

Abstract
Thrombin helps to activate Factor XIII (FXIII) by hydrolyzing the R37-G38 peptide bond. The resultant transglutaminase introduces cross-links into the fibrin clot. With the development of therapeutic coagulation factors, there is a need to better understand interactions involving FXIII. Such knowledge will help predict ability to activate FXIII and thus ability to promote/hinder the generation of transglutaminase activity. Kinetic parameters have been determined for a series of thrombin species hydrolyzing the FXIII (28′41) V34X activation peptides (V34, V34L, V34F, and V34P). The V34P substitution introduces PAR4 character into the FXIII, and the V34F exhibits important similarities to the cardioprotective V34L. FXIII activation peptides containing V34, V34L, or V34P could each be accommodated by alanine mutants of thrombin lacking either the W60d or Y60a residue in the 60-insertion loop. By contrast, FXIII V34F AP could be cleaved by thrombin W60dA but not by Y60aA. FXIII V34P is highly reliant on the thrombin W215 platform for its strong substrate properties whereas FXIII V34F AP becomes the first segment that can maintain its Km upon loss of the critical thrombin W215 residue. Interestingly, FXIII V34F AP could also be readily accommodated by thrombin L99A and E217A. Hydrolysis of FXIII V34F AP by thrombin W217A/E217A (WE) was similar to that of FXIII V34L AP whereas WE could not effectively cleave FXIII V34P AP. FXIII V34F and V34P AP show promise for designing FXIII activation systems that are either tolerant of or greatly hindered by the presence of anticoagulant thrombins.
Keywords: Factor XIII, transglutaminase, thrombin, fibrinogen, kinetics, coagulation
1. Introduction
Three important players in the later stages of blood coagulation include thrombin (IIa), Factor XIII (FXIII) and fibrinogen (Fbg)1. The serine protease IIa [1] converts Fibrinogen (AαBβγ)2 to fibrin by hydrolyzing the Aα and Bβ chains located in the N-terminal E-domain of fibrinogen thereby releasing fibrinopeptides A (FpA) and B (FpB). Fibrin polymerization sites are then exposed that promote formation of a noncovalently associated fibrin clot network [2, 3]. To help generate a proteolytically stable clot, thrombin cleaves the N-terminal activation peptide (AP) segment of FXIII at the R37-G38 peptide bond. In the presence of calcium, the activated FXIIIa catalyzes formation of γ-glutamyl ε-lysyl covalent crosslinks in the fibrin network leading to the formation of a proteolytically stable clot [4]. Besides participating in coagulation, thrombin also exhibits other physiological roles. Thrombin contributes to anticoagulation by activating protein C (PC) in the presence of thrombomodulin [1, 5]. In addition, thrombin-dependent activation of protease-activated receptors (PARs) leads to platelet aggregation/secretion and cellular permeability effects observed during inflammation [6].
Thrombin is a multifunctional enzyme that utilizes several features to control substrate specificity. This protease is a sodium-activated type II enzyme characterized to switch between a sodium-free (E) and a sodium-bound (E-Na+) form [1, 7]. Recent studies have focused on better understanding the presence of an earlier E* form that is inactive and cannot bind Na+ [1, 8]. Thrombin also contains an extended active site region, a series of insertion loops, and two anion binding exosites (Figure 1A) [1, 9, 10]. The insertion loops limit substrate access to the serine protease active site cleft. Of particular note is the 60-insertion or β-insertion loop (Y60a – K60f)2 that includes the key amino acids Y60a and W60d (Figure 1B). W60d is located in the center of the loop and serves as a clamp over the active site. Y60a is positioned at the start of the loop and its placement influences more distant substrate binding.
Figure 1. X-ray crystal structures highlighting major regions of thrombin.

(A) A contour representation of thrombin (PDB entry 1PPB) is displayed. The β- or 60-insertion loop hangs over the serine protease active site and restricts substrate access. Anion binding exosite II is located to the left of the active site whereas anion binding exosite I is located to the right. The sodium binding site serves an allosteric role. (B) The same thrombin structure is displayed in cartoon format. The five residues that are mutated are highlighted in stick form. W60d andY60a are members of the 60-insertion loop and L99 is part of the S2 pocket. W215 serves as an important platform for substrates. E217 participates in the Na+ allosteric network and stabilizes the S1 site.
There are several other important residues within the thrombin active site region to consider (Figure 1B) [9, 10]. Located near Y60a is L99, a residue which is part of the S2 site within the apolar site. Thrombin residue W215 serves as a platform to support several thrombin substrates. The Na+ binding site is located within the 220-loop and is also regulated by the 186-loop [11]. E217 is part of the Na+ allosteric network and participates in stabilizing the S1 specificity pocket3. W215 is critical for binding and hydrolysis of the Fbg Aα chain whereas PAR1 and PC have less of a reliance on this platform [12, 13]. Replacement of E217 also generates a thrombin species with less procoagulant activity [14–16]. The double mutant W215A/E217A (WE) has shown much promise as a potent anticoagulant, and X-ray crystallography has revealed that this mutant exhibits a collapsed primary specificity pocket [12, 17, 18]. The structural features of this double mutant are consistent with those of the E* form. Another proposal denotes that WE exhibits a degree of zymogen-like character that exists along the continuum from inactive zymogen to stabilized proteinase [19].
