

Abstract
Bacterial chemotaxis receptors are posttranslationally modified by carboxyl methylation of specific glutamate residues within their cytoplasmic domains. This highly regulated, reversible modification counterbalances the signaling effects of ligand binding and contributes to adaptation. Based on the crystal structure of the γ-glutamyl-methyltransferase CheR, we have postulated that positively charged residues in helix α2 in the N-terminal domain of the enzyme may be complementary to the negatively charged methylation region of the methyltransferase substrates, the bacterial chemotaxis receptors. Several altered CheR proteins, in which positively charged arginine or lysine residues were substituted with alanines, were constructed and assayed for their methylation activities toward wild-type receptor and a series of receptor variants containing different glutamates available for methylation. One of the CheR mutant proteins (Arg53Ala) showed significantly lower activity toward all receptor constructs, suggesting that Arg53 may play a general role in catalysis of methyl transfer. The rest of the mutant proteins exhibited different patterns of relative methylation rates toward different receptor substrates, indicating specificity, probably through interaction of CheR with the receptor at sites distal to the specific site of methylation. The findings imply complementarity between positively charged residues of the α2 helix of CheR and the negatively charged glutamates of the receptor. It is likely that this complementarity is involved in discriminating different methylation states of the receptors.
In enteric bacteria, chemotaxis is mediated by several homologous transmembrane receptors that sense changes in concentrations of attractant and repellent molecules (reviewed in refs. (1–4)). Ligand binding to the periplasmic domains of the receptors generates conformational signals that are transmitted to the cytoplasmic domains that regulate an associated two-component phosphotransfer signal transduction system. Alterations in ligand concentration evoke a cellular response involving a change in direction of flagellar rotation. Following a transient response, cells return to pre-stimulus behavior, a process known as adaptation. Adaptation is in part mediated by reversible covalent modification of the chemoreceptors. Changes in receptor ligand concentration are accompanied by changes in the level of methylation at several specific glutamate residues in the cytoplasmic domains of the chemoreceptors. The signaling states of the chemotaxis receptors are influenced by both ligand occupancy of the periplasmic domains of the receptors and by the levels of methylation of the receptor cytoplasmic domains.
The specific glutamate residues that are methylated are spaced seven residues apart on two helical segments of the coiled-coil structure of the receptor methylation region (5–7). Each of the methylatable glutamate residues is located within a consensus sequence Glu-Glu-X-X-Ala-Ser/Thr, defined by the two most highly methylated sites (8, 9), where X represents any amino acid and the second conserved glutamate (shown in bold) is the residue that is methylated. Throughout the manuscript, the consensus sequence will be referred to as a “methylation site”. In the aspartate receptor Tar, there are four methylation sites, two of which contain glutamine residues that are posttranslationally converted to glutamates by methylesterase CheB-catalyzed deamidation (10). The methylatable residues Gln295, Glu302, Gln309 and Glu491, referred to as sites one through four respectively, exhibit different rates of methylation (8). Furthermore, the rates of methylation at one glutamate are affected by the type of residue two helical turns away in the C-terminal direction (11, 12).
Reversible methylation of chemotaxis receptors involves two enzymes: a methylesterase/deamidase CheB and an AdoMet-dependent methyltransferase CheR. Methylesterase CheB, a response regulator protein, is activated by phosphorylation via a two-component phosphotransfer pathway (13). Unlike CheB, methyltransferase CheR is constitutively active. CheR binds with micromolar affinity to a C-terminal 5-residue binding site in specific receptors (14). Tethering of methyltransferase CheR to the receptors is required for efficient methylation of glutamate residues located within the methylation regions, and methylation of receptors lacking the CheR binding site occurs through an interdimer mechanism (15–17).
Crystal structures of CheR have provided a structural basis for the activities of this receptor modification enzyme (Figure 1A). CheR is composed of a small N-terminal domain and a larger C-terminal domain with a seven-stranded modified Rossmann nucleotide-binding fold characteristic of class I methyltransferases (18, 19). A small β-subdomain is inserted into the C-terminal domain of CheR. This subdomain is a unique feature of chemotaxis protein methyltransferases and mediates interaction with the pentapeptide binding sequence at the C termini of specific receptors (18, 20). The binding site for AdoMet is located at one edge of the nucleotide-binding domain, formed by residues from both the N- and C-terminal domains as well as from the linker region. Adjacent to the co-factor binding site, a large opening formed by surfaces of the N- and C-terminal domains, and most notably a basic region contributed by the exposed face of helix α2, was postulated to be the recognition cleft for interaction with the methylation region of the receptor. Experimental evidence supporting the role of this region in receptor binding has been provided by recently reported mutagenesis and cross-linking studies (21).
Figure 1.

Methyltransferase CheR and its substrate chemoreceptor. (A) Structure of CheR [(18), pdb code 1AF7]. The N-terminal tail and helix α2, that is postulated to interact with the receptor methylation region, are highlighted in black. S-adenosylhomocysteine (AdoHcy), a product and inhibitor of the methylation reaction, is shown in stick representation. (B) Receptor methylation region [generated from the crystal structure of Tsr (5–7), pdb code 1QU7]. Methylation occurs at four glutamate residues (*1–*4) within the coiled-coil of the cytoplasmic domains of chemoreceptors. The methylatable glutamates/glutamines, as well as the conserved residues that precede the methylation sites, are shown in ball-and-stick representation. A consensus sequence derived from alignment of six E. coli and Salmonella chemotaxis receptors is shown with completely conserved residues underlined, residues conserved in four or five of the six receptors not underlined, and less conserved residues indicated by X. (C) Helix α2 of CheR. The side chains of the six positively charged residues in α2 are shown in ball-and-stick representation. The conservation of these residues is indicated as the % identity within an alignment of fifty bacterial CheR proteins. CheR proteins identified by Shiomi et al. (20) and additional CheR proteins from more recently sequenced genomes were used in this analysis.
