414. doi: 10.1016/j.tins.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA. Nicotine activation of alpha4* receptors: sufficient for reward, tolerance, and sensitization. Science. 2004;306:1029–1032. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- Touroutine D, Fox RM, Von Stetina SE, Burdina A, Miller DM, 3rd, Richmond JE. acr-16 encodes an essential subunit of the levamisole-resistant nicotinic receptor at the Caenorhabditis elegans neuromuscular junction. J Biol Chem. 2005;280:27013–27021. doi: 10.1074/jbc.M502818200. [DOI] [PubMed] [Google Scholar]
- Tuesta LM, Fowler CD, Kenny PJ. Recent advances in understanding nicotinic receptor signaling mechanisms that regulate drug self-administration behavior. Biochem Pharmacol. 2011;82:984–995. doi: 10.1016/j.bcp.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashlishan AB, Madison JM, Dybbs M, Bai J, Sieburth D, Ch'ng Q, Tavazoie M, Kaplan JM. An RNAi screen identifies genes that regulate GABA synapses. Neuron. 2008;58:346–361. doi: 10.1016/j.neuron.2008.02.019. [DOI] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]