

Abstract
With the recent successes in determining membrane protein structures, we explore the tractability of determining representatives for the entire human membrane proteome. This proteome contains 2,925 unique integral α-helical transmembrane domain sequences that cluster into 1,201 families sharing more than 25% sequence identity. Structures of 100 optimally selected targets would increase the fraction of modelable human α-helical transmembrane domains from 26% to 58%, thus providing structure/function information not otherwise available.
Keywords: target selection, human membrane proteins, structural genomics, protein structure initiative, homology modeling
Introduction
Integral membrane proteins are classified into two broad categories based on the nature of their interaction with the membrane: integral monotopic proteins are attached to the lipid membrane from only one side, while the integral bitopic and polytopic proteins, also known as transmembrane proteins, span the lipid bilayer once and more than once, respectively. The transmembrane proteins typically have either an α-helical fold or a multi-stranded β-barrel fold.
Several reliable methods to predict α-helical transmembrane domains from sequence are available (eg, MEMSAT, Phobius, PolyPhobius, Scampi_multi, Spoctopus, Thumbup1.0, TMHMM2.0)1–3. Using such methods, a recent survey predicted that approximately a quarter of the human proteome is comprised of proteins with at least one transmembrane α-helix4. The α-helical transmembrane domains are significantly more abundant than the β-barrel transmembrane domains, and also appear to be functionally more diverse5. For example, α-helical proteins are found in all biological membranes, while β-barrel transmembrane proteins only span the mitochondrial membrane in humans, the outer membranes of Gram-negative bacteria, and eukaryotic chloroplast membranes.
Integral membrane proteins are essential for cellular function. The α-helical transmembrane proteins comprise critical functional groups, including receptors, transporters, transceptors, ion channels, enzymes, and others4,6. In particular, approximately 60% of current drug targets are membrane proteins7, with G protein-coupled receptors (GPCRs) and ion channels alone accounting for 30% and 10% of primary drug targets, respectively. In addition, membrane transporters are important secondary drug targets for regulating the absorption, distribution, metabolism, and excretion (ADME) of drugs targeting other proteins8.
Structures of membrane proteins are a powerful tool for understanding their diverse functions and discovering new drugs. However, in stark contrast to their frequency and importance, only 1,035 of the approximately 85,000 entries in the Protein Data Bank (PDB; 2nd October 2012) describe α-helical transmembrane protein structures9,10, due to extraordinary technical challenges involved in their purification and structure determination by X-ray crystallography, NMR spectroscopy, or electron microscopy.
Progress has recently been made in de novo structure prediction of α–helical membrane protein sequences by relying on a large number of homologous sequences determined by genomic sequencing11. Nevertheless, comparative or homology protein structure modeling, which relies on experimentally determined structures of homologous proteins12–14, remains the most accurate method for computing 3D models of membrane protein sequences. Although human membrane proteins can sometimes be modeled with useful accuracy by comparative modeling based on structures of their homologs from other organisms, the utility of prokaryotic structures for modeling of their human homologs is often limited15, due to low sequence and structure similarity.
To significantly increase the number of proteins with characterized structures, NIH established the Protein Structure Initiative (PSI)16, which includes in the current PSI:Biology stage four large-scale centers focused on globular proteins (http://sbkb.org/kb/psi_centers.html) and nine specialized centers focused on membrane proteins (http://sbkb.org/kb/membprothub.html). The PSI has maximized structural characterization of the protein sequence space by an efficient combination of experimentation and prediction. Thus, selection of target proteins is key to maximizing coverage of the protein universe.
A number of target selection schemes have been used in the past, ranging from focusing on only novel sequences, large families with no structural representative, to selecting all proteins in a model genome5,17–21. These schemes 5 organize the proteins of interest into clusters of related sequences, generally expanded by including additional homologs from other organisms. Any member of a cluster can then be a target for structure determination, because it is by definition sufficiently similar to the remaining cluster members to allow its experimental structure to serve as a template for comparative modeling12. As the sequence similarity between the target and its homologs increases, the accuracy of the resulting models also increases, but at the cost of having to determine a larger number of target structures. Consequently, a target selection scheme needs to balance the effort needed for structure determination of all targets with the accuracy of resulting comparative models.