We have postulated that several positively charged residues in helix α2 that forms part of the receptor interaction surface of methyltransferase CheR may pair specifically with the negatively charged glutamates in the methylation region of the receptor. The positively charged residues of CheR that could form salt bridges with glutamates, but not with methylglutamates, may be involved in distinguishing different methylation states of the receptors. To investigate this hypothesis, we have produced several single-site Arg or Lys to Ala substitutions within Salmonella enterica serovar Typhimurium methyltransferase CheR and have characterized the methylation activities of these CheR proteins toward several receptor substrates containing different glutamates available for methylation. The different receptor substrates exhibited different patterns of relative methylation rates when assayed with the set of CheR mutant proteins. These findings imply specific complementarity between the positively charged residues of CheR and the negatively charged methylatable glutamates of the receptor, and support the hypothesis that these residues are involved in the specific recognition of receptor methylation regions.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Plasmids
Escherichia coli DH5α (22) was used as the host strain for all molecular biological manipulations. E. coli E507 (23) containing pJES307-derived plasmids (24), as well as salt inducible strain GJ1158 (25) was used for expression of all S. enterica CheR proteins. Plasmid pME43 (26) was used as template for PCR mutagenesis of cheR. Plasmid pME98 (27), containing the coding sequence and regulatory region of the S. enterica tar gene, was used as template for PCR mutagenesis and as a vector for expression of all tar genes in E. coli HCB437 (Δ(tsr)7021 Δ(trg)100 zbd::Tn5 Δ(cheA-cheZ)2209 metF159(Am)) (28).
Mutagenesis of cheR and tar
All DNA manipulations were performed by standard techniques. Oligonucleotide-directed mutagenesis of cheR was performed using a two-step PCR strategy (29). Alanine substitutions were introduced into CheR by changing the codons for R46, R47, and R57 to GCG and the codons for R53, R56, and R59 to GCA. The CheR N-terminal deletion (Δ1-19) was constructed using a 5′ primer designed to introduce an initiator methionine codon directly preceding nucleotides encoding amino acid residue 20. Following amplification of each full-length or truncated gene flanked by restriction sites for NdeI and HindIII, the NdeI-HindIII cheR fragment was inserted directly downstream of the T7 promoter in vector pJES307, resulting in cheR expression plasmids pND11 (K46A), pND21 (R47A), pND31 (R53A), pND41 (R56A), pND52 (R57A), pND61 (R59A), and pEP01 (Δ1-19). The mutations were confirmed by dideoxynucleotide sequencing using Sequenase polymerase (United States Biochemical).
Mutations were introduced into S. enterica tar using GAG and CAG codons to specify glutamine and glutamate, respectively. Three Tar variants, Tar QEQQ (E2), QQQE (E4), and QEEE (see Results for description of nomenclature), containing the single substitutions E495Q, E302Q, and Q309E, respectively, were generated by a two-step PCR strategy using plasmid pME98, encoding WT Tar (QEQE), as template. The Tar variant EEEE (4E) containing mutations Q295E/Q309E was generated using the previously constructed plasmid encoding QEEE as template. Two Tar variants, Tar EQQQ (E1) and QQEQ (E3), containing mutations Q295E/E302Q/E491Q and E302Q/Q309E/E491Q, were generated using the plasmid encoding QQQE as template. In each of these cases, two mutations were introduced by a modification of a basic two-step PCR protocol. Briefly, three PCR fragments were generated for each gene: an upstream fragment containing the 5′ region of tar including the upstream mutation, a downstream fragment containing the 3′ region of tar including the downstream mutation, and a middle fragment containing both mutations. Full-length tar genes were generated by PCR using a combination of all three fragments as template. Following amplification, fragments were cleaved at BspE1 and HindIII sites encoded in the 5′ and 3′ termini and the inserts were ligated into BspE1/HindIII-digested pME98 resulting in tar expression plasmids pNGD-8-1 (4E), pNGD-8-2 (E1), pNGD-8-3 (E2), pNGD-8-4 (E3), and pNGD-8-5 (E4). The mutations were confirmed by automated sequencing (UMDNJ Biochemistry Department Facility).
Expression and Purification of CheR Proteins
Cells harboring plasmids with inserts encoding CheR or mutant CheR proteins were used for preparation of partially purified cell-free extracts enriched in CheR. Cells harboring a plasmid lacking an insert were used for preparation of a CheR-free control extract. Yields of soluble CheR protein were variable due to inclusion body formation. Use of the salt inducible T7 system in GJ1158 cells generally provided higher yields of soluble CheR. Cells were grown to mid-logarithimic phase at 30 °C in 1.5 L Luria broth containing 50 μg/mL ampicillin. Expression of T7 polymerase was induced with 0.5 mM IPTG (or 0.3 M NaCl) 3 h prior to harvesting cells. Purification was performed as previously described (26) except that purification was stopped after the DE52 anion exchange chromatography step. Partially purified CheR proteins were stored at −20 °C in 50 mM potassium phosphate, 1 mM EDTA, 1 mM βME, 20% (vol/vol) glycerol, pH 7.0. Two different methods were used to determine CheR concentrations; both yielded similar results. In the first method, aliquots of CheR preparations were separated by electrophoresis on SDS 15% polyacrylamide gels and the gels were stained with Coomassie Blue. CheR was quantitated using a laser densitometer (Bio-Rad Model GS-670) by comparison to a standard curve generated with known amounts of highly purified (>98% purity) CheR protein. In the second method, HPLC was performed on a 6000 series system from Hitachi (L6200A Intelligent Pump, L3000 Photodiode Array detector, D6000 Interface, and D7000HPLC system manager). Samples were injected onto a C18 reverse-phase column (Varian #R0083203F5) equilibrated in 0.001% TFA in water (solvent A) and eluted with a linear gradient of 0 to 98% solvent B (0.001% TFA in acetonitrile) at a flow rate of 1 mL/min over 30 min. WT and mutant CheR proteins eluted at 21 min. The area of this peak was integrated (A220 sec) and CheR concentrations were calculated using a standard curve generated with highly purified CheR protein.