Individual substrate residues are also critical in evaluating thrombin specificity. Substrate amino acids located N-terminal to the scissile bond make important contributions to binding and hydrolysis rates (Table 1). The Factor XIII activation peptide, PAR1, and PAR4 each take advantage of the P4 and P2 positions3 to target interactions within the thrombin active site region. (Figures 2A–C) [20–23]. By contrast, the fibrinogen A chain is highly reliant on the P9 position to interact effectively with thrombin [24]. Unlike fibrinogen, FXIII does not contain a sequence that can take advantage of thrombin anion binding exosite I. The intact FXIII A2 structure thus only helps direct the AP segment to the thrombin active site region.
Table 1.
Substrate sequences that target the thrombin active sitea
| P9 … … … … P4 … … P1 … | |
|---|---|
| Factor XIII (28–41) V34 AP | 28TVELQGVVPRGVNL41 |
| Factor XIII (28–41) V34L AP | 28TVELQGLVPRGVNL41 |
| Factor XIII (28–41) V34P AP | 28TVELQGPVPRGVNL41 |
| Factor XIII (28–41) V34F AP | 28TVELQGFVPRGVNL41 |
| Thrombin Receptor PAR1 (32–45) | 32KATNATLDPRSFLL45 |
| Thrombin Receptor PAR4 (38–51) | 38STPSILPAPRGYPG51 |
| Fibrinogen Aα (7–20) | 7DFLAEGGGVRGPRV20 |
| Fibrinogen Bβ (5–18) | 5DNEEGFFSARGHRP18 |
Human sequences of FXIII, PAR1, PAR4, and fibrinogens Aα and Bβ are displayed.
Figure 2. Conformational features associated with segments of FXIII AP, PAR1, and PAR4 bound to the active site of thrombin.

(A) Selected residues of human thrombin that surround FXIII AP (34–37), (B) selected residues of human thrombin that surround PAR1 (38–41), and (C) selected residues of murine thrombin that surround murine PAR4 (56–59). The X-ray PDB codes include 1DE7 (human), 3LU9 (human), and 2PV9 (murine), respectively. For each panel, the thrombin residues are in cyan, P4 residue of the substrate is in orange, and the P3-P1 residues of the substrate are in purple.
FXIII V34L is a common polymorphism where the Val at the P4 position is replaced with a Leu [25]. FXIII V34L is found in approximately 25% of the Caucasian population and has been correlated with protection from myocardial infarction [4, 26–29]. The extra methylene group on Leu 34 produces a FXIII species which is more easily activated by thrombin [20, 30–32]. Ariens et al. demonstrated that the V34L polymorphism influences fibrin clot structure and lysis [30, 33]. Under high fibrin(ogen) conditions, FXIII V34L is associated with thinner fibrin chains and a more permeable clot structure [33]. Kinetic studies with synthetic models of the FXIII V34 and V34L activation peptide segment have revealed that V34L leads to enhancements in kcat that contribute to improved hydrolysis rates [20, 21, 34–36]
Fibrin clot structure is largely controlled by IIa catalyzed cleavage of fibrinogen chains and the FXIII activation peptide segment [2, 3]. Although much is already known about how to regulate interactions between thrombin and fibrinogen, less is understood about the interactions between thrombin and FXIII. Our earlier studies probed the extent to which recombinant human thrombin mutants W215A, E217A, WE, and L99A could bind and hydrolyze FXIII (28–41) AP sequences containing V34 or L34 [37].
For this project, additional features of thrombin and FXIII AP were examined. FXIII (28–41) V34 and V34L AP kinetic studies were performed with thrombin mutants Y60aA and W60dA to assess how these key 60-insertion loop residues influence FXIII AP interactions within the thrombin active site region. In another series of studies, hydrolysis of FXIII (28–41) V34F and V34P AP was performed in the presence of wild type thrombin and the thrombin mutants Y60aA, W60dA, L99A, W215A, E217A, and WE. FXIII (28–41) V34P provides an opportunity to introduce PAR4 character into the FXIII AP segment. The two prolines at the P4 and P2 positions of PAR4 supply critical anchor points on to the thrombin surface and enhance kinetic properties. Earlier kinetic and NMR studies with FXIII (28–41) V34F and thrombin revealed that the aromatic F is the only other residue thus far that can achieve the stabilizing P4-P2 through space interaction observed with L34 [35].
With an interest in designing FXIII AP segments with altered activation properties, the P and F are intriguing candidates. Both residues can help promote contact with the active regions, but their orientations within the FXIII AP segment and the thrombin active site region are proposed to be different. F34 is likely directed toward the 60-insertion loop whereas the P34 is likely oriented in the opposite direction more toward W215 and the thrombin autolysis-loop. With the current interest in utilizing thrombin mutants for therapeutic purposes, it will also be important to understand how different FXIII AP segments respond to both wild-type and mutant thrombin species.
The thrombin mutants that were examined in the current work reacted to varying degrees with the FXIII V34X activation peptides. With WT-thrombin, FXIII V34P AP exhibited the best Km and FXIII V34L the best kcat. FXIII peptides containing V34, V34L, or V34P could each be accommodated by thrombin W60dA or Y60aA. Hydrolysis of FXIII V34F AP was greatly hindered in the presence of Y60aA suggesting that this thrombin residue is critical for FXIII F34 placement. By contrast, FXIII V34F AP was readily hydrolyzed by thrombins W215A, E217A, L99A, and W60dA, often with little change to Km. FXIII V34F AP becomes the first activation peptide whose Km is preserved upon loss of W215. Unlike V34F, FXIII V34P AP was highly reliant on W215 and exhibited greater kinetic differences among the thrombin mutants. With thrombin W215A/E217A, the kcat/Km for FXIII V34F AP was similar to that of the cardioprotective FXIII V34L AP.