In general, high-accuracy comparative models are based on more than 50% sequence identity to their templates, and tend to have approximately 1 Å rootmean- square (RMS) error for the main-chain atoms22. Medium-accuracy models are based on 30% to 50% sequence identity, and tend to have about 90% of the main-chain atoms modeled with 1.5 Å RMS error. Common errors in these models are side-chain packing, core distortion, and loop modeling errors, with occasional alignment errors. Comparative models based on less than 30% sequence identity are commonly considered low-accuracy comparative models, because alignment errors tend to increase rapidly below this sequence identity threshold. However, even at this low level of target-template similarity, the models can still be useful for some of the most demanding applications. For example, virtual screening against comparative models of a GPCR23 and a Solute Carrier (SLC) transporter24 based on templates with less than 25% sequence identity were instrumental in discovering chemically novel small molecule ligands. Thus, a useful threshold on sequence identity for selecting targets for experimental structure determination may be as low as 25%.
A fitting and unifying potential goal for the nine PSI membrane protein centers involved in PSI:Biology is a comprehensive structural characterization of the human α-helical transmembrane domains. However, even for a large-scale effort, determining the structures of all human α-helical transmembrane proteins by Xray crystallography, NMR spectroscopy, or electron microscopy is not feasible in the foreseeable future. Therefore, an efficient target selection strategy is useful and will have the largest impact on the broad scientific community. Here, we analyze several candidate target selection schemes, thus assessing the feasibility of a comprehensive structural description of the human α-helical transmembrane proteome.
Analysis
The analysis was performed in five stages (Fig. 1; Supplementary Notes 1–3; http://salilab.org/membrane/). First, we identified the human α-helical transmembrane domain sequences (Supplementary Fig. 1a). Second, we assessed how many of them can currently be modeled by comparative modeling (Supplementary Fig. 1b). Third, we clustered the sequences (Supplementary Fig. 2; Supplementary Table 1). Fourth, we quantified the efficiency of two target selection strategies (Supplementary Fig. 3; Supplementary Table 2), and compared the results with the current target lists of the nine PSI membrane protein centers (Supplementary Table 3), to assess the degree to which structure determination following these strategies and lists would allow comparative modeling of the human membrane proteome. Fifth, to prepare lists of proteins that would enable structure determination of the most advantageous target proteins, we expanded the list of human domain targets by adding their homologs from the UniProtKB database25 (Supplementary Fig. 4).
Figure 1. Flowchart of the analysis (Supplementary Notes 1 and 2).
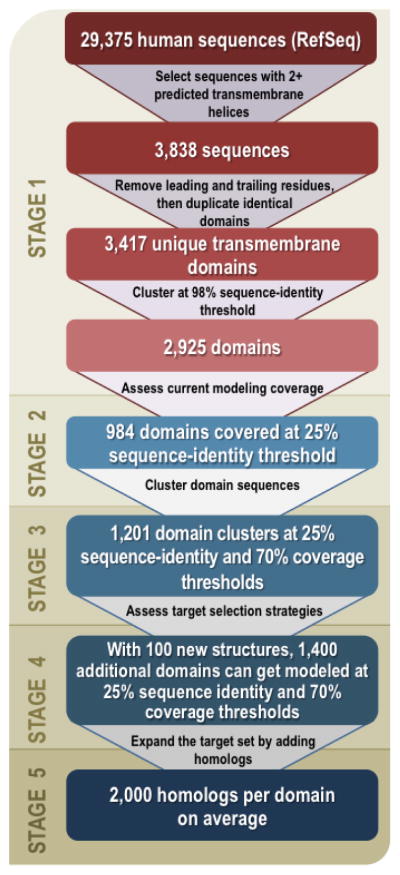
In stage 1,α-helical transmembrane regions of all human sequences from the RefSeq-37 database34 were predicted by TMHMM2.035 and clustered with USEARCH36 at 98% sequence identity, resulting in 2,925 unique α-helical transmembrane domains with at least two predicted transmembrane helices. In stage 2, the current modeling coverage of these domains was assessed by automated comparative modeling using ModPipe37. In stage 3, the 2,925 unique α-helical transmembrane domains were clustered at 25% sequence identity and 70% coverage thresholds using BLASTCLUST38, resulting in 1,201 domain clusters. In stage 4, several target lists, including existing target lists of the nine PSI:Biology centers, were assessed by mapping the number and quality of models as a function of the number of targets; for example, if representative structures for the 100 largest clusters were determined, 1,400 additional α-helical transmembrane domains could be modeled based on at least 25% sequence identity to the closest known structure. In stage 5, the target sequence set was expanded by adding homologous sequences from other organisms extracted from UniProtKB25.