Preparation of Membranes Containing Tar
Cells were grown to stationary phase at 30 °C in 1.5 L Luria broth containing antibiotics. Salt-washed cell membranes containing receptors were prepared using minor modifications of previously described procedures (30). Cells were harvested, resuspended in Buffer A (0.1 M potassium phosphate, 1 mM EDTA, 1.4 mM βME, pH 7.0), collected by centrifugation, resuspended at 3 mL/g wet weight cells in Buffer B (50 mM potassium phosphate, 2 M KCl, 5 mM EDTA, 10% glycerol, 1 mM 1,10-phenanthroline, 0.5 mM PMSF, 0.1 mM p-hydroxymercuribenzoate, pH 7.0), and lysed by sonication. Unbroken cells and debris were removed by centrifugation for 8 min at 10,000 × g and membranes were sedimented by centrifugation for 1 h at 100,000 × g. Membranes were resuspended in Buffer B, sedimented by centrifugation, resuspended in Buffer C (50 mM potassium phosphate, 1 mM EDTA, 10% glycerol, 5 mM 1,10-phenanthroline, 1 mM PMSF, pH 7.0), sedimented by centrifugation, and resuspended at ~7 mg total protein/mL in Buffer C. Membranes were stored in single-use aliquots at −80 °C. The concentration of total protein in the membranes was determined using the Bradford dye-binding method (Bio-Rad Laboratories). Expression levels of Tar proteins in membrane aliquots were examined using Coomassie Blue-stained SDS polyacrylamide gels and were found to be similar for all Tar constructs.
Receptor Methylation Assays
Receptor methylation assays were performed similarly to previously described procedures (26). Each 100-μL reaction mixture contained 40–60 μL of receptor-enriched membranes (membrane preparations contained ~6 μM Tar, estimated by SDS-PAGE, and 6–8 mg total protein/mL, determined by Bradford dye-binding assays) and CheR protein preparation (as specified in the figure legends) in 100 mM potassium phosphate, 100 μM [3H]-S-adenosylmethionine at 162 Ci/mol (specific activity 15 Ci/mmol, NEN Life Science Products, Inc.), 0.2 mM L-aspartate, pH 7.0, unless otherwise indicated. Reactions were initiated by addition of 10–20 μL of CheR to reaction mixtures that had been preequilibrated for 10 min at 30 °C. At appropriate intervals, 10-μL aliquots were removed and applied to 1-cm squares of Whatman 3MM paper and immediately immersed in 10% (w/vol) TCA. The papers were washed twice with 10% TCA and once with ethanol for 15 min each, then air-dried, immersed in 2 mL Ecoscint A (National Diagnostics), and radioactivity was determined by liquid scintillation spectrometry using a Beckman Model LS6500 instrument. For typical assays, 5–8 data points were taken at 4–10 min intervals and initial rates were estimated using linear regression analysis. The cpm measured for the CheR-free control, which remained constant over the time course of the assay, were considered background and were subtracted from all data points. CheR proteins that generated cpm less than three times background level at the end of the time course were considered to have undetectable methyltransferase activity. Concentrations of CheR proteins were adjusted to ensure quantifiable rates that were linear with respect to time. All assays included in the final data had between 15 and 60 mol [3H]methyl incorporated at the last point of the time course. Concentrations of CheR proteins used in assays for which methylation rates were detectable ranged from 8.0 nM to 1.15 μM. For assays that yielded results below detection levels, CheR concentrations up to 3.50 μM were used. Assays with QEQE Tar and WT CheR ranging from 8.4 nM to 830 nM exhibited methyltransferase rates that were linear with respect to enzyme concentration throughout the entire range (data not shown). Additionally, each CheR-receptor pair was assayed at a minimum of two different CheR protein concentrations to confirm linearity with respect to CheR. All methylation rates reported in Figures 2–4 were derived from assays repeated 3–5 times using two independent preparations of both Tar-enriched membranes and CheR proteins.
Figure 2.

Methyltransferase activity of WT and mutant CheR proteins with WT Tar receptor (QEQE) as substrate. Methyltransferase activity was determined as described in Experimental Procedures. (A) CheR proteins containing single-site substitutions in helix α2. Assays were performed using 0.94 pmol WT CheR (○), 2.92 pmol K46A (●), 11.5 pmol R53A (■), 4.86 pmol R56A (□), and 3.34 pmol R59A (×). Representative data from a single set of assays are shown. Relative methyltransferase activities obtained from multiple assays, expressed as a percentage of the activity of WT CheR (1.12 mol [3H]methyl incorporated min−1 mol CheR−1), are: WT CheR (100 ± 8.0), K46A (55.6 ± 9.7), R53A (4.3 ± 0.26), R56A (38.5 ± 2.4), and R59A (41 ± 2.7). (B) CheR lacking N-terminal residues 1-19 (CheRΔ1-19). Assays were performed using 1.09 pmol WT CheR with (○) or without (●) 0.2 mM aspartate, and 1.04 pmol of CheRΔ1-19 with (△) or without (▲) aspartate. Representative data from a single set of assays are shown. Relative methyltransferase activities derived from multiple assays are: WT CheR (100 ± 7.7) and CheRΔ1-19 (84 ± 3.7).
Figure 4.

Relative methyltransferase activities of mutant CheR proteins at different receptor methylation sites. Methyltransferase activities were determined as described in Experimental Procedures. The data shown are average methylation rates and standard errors obtained from multiple assays. For each Tar variant, methylation rates are normalized to the rate obtained with WT CheR (shown in Figure 3).