2. Materials and Methods
2.1. Synthetic Peptides
Peptides based on residues 28–41 of the human FXIII activation peptide were synthesized by New England Peptide (Gardner, MA) or Synbiosci (Livermore, CA). The amino acid sequences of the peptides are as follows: FXIII (28–41) V34 AP, Ac-TVELQGVVPRGVNL-amide; FXIII (28–41) V34L AP, Ac-TVELQGLVPRGVNL-amide; FXIII (28–41) V34P AP, Ac-TVELQGPVPRGVNL-amide; FXIII (28–41) V34F AP, Ac-TVELQGFVPRGVNL-amide.
All the peptides were soluble in de-ionized water to 8 mM. The purity of the peptides was determined by analytical reversed phase HPLC. Matrix assisted laser desorption time-of-flight (MALDI-TOF) measurements on an Applied Biosystems Voyager DE-Pro mass spectrometer were used to verify the peptide m/z values. The concentrations of the peptides in solution were determined by quantitative amino acid analysis (AAA Service Laboratory, Damascus, OR).
2.2. Thrombin Preparation
All human recombinant thrombin was generously provided by Dr. Enrico Di Cera and Ms. Leslie Pelc, Saint Louis University, St. Louis, MO. The expression of these mutants was previously described [22]. The thrombins used in this project include: wild type (WT), W60dA, Y60aA, L99A, W215A, E217A, and the double mutant W215A/E217A (WE). Thrombin concentrations were determined from absorbance measurements at E280 = 1.83 mL/(mg cm) and MW = 36,500. The thrombin species were >90% active as determined by active site titration with hirudin.
2.3. Kinetics Procedure
The HPLC-based kinetic assay methods were described previously by Trumbo and Maurer [20]. Briefly, a solution of peptide and assay buffer (50 mM H3PO4, 100mM NaCl, 0.1% PEG, pH 7.4) was heated to 25 °C in a heat block. Hydrolysis was begun with the addition of WT or mutant human thrombin. At regular intervals, an aliquot of the reaction mixture was removed and quenched in 12.5% H3PO4. Peptide peaks were separated by RP-HPLC using a Brownlee Aquapore octyl RP-300 C8 cartridge column or a Waters X-Bridge C18 Column on a Waters 2695 or Waters 600 series HPLC system. The peptide concentrations for FXIII (28–41) V34, V34L, V34P, and V34F were within the range of 100–3000 μM. Thrombin concentration and time points were chosen such that less than 15% of the peptide concentration was hydrolyzed in 30 min.
The thrombin concentration for the hydrolysis reactions with WT was 33.6 nM for all FXIII peptides. For W60dA thrombin, the enzyme concentration was 0.260 μM for FXIII V34 AP and V34L AP whereas it was 58.5 nM for V34P AP and 112 nM for V34F AP. For Y60aA thrombin, the enzyme concentration was 0.84 μM for FXIII V34 AP and 0.28 μM for both V34L and V34P AP. With FXIII V34F AP, the hydrolysis rates were too low to quantitate even at 1 μM Y60aA thrombin. For L99A thrombin, the enzyme concentration was 33.6 nM for the FXIII V34L and V34F peptides whereas it was 0.7 μM for V34 AP and 0.224 μM for V34P. For W215A thrombin, the enzyme concentration was 33.6 nM for all peptides except FXIII V34P AP for which the concentration was 0.214 μM. For E217A thrombin, the concentration was 33.6 nM for FXIII V34 and V34L AP whereas it was 78.4 nM for both V34P and V34F AP. For WE thrombin, the concentration was 1μM for all peptides except FXIII V34P AP for which the hydrolysis rates were too low to quantitate.
After the thrombin-catalyzed kinetic reactions were run on the HPLC columns, the FXIII AP (28–37) product peaks were integrated and the peak areas converted to concentration using a calibration curve. The slopes of product concentration versus time plots were used to determine the initial velocities (in μM/s) for the different thrombin-catalyzed reactions. The results represent experiments which were done in triplicate. Kinetic values were calculated using nonlinear regression analysis fit to the equation V = Vmax/(1 + Km/[S]) using the Marquardt-Levenberg algorithm in Sigma Plot (Jandel Scientific). Km, Vmax, and kcat were calculated from the coefficients of this equation.
3. Results
An HPLC based assay was successfully employed to monitor thrombin hydrolysis rates for a series of Factor XIII activation peptide segments [FXIII (28–41) V34, V34L, V34P, or V34F]. Kinetic comparisons were made between WT thrombin and the thrombin mutants W60dA, Y60aA, L99A, W215A, E217A, and W215A/E217A (WE). The thrombin cleaved products, FXIII (28–37) V34X AP, eluted from the reverse phase columns as distinct peaks. Nonlinear regression analysis values of Km, kcat, and kcat/Km for the different thrombin and FXIII V34X AP pairs are shown in Table 2. Bar chart comparisons across the peptide and thrombin series are found in Figures 3 and 4.
Table 2.