Target selection for structural genomics of human membrane proteins
Nine PSI membrane protein centers have been funded to improve structure characterization of membrane proteins. Several of these centers focus on human membrane protein families, and almost all centers aim to determine the structures of at least some human proteins or their homologs. This effort, combined with the broader membrane protein structural biology community, should make a significant impact on the structural coverage of the human transmembrane proteome, especially if a coordinated target selection strategy is pursued. A coordinated approach to future target selection for the nine membrane protein centers seems to be reasonable, particularly if the resulting increase in the structural coverage is significant.
For this study, we relied on established methods to predict α-helical transmembrane domains. The relatively low number of known structures of βbarrel transmembrane proteins, combined with a less prominent hydrophobic profile, makes it more difficult to develop reliable computational methods for predicting β-barrel transmembrane domains on a genomic scale2,26. In addition, the estimated fraction of β-barrel transmembrane proteins in proteomes (2–3%)5 is significantly lower than the fraction of α-helical transmembrane proteins (approximately 25%). For these reasons, we analyzed here only the human α-helical transmembrane domain sequences.
Two key parameters of a target selection strategy include the thresholds on the target-template sequence similarity and modeling coverage. Since membrane protein crystallization and structure determination is notoriously difficult, we considered a maximum possible accommodation on these thresholds, to maximize the number of sequences that can be modeled based on a given number of new structures. For the human α-helical transmembrane domain sequences, accepting comparative models covering at least 60% of the domain sequence based on at least 25% sequence identity to the closest template structure, 100 structures selected by the guided target selection would increase the number of modelable human α-helical transmembrane domains by more than a factor of two, from 26% to 58%.
Relevance and biological impact
To increase our confidence in the automatically computed clusters of sequences, we examined three well-defined superfamilies as examples – GPCRs, SLC membrane transporters, and claudins. Based on these examinations, we conclude that our automated clustering procedure reproduces previous detailed annotations and thus the analysis of target selection is likely to be statistically robust.
G-protein Coupled Receptors
GPCR sequences were collected from GPCRDB26. This superfamily forms the largest cluster (616 sequences), with the remaining 128 sequences forming several smaller clusters. Although a number of structures have recently been determined for this important class of membrane proteins27, it is the diversity that is perhaps most intriguing and in need of further investigation. For example, the opioid receptors that have recently been structurally characterized differ only in a few residues throughout the entire sequence, but have drastically different pharmacological effects28. The key question from a structural genomics perspective is what level of granularity (i.e. how many structures) is needed to create reliable models of additional GPCRs. Although not an ideal target for α-helical transmembrane novelty, it is critically important to know how many structures are needed for computational approaches to be reliable for basic and applied science. This superfamily serves as a control and a test case for answering the question about required structure mapping granularity.
SLC transporters
Unlike the GPCRs, this important family of transporters is a very diverse set of sequences that are not all related by a common ancestor29; nevertheless, some members can share similar structural features despite weak sequence similarities. 340 SLC sequences are included in 116 domain clusters with 1 – 14 cluster members. The majority of the sequences in the clusters have been previously annotated as SLC members, with 5% being annotated as hypothetical protein, uncharacterized, fragment, or similar. As with the GPCR superfamily, there is an important question about the granularity of the experimental structure set that is needed before one can reliably model other transporters.