Computational Modeling of CheR and Chemoreceptor Interaction
The pentapeptide-bound structure of CheR (PDB code: 1BC5) and the dimeric chemoreceptor Tsr (PDB code: 1QU7; edited to remove the kinase docking region, residues 348–427 of each monomer, and to convert the 4 methylation sites to the WT QEQE sequence), both lacking all heteroatoms, were docked together using the program FTDOCK (31). This program generates all possible rotational orientations (12° intervals) of CheR or Tsr and then docks the two proteins together as rigid bodies. The docked complexes are scored on the basis of shape complementarity and electrostatic considerations. Distance constraints were then applied in order to screen the ~10,000 predicted complexes for those that had at least one atom each from Arg53, Asn92, Leu93, and Arg98 (residues in close proximity to the predicted position of the methyl group of AdoMet) all within a specified distance of each methylatable Glu/Gln residue (Gln297, Glu304, Gln311). Constraints of 6, 7, 8, and 10 Å were applied. This filtering step yielded six complexes for methylation site 1 with constraints ≤7 Å, three complexes for site 2 with constraints ≤10 Å, and seven complexes for site 3 with constraints ≤10 Å. The program MULTIDOCK (32) was used for refinement and rigid-body energy minimization of side chain conformations at the protein-protein interface. The fifteen complexes cited above (one of which was common to both sites 2 and 3) were examined using the molecular graphics display program O (33). Complexes for which the methylatable residues were facing the opposite direction from the proposed catalytic site of CheR (AdoMet binding pocket) and those in which the α2 helix of CheR was positioned away from the methylation sites were eliminated. This left one model (no. 8152) that serves as a basis for discussion in this study.
RESULTS
Interactions Between the Chemotaxis Receptors and the Receptor Modifying Enzyme CheR
Biochemical and crystallographic studies have established that recruitment of the methyltransferase to the membrane-associated receptors occurs through binding of the C-terminal pentapeptide of the receptor to the β-subdomain, a specific structural unit of CheR (14, 20). From this tethering point, CheR can interact with methylation regions of surrounding receptors through an inter-dimer mechanism (15, 16). The methylation regions of the cytoplasmic domains of the chemotaxis receptors that must come into close contact with the active site of CheR during methyl transfer are negatively charged due to the presence of carboxylate side chains within, and adjacent to, the methylatable residues (Figure 1B). Based on the crystal structure of CheR and molecular surface electrostatic potential calculations, we identified a highly positively charged region, helix α2 in the N-terminal domain of CheR, that we proposed to be involved in the recognition and positioning of the receptor methylation domain (18). This helix, that forms one face of the putative receptor-binding opening, contains one lysine and five arginine residues, with the majority of them being solvent exposed (Figure 1C).
Effects of Substitution of Positively Charged Residues in Methyltransferase CheR on Methylation of Wild-Type Receptor Tar
To assess the relevance and the role of the positively charged residues in CheR, we generated six single-site substituted CheR proteins and purified and assayed four in which conserved lysine or arginine residues in helix α2 were replaced by alanine. All cheR genes were expressed in E. coli at 30 °C under the control of the T7 polymerase promoter, ensuring high levels of expression of the proteins. Protein expression and purification were carried out in parallel for all WT and mutant CheR proteins and as a control with cells that contained an expression vector without the cheR insert. In contrast to WT CheR and the majority of the mutant proteins, two of the mutant proteins, K47A and R57A, did not express well and all of the CheR protein was found in inclusion bodies; further characterization of these proteins was not pursued. Both of these residues are oriented toward the interior of the protein and are involved in salt bridge interactions with glutamates from other parts of the N-terminal domain of CheR, hence mutations at these sites probably result in folding defects. During expression and purification, the other four mutant proteins were indistinguishable from the WT enzyme. A single anion chromatography step was sufficient to produce CheR preparations with greater than 80% homogeneity. SDS-PAGE analysis of each of the purified preparations showed identical patterns and proportional amounts of minor contaminants that correlated well with the preparation that was obtained from the control cells that did not contain CheR.
Methyltransferase activity of all CheR proteins was determined using as substrate WT Tar-enriched membranes produced in an esterase/amidase-deficient cheB strain (Figure 2A). Initial rates of methylation were determined with saturating concentrations of Tar-containing membranes, and at CheR concentrations for which rates were linearly proportional to enzyme concentrations. The CheR-free control preparation showed no additional incorporation of methyl groups relative to that observed with buffer, indicating that the CheR-enriched preparations were sufficiently pure to specifically assess methyltransferase CheR activity. Relative to WT CheR, all mutant proteins showed reduced methyltransferase activity. Three of the mutant proteins had similar methyltransferase activity with only moderately reduced activity ranging from ~40 to 60% that of WT CheR, while one of the mutant proteins, R53A, exhibited significantly lower methyltransferase activity, only 4% that of WT CheR.
Effects of N-terminal Deletion of Methyltransferase CheR on Methylation of Wild-Type Receptor Tar
Analysis of different crystal structures of CheR have shown different conformations of the N-terminal tail (residues 1-19), a region suspected to be disordered in solution (18, 20). To explore the possible role of the N terminus in methylation, a mutant protein lacking nineteen N-terminal residues was generated and the methyltransferase activity of this truncated CheR, CheRΔ1-19, was assessed using WT Tar as substrate (Figure 2B). The activity of CheRΔ1-19 was only slightly reduced (85%) relative to WT CheR. Also, as expected, and similar to WT CheR, the methylation rate of CheRΔ1-19 was approximately doubled in the presence of 0.2 mM aspartate.
Methylation Rates of Wild-Type CheR with Variant Tar Substrates
The four glutamate residues in WT Tar are methylated at significantly different rates, with site 3 methylated most rapidly, and sites 2, 1, and 4 methylated at about one-half, one-tenth, and one-fiftieth the rate of site 3, respectively (8). Additionally, rates of methylation at one site in Tar have been shown to be significantly altered by substitution of residues at a position seven residues in the C-terminal direction, two helical turns away, from the site of methylation (11, 12). To begin to address the specificity of interaction of CheR with different methylation sites, a set of variant Tar proteins that contained different glutamate residues available for methylation were constructed and used as substrates in methylation assays with WT CheR.