Kinetic Constants for hydrolysis of FXIII (28–41) V34, V34L, V34P, and V34F Activation Peptides with different IIa mutantsa
| FXIII (28–41) V34 AP | Km (μM) | kcat (s−1) | kcat/Km (μM−1.s−1) |
|---|---|---|---|
| WT IIab | 298 ± 42 | 2.57 ±0.005 | 8.6 × 10−3 ± 1.0 × 10−3 |
| W60dA IIa | 1375 ± 422 | 0.86 ±0.046 | 6.3 × 10−4 ± 1.9 × 10−4 |
| Y60aA IIa | 678 ±120 | 0.048 ± 0.0032 | 7.1 × 10−5 ± 1.3 × 10−5 |
| L99A IIab | 2074 ± 414 | 0.89 ± 0.073 | 4.3 × 10−4 ± 1.0 × 10−4 |
| W215A IIab | 2374 ± 372 | 5.53 ± 0.017 | 2.3 × 10−3 ± 4.0 × 10−4 |
| E217A IIab | 1242 ± 392 | 2.84 ± 0.014 | 2.3 × 10−3 ± 7.0 × 10−4 |
| WE (W215A/E217A) IIab | 745 ± 98 | 0.117 ± 0.008 | 1.5 × 10−4 ± 2.0 × 10−5 |
| FXIII (28–41) V34L AP | Km (μM) | kcat (s−1) | kcat/Km (μM−1.s−1) |
|---|---|---|---|
| WT IIab | 315 ± 42 | 22.9 ± 0.003 | 7.3 × 10−2 ± 1.2 × 10−2 |
| W60dA IIa | 1000 ± 201 | 5.07 ± 0.021 | 5.1 × 10−3 ± 1.0 × 10−3 |
| Y60aA IIa | 696 ± 161 | 0.37 ± 0.012 | 5.3 × 10−4 ± 1.2 × 10−4 |
| L99A IIab | 1930 ± 354 | 5.94 ± 0.02 | 3.1 × 10−3 ± 2.0 × 10−4 |
| W215A IIab | 3619 ± 470 | 5.31 ± 0.002 | 1.5 × 10−3 ± 2.0 × 10−4 |
| E217A IIab | 1025 ± 121 | 9.93 ± 0.02 | 9.7 × 10−3 ± 1.0 × 10−3 |
| WE (W215A/E217A) IIab | 142 ± 28 | 0.052 ± 0.003 | 3.6 × 10−4 ± 1.0 × 10−4 |
| FXIII (28–41) V34P AP | Km (μM) | kcat (s−1) | kcat/Km (μM−1.s−1) |
|---|---|---|---|
| WT IIa | 133 ± 10 | 2.17 ± 0.0021 | 1.6 × 10−2 ± 1.3 × 10−3 |
| W60dA IIa | 605 ± 97 | 1.61 ± 0.127 | 2.7 × 10−3 ± 7.9 × 10−4 |
| Y60aA IIa | 810 ± 144 | 0.085 ± 0.002 | 1.0 × 10−4 ± 2.0 × 10−5 |
| L99A IIa | 749 ± 118 | 0.111 ± 0.010 | 2.0 × 10−4 ± 3.0 × 10−5 |
| W215A IIa | 2662 ± 193 | 0.947 ± 0.0365 | 3.6 × 10−4 ± 3.0 × 10−5 |
| E217A IIa | 947 ± 93 | 1.69 ± 0.079 | 1.8 × 10−3 ± 1.9 × 10−4 |
| WE (W215A/E217A) IIa | N/A | N/A | N/A |
| FXIII (28–41) V34F AP | Km (μM) | kcat (s−1) | kcat/Km (μM−1.s−1) |
|---|---|---|---|
| WT IIa | 442 ± 93 | 6.27 ± 0.54 | 1.4 × 10−2 ± 3.0 × 10−3 |
| W60dA IIa | 435 ± 98 | 1.46 ± 0.15 | 3.4 × 10−3 ± 8.0 × 10−4 |
| Y60aA IIa | N/A | N/A | N/A |
| L99A IIa | 570 ± 125 | 2.73 ± 0.3 | 5.0 × 10−3 ± 1.0 × 10−3 |
| W215A IIa | 437 ± 81 | 1.97 ± 0.14 | 4.5 × 10−3 ± 9.0 × 10−4 |
| E217A IIa | 454 ± 102 | 3.36 ± 0.34 | 7.4 × 10−3 ± 2.0 × 10−3 |
| WE (W215A/E217A) IIa | 190 ± 43 | 0.067 ± 0.005 | 3.5 × 10−4 ± 8.0 × 10−5 |
Kinetic constants for the thrombin-catalyzed hydrolysis reactions were determined from an HPLC assay as described in Materials and Methods. The results shown here represent averages of at least three independent experiments. Kinetic values were calculated using nonlinear regression analysis methods using SigmaPlot. The error values correspond to standard error of the mean (SEM).
Isetti and Maurer [37].
Figure 3. Bar charts displaying Km and kcat values for hydrolysis of FXIII V34X peptides by wild-type thrombin.
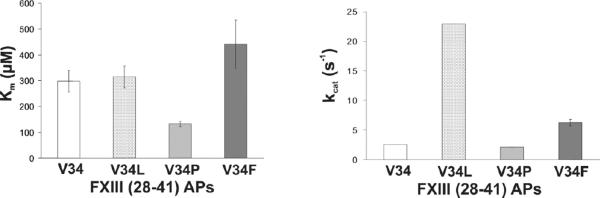
(A) Visual comparison of the Km values for FXIII (28–41) V34 (white), V34L (dotted), V34P (gray), and V34F (dark gray) AP in the presence of thrombin. (B) Visual comparison of the kcat values for FXIII (28–41) V34 (white), V34L (dotted), V34P (gray), and V34F (dark gray) AP in the presence of thrombin. (B) Visual comparison of the kcat values for FXIII (28–41) V34 (white), V34L (dotted), V34P (gray), and V34F (dark gray) AP in the presence of thrombin. An HPLC assay described in Experimental Procedures was used to obtain data to calculate these individual kinetic parameters. The results shown represent averages from at least three independent experiments.