Claudins
The claudin family cluster illustrates the novel impact that structural genomics can have on biology and medicine. The family of claudins in humans consists of 23 proteins, and 3 additional members were recently proposed30. Almost all of the 23 known human claudin sequences (20–27 kDa in size) form a single cluster31 and contain 4 transmembrane helices with their 2 extracellular loops important for the formation of cell/cell interactions in tight junctions32,33. Despite the high importance of claudins, no structure has yet been determined for any claudin protein. Numerous studies of mutants, as well as freeze-fracture studies, have revealed a model of the function of individual domains33. The Nterminus is located at the intracellular side generally containing only 7 residues. Despite the small size of the claudin monomers, claudins are challenging targets for structure determination because their transmembrane domains form multimeric complexes within the membrane. Even a single experimentally determined claudin structure is likely to result in a significantly increased understanding of the claudin function in molecular terms. Such a structure would also allow the modeling of the other claudins, further increasing its impact.
Proposed target selection strategy
To most efficiently bridge the gap between the human genomic sequences and membrane protein structure knowledge, we propose to collectively pursue structural studies from the largest 100 clusters in need of structural coverage. Such an effort would lead to a structural characterization of 100 additional protein families and increase the coverage of the human α-helical transmembrane proteome to 58% of all sequences. However, the mandate for the PSI:Biology membrane protein centers is broader, encouraging the centers to concentrate on a variety of important biological questions. The approach integrates the goals of maximizing structural characterization with the biological focus of the individual centers.
Since determining structures of eukaryotic membrane proteins continues to be technically challenging, a target with several homologs from bacteria or archaea may be a good initial or alternate target, provided it can be used to further biological research or to compute sufficiently accurate models of its eukaryotic homologs. Thus, the centers will routinely process several related homologous sequences through their structure determination methods to maximize the likelihood of determining a structure from a family.
Lastly, coordination of the PSI:Biology targets with the structural biology and broader scientific community will help to more efficiently advance the field where follow up studies are conducted to understand specific systems in more depth. This analysis provides a comprehensive assessment of the human α-helical transmembrane proteome, where we are today as a field, and what the field can accomplish in the next few years. By identifying sequence families that can be better characterized through the solution of the structures of a few homologs (from either eukaryotic or prokaryotic sources), a membrane biologist can select the best templates and models for any membrane protein sequence of as yet undetermined structure or understand the reliability of the model with sufficient structural coverage. Structural biologists can readily identify the impact that further investigations of specific structures can have or that any particular new membrane protein structure will have on the knowledge of human membrane proteome.
Availability
A computational resource, the Membrane Protein Hub (http://sbkb.org/kb/membprothub.html), has recently been established as part of the Structural Biology Knowledgebase in collaboration between PSI and the journal Nature. The purpose is to disseminate the results of the nine PSI membrane protein centers. All results of the current study are accessible in its own knowledgebase through the Membrane Protein Hub home page (link: human TM proteome) and through http://salilab.org/membrane (Supplementary Note 3).
Supplementary Material
Acknowledgments
We thank Drs. Ian Wilson, Jean Chin and Peter Preusch for critical comments on the manuscript. Research was supported by the National Institutes of Health PSI:Biology grants U54 GM094662 (AS, UP), U54 GM094618 (RCS), U54 GM094625 (RMS, AS, UP), U54 GM094584 (BGF), U54 GM094599 (PF), U54 GM094611 (MW, MED, MGM), U54 GM094610 (GAC, DCR, MHBS), U54 GM094608 (JJC), U54 GM095315 (WAH, BR, EK), and U54 GM094598 (DLS).