Within Tar, two of the four methylatable residues are encoded as glutamines. The four methylation sites, represented by a one-letter code designating the methylatable residue, can be summarized as QEQE. In a WT cell, methylesterase/amidase CheB can deamidate the glutamine side chains, converting the residues to glutamates that can subsequently function within the reversible methylation cycle. However, to ensure homogeniety at each methylation site, the Tar substrates used for in vitro assays were expressed in a methyltransferase/methylesterase-deficient strain so that residues remained in their encoded states. Four Tar variants, each containing a single glutamate available for methylation, EQQQ (E1), QEQQ (E2), QQEQ (E3), and QQQE (E4), and one Tar with four glutamates, EEEE (4E), were constructed as described in Experimental Procedures. Substitutions within the methylation sites had no apparent effect on expression of the proteins, and for each of the Tar variants, membranes were prepared in which Tar constituted approximately 20% of the total membrane protein, a level similar to that observed for WT Tar (QEQE).
Initial rates of methylation of these Tar variants catalyzed by WT CheR were determined (Figure 3). WT Tar is methylated at a much greater rate than the other Tar constructs. Tar 4E, with all four glutamates available for methylation, is methylated at a rate only 15% that of WT Tar. These results were found to be dissimilar to a previous report which showed Tar 4E to be methylated at a higher rate relative to WT Tar (34). However, our results are consistent with the previous observation that a negatively charged residue, two helical turns away from the site of methylation lowers methylation rates (12). In all cases, methylation rates were higher in the presence of aspartate (data not shown), confirming that these receptor variants were competent in transmembrane signaling.
Figure 3.

Methyltransferase activities at different receptor methylation sites. Initial rates of methylation were determined as described in Experimental Procedures using receptor variants described in the text as substrates. Assays were performed using 0.81 pmol WT CheR with WT Tar (QEQE). Greater quantities of WT CheR were used with the Tar variants to achieve quantifiable rates, with values ranging from 3.8 pmol WT CheR with EEEE Tar to 15.1 pmol WT CheR with QQQE Tar. The data shown represent the average methylation rates and standard errors obtained from multiple experiments. Methylation rates are normalized to the rate of WT Tar (QEQE) (1.12 mol [3H]methyl incorporated min−1 mol CheR−1).
Methylation Rates of the Individual Tar Methylation Sites Catalyzed by Different Variants of CheR
To investigate which of the residues in CheR contribute to formation of the methylation catalytic site, and the extent to which each of these residues participates in differentiation of the methylation sites, the mutant CheR proteins in which positively charged residues were replaced by alanines were examined for methyltransferase activity toward the set of different Tar receptors. For each of the Tar variants, assays were performed at the same time with all CheR proteins and methylation rates were normalized to the rate obtained with WT CheR. The results from these experiments, grouped according to the Tar receptor used as a substrate, are shown in Figure 4. For comparative purposes, for each Tar substrate, the rate of methylation with WT CheR is set to unity. Rates of methylation with Tar variants as substrates were significantly lower than with WT Tar as shown in Figure 3.
With all Tar variants, CheR mutant Arg53Ala showed extremely low activity ranging from 8% for the WT Tar to undetectable for Tar E2 and E3. The other three CheR mutant proteins showed much greater variability in their methyltransferase activities depending on the specific substrate. The most dramatic specificity was observed for the methylation of Tar E1 and E4 for which different CheR residues had very different effects on the rates of methylation. Specifically, CheR mutant Lys46Ala had very low activity toward Tar E1 whereas its activity toward Tar E4 was nearly identical to that of WT CheR. Interestingly, patterns of methylation rates exhibited by mutant CheR enzymes with Tar E2 and E3 were very similar to the pattern of relative rates obtained with the all-glutamates Tar 4E receptor. This correlates with previous reports that the main contributors to the methylation of Tar are methylation sites 2 and 3 (8, 11), hence the mutant CheR enzymes act on Tar 4E similarly as on isolated methylation sites 2 or 3 alone.
DISCUSSION
The active site for methylation of chemotaxis receptors is formed and influenced by both the enzyme (AdoMet-dependent methyltransferase CheR) and the substrate (receptor). Since methylation occurs through direct transfer of the methyl group from the CheR-bound AdoMet to the specific nucleophilic methyl-accepting glutamate residue on the receptor (35, 36), the rate of transfer is strongly dependent on the distance and orientation of these reactive groups. This study addresses the role and involvement of specific positively charged residues in the N-terminal domain of CheR in the processes of formation of the methylation active site and discrimination between various methylation sites of the chemotaxis receptor Tar.
The methylation rates at specific methylation sites have been shown to be influenced by the nature of the residue at adjacent methylation sites (11, 37). Based on these data and the helical structure of the receptor methylation region, Shapiro et al. proposed that methyltransferase CheR simultaneously contacts the receptor at two sites: the site of methylation and a residue seven amino acids to the C-terminal side of the site of methylation (11, 37). Subsequently, the crystal structure of methyltransferase CheR provided insight to the surfaces of CheR involved in these interactions with the receptor (18). The calculated electrostatic potential revealed a highly positively charged surface corresponding to helix α2 in the N-terminal domain and the high conservation of basic residues Lys46 and Arg53 among CheR orthologs provided further support for the functional significance of this region. Recently, evidence of interaction of this region of methyltransferase CheR with the receptor methylation region has been obtained from cross-linking studies that demonstrated disulfide bond formation between cysteine-substituted residues 46, 56, 59, and 60 of methyltransferase CheR and cysteine-substituted Glu308 within the methylation region of Tar (21).