Figure 4. Km and kcat values for hydrolysis of FXIII V34X peptides by thrombin mutants.
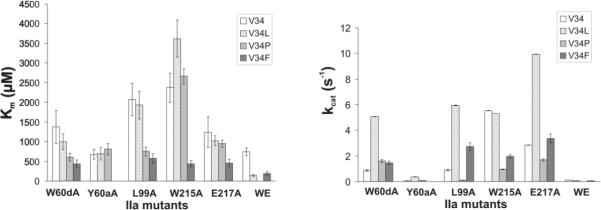
(A) Visual comparison of the Km values for FXIII (28–41) V34 (white), V34L (dotted), V34P (gray), and V34F (dark gray) AP in the presence of the thrombin mutants W60dA, Y60aA, W215A, E217A, and WE. (B) Visual comparison of the kcat values for FXIII (28–41) V34 (white), V34L (dotted), V34P (gray), and V34F (dark gray) AP in the presence of the thrombin mutants W60dA, Y60aA, W215A, E217A, and WE. An HPLC assay described in Experimental Procedures was used to obtain data to calculate these individual kinetic parameters. The results shown represent averages from at least three independent experiments.
3.1. Kinetics Involving FXIII (28–41) V34X AP Hydrolyzed by WT Thrombin
The kinetic values for thrombin-catalyzed hydrolysis of FXIII V34 AP and V34L AP revealed that FXIII V34L AP was a better substrate for WT thrombin than FXIII V34 AP (Table 2 and Figure 3) with kcat being a key contributor [37]. In the current work, FXIII V34P AP promoted a 2-fold improvement in Km leading this peptide sequence to exhibit the best enzyme-substrate interactions of the series. By contrast, the kcat value for FXIII V34P AP was comparable to that of FXIII V34 AP. Unlike FXIII V34P AP, FXIII V34F AP resulted in a moderate increase in Km relative to FXIII V34 AP. The bulkier aromatic residue had greater difficulty promoting effective enzyme-substrate interactions. Once bound, the kcat value, however, improved 2.4-fold over the FXIII V34 AP sequence. Taking the kcat and Km values into account, both FXIII V34P AP and FXIII V34F AP exhibited some improvements in kcat/Km relative to FXIII V34 AP.
3.2 Kinetics Involving FXIII (28–41) V34X AP Hydrolyzed by W60dA Thrombin
W60d is located in the center of the thrombin 60 insertion loop and its alanine mutant W60dA has not been extensively probed with FXIII activation peptides [9, 38]. In the presence of this mutant, the Km for hydrolysis of FXIII V34 AP increased 4.6-fold and the kcat decreased 3-fold relative to WT thrombin (Table 2 and Figure 5). For the FXIII V34L AP sequence, the Km increased 3.2-fold and the kcat decreased 4.6-fold. These kinetic changes caused the kcat/Km values for both peptides to decrease almost 14-fold.
With the FXIII V34P AP, the Km underwent a 4.5-fold increase relative to hydrolysis by WT thrombin whereas the kcat underwent a smaller 1.3-fold decrease. Overall, a 6-fold decrease in kcat/Km had occurred. Interestingly, the Km for hydrolysis of FXIII V34F AP by W60dA thrombin was comparable to that of WT thrombin; yet, the kcat was hindered by 4-fold. FXIII V34F AP binding to thrombin could handle the loss of the W60d residue but the ability to be hydrolyzed had been compromised.
3.3. Kinetics Involving FXIII (28–41) V34X AP Hydrolyzed by Y60aA Thrombin
Residue Y60a is located at the start of the thrombin 60-insertion loop and regulates entrance into the thrombin active site region [9]. The current work presents the first studies on examining the influences of the Y60aA substitution on FXIII AP cleavage. With the Y60aA mutant, hydrolysis of FXIII V34 AP led to a 2-fold increase in Km and a dramatic 54-fold loss in kcat. Overall, the kcat/Km value had decreased 121-fold. For the common polymorphism FXIII V34L AP, a 2-fold increase occurred to Km, a 62-fold decrease occurred to kcat, leading overall to a 138-fold loss in kcat/Km. Replacement of the Y60a residue of thrombin with an alanine clearly had an impact on the ability of the two physiological FXIII APs to orient effectively for hydrolysis.
The FXIII V34P AP and V34F peptides were also greatly influenced by the loss of this 60-insertion loop residue. For the FXIII V34P AP, there was a 160-fold reduction in kcat/Km due to a 6-fold increase in Km and a 25-fold decrease in kcat. Interestingly, very little hydrolysis of the FXIII V34F AP sequence could be observed at the concentrations of Y60aA thrombin available for this project (highest tested 1μM). As a result, individual kinetic parameters could not be calculated for hydrolysis of FXIII V34F AP by Y60aA thrombin.