References
- 1.von Heijne G. Journal of internal medicine. 2007;261:543–57. doi: 10.1111/j.1365-2796.2007.01792.x. [DOI] [PubMed] [Google Scholar]
- 2.Punta M, et al. Methods. 2007;41:460–74. doi: 10.1016/j.ymeth.2006.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Almen MS, Nordstrom KJ, Fredriksson R, Schioth HB. BMC biology. 2009;7:50. doi: 10.1186/1741-7007-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fagerberg L, Jonasson K, von Heijne G, Uhlen M, Berglund L. Proteomics. 2010;10:1141–9. doi: 10.1002/pmic.200900258. [DOI] [PubMed] [Google Scholar]
- 5.Punta M, et al. Journal of structural and functional genomics. 2009;10:255–68. doi: 10.1007/s10969-009-9071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nucleic acids research. 2012;40:D71–5. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hopkins AL, Groom CR. Nature reviews Drug discovery. 2002;1:727–30. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 8.Giacomini KM, et al. Nature reviews Drug discovery. 2010;9:215–36. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rose PW, et al. Nucleic acids research. 2011;39:D392–401. doi: 10.1093/nar/gkq1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kloppmann E, Punta M, Rost B. Current opinion in structural biology. 2012;22:326–32. doi: 10.1016/j.sbi.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hopf TA, et al. Cell. 2012 Epub ahead of print. [Google Scholar]
- 12.Baker D, Sali A. Science. 2001;294:93–96. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- 13.Dunbrack RL., Jr Current opinion in structural biology. 2006;16:374–84. doi: 10.1016/j.sbi.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 14.Liu T, Tang GW, Capriotti E. Combinatorial chemistry & high throughput screening. 2011;14:532–47. doi: 10.2174/138620711795767811. [DOI] [PubMed] [Google Scholar]
- 15.Granseth E, Seppala S, Rapp M, Daley DO, Von Heijne G. Molecular membrane biology. 2007;24:329–32. doi: 10.1080/09687680701413882. [DOI] [PubMed] [Google Scholar]
- 16.Norvell JC, Berg JM. Structure. 2007;15:1519–22. doi: 10.1016/j.str.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Vitkup D, Melamud E, Moult J, Sander C. Nat Struct Biol. 2001;8:559–66. doi: 10.1038/88640. [DOI] [PubMed] [Google Scholar]
- 18.Marsden RL, Orengo CA. Methods in molecular biology. 2008;426:3–25. doi: 10.1007/978-1-60327-058-8_1. [DOI] [PubMed] [Google Scholar]
- 19.Raftery J. Methods in molecular biology. 2008;426:37–47. doi: 10.1007/978-1-60327-058-8_3. [DOI] [PubMed] [Google Scholar]
- 20.Bussow K, et al. Microbial cell factories. 2005;4:21. doi: 10.1186/1475-2859-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelly L, et al. J Struct Funct Genomics. 2009;10:269–280. doi: 10.1007/s10969-009-9069-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marti-Renom MA, et al. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 23.Carlsson J, et al. Nature chemical biology. 2011;7:769–78. doi: 10.1038/nchembio.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schlessinger A, et al. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:15810–5. doi: 10.1073/pnas.1106030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nucleic acids research. 2010;38:D142–8. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vroling B, et al. Nucleic acids research. 2011;39:D309–19. doi: 10.1093/nar/gkq1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katritch V, Cherezov V, Stevens RC. Trends in pharmacological sciences. 2012;33:17–27. doi: 10.1016/j.tips.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Granier S, et al. Nature. 2012;485:400–4. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schlessinger A, et al. Protein science: a publication of the Protein Society. 2010;19:412–28. doi: 10.1002/pro.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mineta K, et al. FEBS Lett. 2011;585:606–12. doi: 10.1016/j.febslet.2011.01.028. [DOI] [PubMed] [Google Scholar]
- 31.Escudero-Esparza A, Jiang WG, Martin TA. Frontiers in bioscience: a journal and virtual library. 2011;16:1069–83. doi: 10.2741/3736. [DOI] [PubMed] [Google Scholar]
- 32.Angelow S, Ahlstrom R, Yu AS. Am J Physiol Renal Physiol. 2008;295:F867–76. doi: 10.1152/ajprenal.90264.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krause G, et al. Biochim Biophys Acta. 2008;1778:631–45. doi: 10.1016/j.bbamem.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 34.Larsson TP, Murray CG, Hill T, Fredriksson R, Schioth HB. FEBS Lett. 2005;579:690–8. doi: 10.1016/j.febslet.2004.12.046. [DOI] [PubMed] [Google Scholar]
- 35.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Journal of molecular biology. 2001;305:567–80. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 36.Edgar RC. Bioinformatics. 2010;26:2460–1. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 37.Pieper U, et al. Nucleic Acids Research. 2011;39:D465–74. doi: 10.1093/nar/gkq1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson M, et al. Nucleic acids research. 2008;36:W5–9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.