Our data show the rate of methylation to be highly sensitive to the nature of the substrate. The differences in methylation rates at the individual methylation sites of Tar may imply that they are recognized differently by the methyltransferase, however the contribution of CheR to the catalytic site presumably remains the same in all reactions. Alternatively or additionally, the observed rates may be the consequence of differences in accessibility of individual glutamate residues to the methyltransferase. The latter consideration may be relevant to the methylation rate for the E2 Tar variant, which contains a single substitution relative to WT Tar (QEQE), specifically, the substitution of Glu491 (methylation site 4) with glutamine (QEQQ). This receptor is methylated by WT CheR at only 6% the rate of WT Tar even though the substitution at site 4 occurs on the α helix that runs antiparallel to the site of methylation, and the local sequence of the α helix that contains the methylatable glutamate is unchanged between the two receptor variants.
The decreased methylation rates and distinct patterns of methyltransferase activities exhibited by different mutant CheR proteins toward different Tar variants support the hypothesis that positively charged residues in the α2 helix of methyltransferase CheR interact with the receptor methylation region and play a role in differentiation of individual methylation sites. Substitutions of basic residues in helix α2 influenced methyltransferase activity in two ways: three out of four substitutions affected the methylation rates in a Tar variant-dependent manner, whereas one of the mutations (Arg53Ala) resulted in a general and significant decrease in the enzyme activity toward all of the different receptors. It has been reported previously that substitution of Arg53 with Ala resulted in a CheR protein that lacked methyltransferase activity and was unable to support chemotaxis (21). The generally reduced activity of Arg53Ala CheR suggests involvement of Arg53 in the mechanism of methyl-transfer that is common to all methylation sites. The positively charged Arg53 side chain may act by stabilizing the γ-carboxylate ion of the glutamate that is to be methylated, similarly to the proposed role of the lysine residue in the active site of O-catechol methyltransferase (38). Alternatively, Arg53 may function in properly positioning and orienting the methylatable glutamate through an ion-pair interaction with the negatively charged methylation site, or through such a salt-bridge or other electrostatic interactions, Arg53 may enhance binding affinity between the enzyme and substrate. In any of these roles, the contribution of Arg53 would be expected to be similar at each methylation site. The role of Arg56 appears to be somewhat similar, but less crucial than Arg53. In the crystal structures, the guanidinium of Arg56 interacts with the backbone carbonyl oxygen of Arg53 and thereby potentially affects the position of Arg53 and the overall conformation of helix α2.
The other two substitutions of basic residues within helix α2 impart more specific effects, with rates dependent on the specific substrate Tar receptor. This is particularly evident with the E1 and E4 Tar constructs. Substitution of Lys46 with Ala almost completely abolishes methyltransferase activity toward E1 while it has a minimal effect on the methylation rate of E4. Opposite to that is the specificity of the Arg59Ala substitution that only moderately decreases the methylation rate at E1 while causing a complete loss of activity toward E4. These observations are consistent with the hypothesis that the two residues, Lys46 and Arg59, interact with Tar at sites distant from the site of methylation. The contribution of these residues to the reaction rate at any specific site will depend on the nature of the different flanking regions they probe.
Specificity between positively charged residues on one face of helix α2 of CheR with negatively charged glutamate residues on one face of the helical methylation region of Tar might be envisioned most simply as a set of ion pairs between the two helices within the heteromeric complex. Experimental data suggest a methylation competent complex in which Arg53 of methyltransferase CheR is positioned at the active site with the helices of α2 of CheR and the methylation region of the receptor oriented roughly antiparallel to one another. In such an orientation, Lys46 and Arg59 are positioned to the C- and N-terminal sides of the receptor methylation site, respectively. Specifically, during methylation of sites 1, 2, or 3, Lys46 would be near Glu or Gln residues in methylation sites 2 or 3, or the conserved Gln/Asn pair two turns of the helix to the C-terminal side of site 3 (Figure 5). The proposed significance of these interactions correlates well with the relatively large decrease in methylation rate seen at sites 1-3 (receptors EQQQ, QEQQ, and QQEQ) with the Lys46Ala CheR protein relative to WT CheR. This model is also consistent with Shapiro’s observations that the identity of residues two helical turns to the C-terminal side of the site of methylation influences the rate of methylation at that site.
Figure 5.
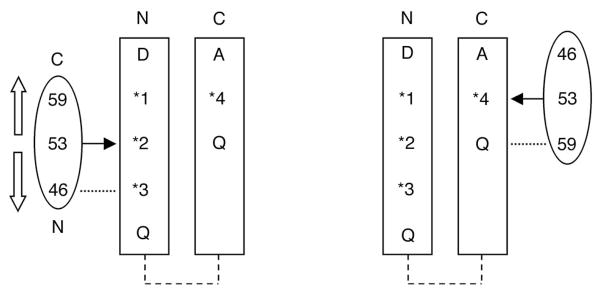
Schematic representations of the proposed interaction modes of helix α2 of CheR with the receptor methylation region. Biochemical data support a complex in which the helices of methyltransferase CheR and the receptor are oriented antiparallel to each other. Helix α2 of CheR is represented as an oval with numbers corresponding to the positively charged residues. The two helices of the receptor are shown as rectangles with methylation sites numbered *1–*4 and residues that lie 7 residues upstream or downstream indicated by one-letter code. The arrow located at residue 53 represents the vicinity of the active site. (A) When CheR is positioned at site 2, residue 46 is proposed to interact (dotted line) with the conserved glutamine located 7 amino acids in the C-terminal direction. Open arrows indicate the movements of this helix that would allow methylation at sites 1 and 3. In all positions, residue 46 would contact a glutamine or glutamate. (B) At site 4, if the same antiparallel orientation is maintained, residue 59, rather than residue 46, is in close proximity (dotted line) to the conserved glutamine located to the N-terminal side of the methylation site. These models account for the biochemical data that suggest an important role for residue 46 in methylation of sites 1–3 and for residue 59 in methylation of site 4.