3.4. Kinetics Involving FXIII (28–41) V34X AP Hydrolyzed by L99A Thrombin
L99 is part of the thrombin apolar binding site and contributes to the S2 enzyme subsite [9]. This leucine acts in concert with Y60a to encompass incoming substrates. With the FXIII V34 AP [37], the Km value increased 7-fold, the kcat value decreased 3-fold, and the kcat/Km value decreased 20-fold relative to WT thrombin. With the FXIII V34L AP, similar increases in Km and decreases in kcat occurred relative to WT thrombin [37]. A still strong kcat allowed the FXIII V34L AP to remain the better substrate sequence for the L99A mutant.
The current work introduced the effects of the V34P and V34F substitutions. For the FXIII V34P AP, the Km increased 5.6-fold and the kcat decreased 19.5-fold. As a result, there was an 80-fold decrease in kcat/Km relative to hydrolysis studies with WT thrombin. The hindrances in Km and kcat were comparable to what was observed with the Y60aA thrombin mutant. In contrast to the Y60aA thrombin studies, the FXIII V34F AP could be accommodated by the L99A mutant. Modest increases in Km were observed relative to WT thrombin and, furthermore, the kcat decreased 2-fold.
3.5. Kinetics Involving FXIII (28–41) V34X AP Hydrolyzed by W215A Thrombin
W215 provides a critical platform that supports a number of thrombin substrates [9]. Since W215A thrombin is already known to exhibit greatly hindered ability to hydrolyze the key procoagulant substrate Fbg Aα, there was strong interest in exploring the influences of this thrombin mutant on hydrolysis of FXIII V34X APs [12, 13]. Replacement of the aromatic W215 residue with an alanine leads to an 8-fold increase in Km for FXIII V34 AP and an 11-fold increase for FXIII V34L AP [37]. Both peptides exhibit similar kcat values with the FXIII V34L AP segment more affected by the W215A substitution than the FXIII V34 AP. As a result, the kcat/Km value decreased 4-fold for FXIII V34 AP and 49-fold for FXIII V34L AP relative to WT thrombin [37].
The influence of this thrombin W215A mutation on hydrolysis of FXIII V34P and V34F AP was also interesting to consider. FXIII V34P AP displayed a 20-fold increase in Km and a 2-fold decrease in kcat relative to wild-type thrombin. By contrast, FXIII V34F AP exhibited virtually no difference in Km values between wild-type and W215A thrombin. For the first time with a FXIII activation peptide, a segment had been found that was not reliant on W215A for further promoting binding within the active site region. Although binding was preserved, there was a 3-fold loss in kcat. Overall, the kcat/Km values for FXIII V34P AP and FXIII V34F AP decreased 44-fold and 3-fold, respectively.
3.6. Kinetics Involving FXIII (28–41) V34X AP Hydrolyzed by E217A Thrombin
The next thrombin amino acid to be probed was the neighboring E217 which is part of the sodium-dependent allosteric network [1, 11]. This residue plays a lesser role than W215A thrombin in binding and orienting FXIII V34 and V34L AP for hydrolysis [37]. The Km values for FXIII V34 AP and V34L AP in the presence of E217A thrombin increased 3–4 fold, and the kcat was hindered at most by 2-fold [37]. In the current work, FXIII V34P AP was also able to tolerate E217A thrombin better than W215A. The Km for hydrolysis of FXIII V34P AP by E217A thrombin increased 7-fold relative to WT thrombin, and there was only a minor decrease in kcat. Further kinetic studies revealed that the Km for hydrolysis of FXIII V34F AP by E217A thrombin remained comparable to the values for WT and W215A thrombin. The E217A mutant exhibited only a 2-fold loss in kcat thus showing improvements relative to W215A.
3.7. Kinetics Involving FXIII (28–41) V34X AP Hydrolyzed by W215A/ E217A (WE) Thrombin
The last thrombin species that was examined was the double mutant W215A/ E217A. Removal of these two residues leads to collapse of the active site cleft and the resultant mutant has shown much promise as an anticoagulant (Figure 2) [12, 17] The replacement of these two residues with alanines led to 2.5-fold increase in Km for FXIII V34 AP and a 2-fold decrease in Km for FXIII V34L AP relative to WT thrombin [37]. By contrast, the kcat for FXIII V34 AP decreased 22-fold and the FXIII V34L AP decreased 440-fold, both relative to wild-type [37]. FXIII V34P AP could be accommodated by the single mutants W215A and E217A but could not be tolerated by the double mutant WE. The limited amount of hydrolyzed product hindered ability to determine individual kinetic parameters. Similar to FXIII V34L AP, hydrolysis of FXIII V34F AP by WE thrombin displayed a 2-fold improvement in Km relative to wild-type thrombin. The kcat value for FXIII V34F AP was 94-fold reduced and together with the Km value caused the kcat/Km value to decrease 40-fold. Interestingly, the kcat/Km values were comparable for FXIII V34L AP and V34F AP.
4. Discussion
The kinetic studies performed have provided an opportunity to study substrate specificity from the perspective of both the enzyme thrombin and the substrate FXIII (28–41) V34X AP. The new knowledge gained may be used to design coagulation systems exhibiting distinct characteristics.
4.1. Interactions between Wild-type IIa and FXIII AP (28–41) V34, V34L, V34P, and V34F
Previous kinetic studies have shown that the common polymorphism V34L leads to a FXIII that is more readily activated than FXIII V34 primarily due to an enhancement in kcat [20, 37]. Solution NMR studies have further supported these observations [21, 35, 36]. A hallmark P4 (L34) to P2 (P36) through-space NOE interaction likely contributes to enhanced binding of the FXIII V34L AP segment on to the thrombin active surface. In addition, an improved orientation for thrombin-based cleavage is achieved. Such a P4 to P2 interaction has also been documented for PAR1 (38LDPR41) using NMR and X-ray crystallography (Figure 2B) [39–41]. Computational docking studies on FXIII suggest that an extended conformation is energetically favorable for the FXIII V34L AP segment and may help avoid steric repulsion within the thrombin apolar site between FXIII V29 at the P9 position and FXIII L34 at the P4 position [42].
In the current project, FXIII AP (28–41) peptides containing V34P and V34F were explored with recombinant wild-type thrombin (Table 2 and Figure 3). The pyrrolidine ring of P34 led to the best Km of the different FXIII V34X AP substrates. Interestingly, the human FXIII AP (33GPVPR37) segment resembles that of human PAR4 (43LPAPR47) and murine PAR4 (55KPNPR59) (Table 1 and Figure 2C). Moreover, the Km values for human FXIII AP (28–41) V34P and human PAR4 (38–51) are comparable. By contrast, the kcat value is 5-fold improved for the PAR4 sequence [43]. Both peptide substrates are better able to interact with the thrombin active site surface than the FXIII AP (28–41) V34 segment (33GVVPR37). Introducing PAR4 character into the FXIII AP segment thus provides a strategy for enhancing FXIII interactions with the thrombin active site surface [43, 44]. The improved kcat value for PAR4 over FXIII AP V34P may be due to the presence of PAR4 L43 at the P5 position. Kinetic studies with PAR4 exodomains revealed that a L43A substitution causes a decrease in kcat but does not affect Km [45]. The (43LPAPR47) segment may further promote effective orientation of PAR4 at the thrombin active site. Peptide NMR studies [43] concluded that the two proline residues on PAR4 (38–51) are positioned gauche to one another, and this orientation was later confirmed by X-ray crystallography [46] (Figure 2C). The P34 and P36 residues of FXIII AP (28–41) V34P are proposed to exhibit a similar structural arrangement.
The binding interactions (Km) involving FXIII V34F AP and WT IIa were not as strong as those observed with FXIII V34, L34, and P34 AP. Previous NMR studies, however, revealed that FXIII V34F AP peptide exhibits the same hallmark P4 to P2 through-space interaction as observed with the cardioprotective FXIII V34L AP sequence [35]. The F34 residue is able to find a thrombin binding site that can accommodate its bulky aromatic side chain. The weaker kinetic parameters relative to FXIII V34L AP suggest that F34 binding and orientation with WT-IIa are not as optimal.
4.2. Interactions Involving W60dA IIa and FXIII AP (28–41) V34, V34L, V34P, and V34F
W60d is located in the center of the thrombin 60-insertion loop and its mutants (W60dA or W60dS) markedly hinder cleavage of the Fbg Aα and Bβ chains and also the thrombin receptor PAR4 (Figures 1 and 2) [22, 47]. By contrast, W60dA thrombin has not been extensively probed with FXIII activation peptides. Philippou and coworkers [38] reported that IIa-dependent cleavage of plasma FXIII is reduced by 53% in the presence of IIa W50A (also known with chymotrypsin numbering as W60dA).
The current peptide studies provide an opportunity to determine individual kinetic parameters and examine the influences of different FXIII V34X AP substitutions (Table 2 and Figure 4). The Km values for the FXIII AP series indicate that the two physiologically observed residues (V34 and L34) have the greatest difficulty interacting effectively with the mutated thrombin active site surface, whereas, the V34P AP is more tolerated. Surprisingly, the Km value for hydrolysis of FXIII V34F AP by IIa W60dA is virtually unchanged from that observed with wild-type thrombin. For the kcat values, the V34L sequence continues to dominate followed by F34, P34, and V34 (Table 2 and Figure 4).
If a therapeutic thrombin is designed with residues missing from the upper portion of the 60s-insertion loop (Y60a-K60f), then FXIII V34F could be a promising candidate to help maintain ability to activate FXIII and still generate transglutaminase activity. Although the important central residue of the 60-insertion loop has been removed, the F34 can still find an effective binding site within the thrombin active site region. Di Cera and coworkers have already explored the influences of removing another key insertion loop called the autolysis loop and demonstrated that its loss generates a mutant thrombin with anticoagulant properties [47, 48].
4.3. Interactions Involving Y60aA IIa and FXIII AP (28–41) V34, V34L, V34P, and V34F
Y60aA is located at the start of the thrombin 60-insertion loop and its placement influences such ligands as fibrinogen Aα [47], hirudin [49], and thrombomodulin [50]. An X-ray crystal structure of FXIII AP (28–37) bound to thrombin reveals that V29 at the P9 position and V34 at the P4 position are both boarded by IIa Y60a [51]. FXIII V34 and V34L AP exhibited similar moderate increases in Km values whereas the V34P was not as well tolerated. The dramatic decreases in kcat for hydrolysis of FXIII V34 and V34L AP by thrombin Y60aA suggests that this thrombin loop mutant causes more of a kcat than a Km effect. By contrast, hydrolysis rates of cleavage of FXIII V34F AP were insufficient for determining kinetic constants. Such effects indicate that FXIII V34F AP requires the presence of the Y60a dependent environment. Overall, the Y60aA substitution would produce a thrombin with a greatly hindered ability to activate FXIII or one of its V34X mutants. Thrombin Y60aA is also known to highly diminish FpA release from fibrinogen [47].