A different scenario exists for methylation at site 4 that lies on the opposite strand of the coiled-coil from the strand containing methylation sites 1-3. There are two potential modes of interaction of methyltransferase CheR with site 4, with either a parallel or antiparallel arrangement of helix α2 of CheR and the receptor helix containing the methylation site. The latter arrangement retains the same local helix polarity as postulated for interaction of methyltransferase CheR at methylation sites 1-3, but requires that CheR bind to the receptor in an opposite orientation relative to the plane of the membrane. Interestingly, in this orientation, Arg59 of methyltransferase CheR would be positioned near a pair of conserved receptor Gln residues located 7 residues to the N-terminal side of methylation site 4, while Lys46 would be oriented to the C-terminal side of the methylation site in a region that contains no recognizable consensus residues (Figure 5). Interaction of Arg59 with residues to the N-terminal side of methylation site 4 is consistent with the observation that substitution of Arg59 causes a significant decrease in the methylation rate at site 4 (receptor QQQE) while substitution of Lys46 has only a minimal effect. Thus it appears that in contrast to the situation for methylation sites 1-3, it is the region N-terminal to site 4 that may dictate interactions with methyltransferase CheR.
A more detailed view of the interaction of methyltransferase CheR with receptor methylation sites 1-3 was pursued by computational docking experiments. Crystal structures of methyltransferase CheR (20) and the methylation regions of Tsr (5–7), modified to include the WT methylation site configuration (QEQE), were used to generate a set of models of methylation-competent complexes as described in Experimental Procedures. The modeling confirmed that the receptor substrate could be docked within the active site cleft of methyltransferase CheR in an orientation that aligned the α2 helix of CheR roughly antiparallel to the methylation helix of the receptor. The complex exhibits a remarkable shape complementarity between enzyme and substrate, with the two domains of methyltransferase CheR clamped around the Tsr coiled-coil (Figure 6). In this model, the methyl group of AdoMet is positioned closest (8 Å) to the carboxylate oxygen of Glu311 of methylation site 3. Similar orientations toward other methylation sites can be envisioned by sliding CheR along the receptor parallel to the axis of the coiled-coil. Interestingly, it is not possible to dock the receptor in a reversed orientation with the α2 helix of CheR parallel, rather than antiparallel, to the methylation helix of the receptor. The curvature of the receptor precludes a close approach of the methylatable glutamate residues to the CheR active site without steric collisions.
Figure 6.

Possible mode of interaction of methyltransferase CheR with the receptor methylation region derived from computational modeling. Modeling and filtering were performed as described in Experimental Procedures, yielding a single model complex with an overall orientation similar to that postulated from biochemical data. (A) Shape complementarity of CheR and the receptor substrate. CheR is shown in green with helix α2 highlighted in gold. A pentapeptide corresponding to the C-terminus of the receptor is shown in blue bound to the β-subdomain of CheR. The cofactor AdoMet, placed by superposition with the AdoHcy molecule present in the crystal structure, is shown in stick representation. The monomers of the receptor dimer region used in modeling are colored dark and light blue, with the methylation sites 1–4 highlighted in red. (B) Interactions of helix α2 of CheR with the receptor methylation region. An expanded view of helix α2 and the methylation regions of a receptor monomer, rotated ~90° from the view in (A) shows side chain interactions between the antiparallel helices of CheR and the receptor.
Despite overall similarity to the complex proposed from biochemical data, specific interactions differ. Notably, Arg53 is too far from the active site methyl group to participate directly in catalysis. Thus, barring a major conformational change in CheR, a role in properly positioning the substrate through contacts with flanking regions of the receptor seems more plausible. The model of the complex presents opportunities for numerous salt bridges and hydrogen bonds between side chains of residues within helix α2 of CheR and the receptor methylation helices. However, spatial arrangement of these interactions differs from those proposed from methylation kinetics (Figure 5).
The inability to generate a model that fully accounts for the biochemical observations may reflect the limitations of modeling protein complexes from rigid-body docking methods. The modeling does not allow for the dynamic nature of either the receptor or CheR. Comparison of CheR structures determined from two different crystal lattices in the presence (20) or absence (18) of receptor pentapeptide reveals small differences (~1–2 Å displacement) in the conformation of helix α2 and the following loop. This change in the position of helix α2 relative to the rest of the domain underscores the dynamic nature of this region, a feature that may be important for its role in interacting with the receptor substrate. Likewise, conformational changes induced by ligand binding and methylation are thought to be central to receptor signaling. It should be noted that the receptor model used in the docking studies corresponds to the crystal structure of a receptor in which all methylatable glutamates were substituted with glutamines and the conformation of this receptor likely differs substantially from a receptor with glutamates available for methylation. Thus the rigid-body docking model generated in this study is an approximation at best. If substantial structural alterations are induced by contact of methyltransferase CheR with its receptor substrate, then an understanding of specific interactions will likely require direct structural analysis of the protein complex.
Acknowledgments
We thank Natalia Denissova for construction of the Tar expression vectors and Brian Benoff for assistance in computer modeling.
Abbreviations
- AdoMet
S-adenosylmethionine
- βME
β-mercaptoethanol
- cpm
counts per minute
- EDTA
ethylenediaminetetraacetic acid
- HPLC
high performance liquid chromatography
- IPTG
isopropyl-1-thio-β-D-galactopyranoside
- SDS
sodium dodecyl sulfate
- PCR
polymerase chain reaction
- PAGE
polyacrylamide gel electrophoresis
- PMSF
phenylmethylsulfonyl fluoride
- TCA
trichloroacetic acid
- TFA
trifluoroacetic acid
- WT
wild-type
Footnotes
A.M.S. is an investigator of the Howard Hughes Medical Institute. E.P. is supported by NIH Training Grant T32 GM08360. A.H.W. is a Cottrell Scholar of Research Corporation.
References
- 1.Le Moual H, Koshland DE., Jr J Mol Biol. 1996;261:568–585. doi: 10.1006/jmbi.1996.0483. [DOI] [PubMed] [Google Scholar]
- 2.Falke JJ, Kim SH. Curr Opin Struct Biol. 2000;10:462–469. doi: 10.1016/s0959-440x(00)00115-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Falke JJ, Hazelbauer GL. Trends Biochem Sci. 2001;26:257–265. doi: 10.1016/s0968-0004(00)01770-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bourret RB, Stock AM. J Biol Chem. 2002;277:9625–9628. doi: 10.1074/jbc.R100066200. [DOI] [PubMed] [Google Scholar]
- 5.Kehry MR, Dahlquist FW. J Biol Chem. 1982;257:10378–10386. [PubMed] [Google Scholar]
- 6.Terwilliger TC, Koshland DE., Jr J Biol Chem. 1984;259:7719–7725. [PubMed] [Google Scholar]
- 7.Kim KK, Yokota H, Kim SH. Nature. 1999;400:787–792. doi: 10.1038/23512. [DOI] [PubMed] [Google Scholar]
- 8.Terwilliger TC, Wang JY, Koshland DE., Jr J Biol Chem. 1986;261:10814–10820. [PubMed] [Google Scholar]
- 9.Terwilliger TC, Wang JY, Koshland DE., Jr Proc Natl Acad Sci USA. 1986;83:6707–6710. doi: 10.1073/pnas.83.18.6707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kehry MR, Bond MW, Hunkapillar MW, Dahlquist FW. Proc Natl Acad Sci USA. 1983;80:3599–3603. doi: 10.1073/pnas.80.12.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shapiro MJ, Koshland DE., Jr J Biol Chem. 1994;269:11054–11059. [PubMed] [Google Scholar]
- 12.Shapiro MJ, Panomitros D, Koshland DE., Jr J Biol Chem. 1995;270:751–755. doi: 10.1074/jbc.270.2.751. [DOI] [PubMed] [Google Scholar]
- 13.Stock AM, Wylie DC, Mottonen JM, Lupas AN, Ninfa EG, Ninfa AJ, Schutt CE, Stock JB. Cold Spring Harbor Symp Quant Biol. 1988;53:49–57. doi: 10.1101/sqb.1988.053.01.009. [DOI] [PubMed] [Google Scholar]
- 14.Wu J, Li J, Li G, Long DG, Weis RM. Biochemistry. 1996;35:4984–4993. doi: 10.1021/bi9530189. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Li G, Weis RM. Biochemistry. 1997;36:11851–11857. doi: 10.1021/bi971510h. [DOI] [PubMed] [Google Scholar]
- 16.Le Moual H, Quang T, Koshland DE., Jr Biochemistry. 1997;36:13441–13448. doi: 10.1021/bi9713207. [DOI] [PubMed] [Google Scholar]
- 17.Barnakov AN, Barnakova LA, Hazelbauer GL. J Bacteriol. 1998;180:6713–6718. doi: 10.1128/jb.180.24.6713-6718.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Djordjevic S, Stock AM. Structure. 1997;5:545–558. doi: 10.1016/s0969-2126(97)00210-4. [DOI] [PubMed] [Google Scholar]
- 19.Schubert HL, Blumenthal RM, Cheng X. Trends Biochem Sci. 2003;28:329–335. doi: 10.1016/S0968-0004(03)00090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Djordjevic S, Stock AM. Nat Struct Biol. 1998;5:446–450. doi: 10.1038/nsb0698-446. [DOI] [PubMed] [Google Scholar]
- 21.Shiomi D, Zhulin IB, Homma M, Kawagishi I. J Biol Chem. 2002;277:42325–42333. doi: 10.1074/jbc.M202001200. [DOI] [PubMed] [Google Scholar]
- 22.Hanahan D. J Mol Biol. 1983;166:557. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 23.Studier FW, Moffatt BA. J Mol Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 24.Tabor S, Richardson CC. Proc Natl Acad Sci USA. 1985;84:1074–1078. doi: 10.1073/pnas.82.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhandari P, Gowrishankar J. J Bacteriol. 1997;179:4403–4406. doi: 10.1128/jb.179.13.4403-4406.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simms SA, Stock AM, Stock JB. J Biol Chem. 1987;262:8537–8543. [PubMed] [Google Scholar]
- 27.Simms SA, Keane MG, Stock J. J Biol Chem. 1985;260:10161–10168. [PubMed] [Google Scholar]
- 28.Wolfe AJ, Conley MP, Kramer TJ, Berg HC. J Bacteriol. 1987;169:1878–1885. doi: 10.1128/jb.169.5.1878-1885.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Higuchi R, Krummel B, Saiki RK. Nucleic Acids Res. 1988;16:7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foster DL, Mowbray SL, Jap BK, Koshland DE., Jr J Biol Chem. 1985;260:11706–11710. [PubMed] [Google Scholar]
- 31.Gabb HA, Jackson RM, Sternberg MJE. J Mol Biol. 1997;272:106–120. doi: 10.1006/jmbi.1997.1203. [DOI] [PubMed] [Google Scholar]
- 32.Jackson RM, Gabb HA, Sternberg MJE. J Mol Biol. 1998;276:265–285. doi: 10.1006/jmbi.1997.1519. [DOI] [PubMed] [Google Scholar]
- 33.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 34.Le Moual H, Quang T, Koshland DEJ. Biochemistry. 1998;37:14852–14859. doi: 10.1021/bi980343y. [DOI] [PubMed] [Google Scholar]
- 35.Woodard RW, Tsai MD, Floss HG, Crooks PA, Coward JK. J Biol Chem. 1980;255:9124–9127. [PubMed] [Google Scholar]
- 36.Floss HG, Mascaro L, Tsai M-D, Woodard RW. In: Transmethylation. Usdin E, Borchardt RT, Creveling CR, editors. Elsevier North Holland Inc; New York: 1979. pp. 135–141. [Google Scholar]
- 37.Shapiro MJ, Chakrabarti I, Koshland DE., Jr Proc Natl Acad Sci USA. 1995;92:1053–1056. doi: 10.1073/pnas.92.4.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng YJ, Bruice TC. J Am Chem Soc. 1997;119:8137–8145. [Google Scholar]