4.4. Interactions Involving L99A IIa and FXIII AP (28–41) V34, V34L, V34P, and V34F
The L99 residue is part of the thrombin apolar binding pocket and together with Y60aA serves to cage the P2 residue of incoming substrates. A review of the X-ray crystal structure of FXIII (28–41) V34 AP indicates that the Pro36 at the P2 position interacts with thrombin L99, Y60a, and W215 [51]. Replacement of IIa L99 with the smaller amino acid alanine will generate a larger S2 pocket. Such a widening may impair stabilization of the enzyme-substrate transition state as reported previously for IIa L99G and Fbg Aα [52].
The individual FXIII AP segments responded differently to the loss of the long aliphatic chain of L99. FXIII V34 and V34L AP both exhibited the highest Km values of the IIa L99A series clearly indicating that these residues were taking advantage of this neighboring thrombin active site region to bind effectively [37]. Although a better binding surface could be found for the FXIII V34P AP residue, the substrate orientation was less well optimized for thrombin cleavage as reflected by the decrease in kcat. The P34 must contribute to proper positioning of the 34PVPR37 segment and must have difficulties dealing with the widening of this thrombin cage. The aromatic F34 is also important to consider. In contrast to kinetic studies with Y60aA, thrombin L99A could still bind and hydrolyze FXIII V34F AP with moderate changes to Km and kcat. These results may suggest that F34 is positioned closer to Y60a than L99.
4.5. Interactions Involving W215A IIa and FXIII AP (28–41) V34, V34L, V34P, and V34F
There are numerous examples where W215 serves as a platform to support one or more of the thrombin substrate/inhibitor residues [1, 53]. For example, F8 of the Fbg Aα chain exhibits a critical interaction with thrombin W215 and a Phe to Ala substitution is quite detrimental to Fbg Aα hydrolysis [13, 54]. The FXIII AP segments can handle this loss better. Removing the W215 platform causes the Km values for thrombin catalyzed hydrolysis of both FXIII V34 and V34L AP to increase an average of 9-fold and now both peptides have similar kcat values [37]. A review of X-ray crystal structures [51] suggests that the W215 plays a role in supporting the main chain backbone of FXIII V34 and V35 AP (Figure 2A). Both FXIII V34 and V34L AP can find an appropriate environment for binding with the IIa W215A active site. FXIII V34L AP has a greater dependence on the aromatic platform to orient for proper hydrolysis.
The effects of FXIII V34P and V34F on thrombin W215A binding and hydrolysis should also be evaluated. With the related murine PAR4 segment (56PNPR59), the Pro56 makes contact with thrombin residues L99, I174, and W215 [46]. This Pro56 initiates a strong edge to face interaction with thrombin W215 (Figure 2C). Disruption of such a conformational feature may help to examine why FXIII V34P AP exhibits the weakest kcat/Km of the FXIII AP series probed with IIa W215A. With FXIII V34F AP, replacement of W215 with A215 leads to virtually no change in Km relative to wild-type thrombin. Such results further support the notion that F34 is not highly dependent on W215 and may instead be taking advantage of a binding region within the start of the 60-insertion loop. These observations suggest that FXIII V34F AP might serve as a promising mutant transglutaminase that could still function in the presence of the anti-coagulant thrombin W215A.
4.6. Interactions Involving E217A IIa and FXIII AP (28–41) V34, V34L, V34P, and V34F
Thrombin E217 is part of the sodium dependent allosteric network. The E217A mutant disrupts a valuable H-bond network and destabilizes the S1 pocket [11]. Similar to W215A, E217X mutants have been screened for ability to shift thrombin from procoagulant to anticoagulant substrates [16]. For the FXIII AP series, increases in Km values were observed for FXIII V34 and V34L AP relative to wild-type thrombin, but the losses in binding interaction were not as severe as with W215A [37]. As might be predicted from earlier W215 studies, the FXIII V34P AP is the most hindered. The FXIII V34F AP is once again most tolerant following substitution at the P4 position.
4.7. Interactions Involving W215A/E217A (WE) IIa and FXIII AP (28–41) V34, V34L, V34P, and V34F
With the double mutant WE, important polar interactions involving E217, T172, and K224 are lost leading to collapse of the primary specificity pocket of thrombin [17]. In addition, the D189 that accommodates the P1 Arg is not oriented properly. An X-ray crystal structure of PPACK-WE [17, 18] suggests that an inhibitor can help correct the non-optimal arrangement of the mutant thrombin active site. However, the WE thrombin remains a poor enzyme for procoagulant substrates such as fibrinogen [12]. By contrast, Protein C function remains possible. WE has thus been tested as an anticoagulant in non-human primates and demonstrated to have desirable properties associated with potency and efficacy [55–57]. Furthermore, WE exhibits cytoprotective effects [58], acts as an antithrombotic by antagonizing GpIb interactions with von Willebrand factor [59], and suppresses development of collagen-induced arthritis in mice [60].
With the ongoing therapeutic interest in W215A, WE, and their recently improved versions [47], it will also be important to know how FXIII responds to WE. There may be scenarios where decreased levels of both fibrin formation and fibrin cross-linking are desired. By contrast, there may be cases where FXIII dependent cross-linking of other substrates may still be needed. Curiously, the Km values for WE-dependent hydrolysis of FXIII V34 and V34L AP improve significantly relative to the W215A studies [37]. The V34L Km is actually lower than the wild-type thrombin suggesting that the extra methylene group on Leu has helped this peptide segment to find an effective and likely alternative binding surface on the collapsed thrombin. The kcat values for both sequences undergo a major drop with L34 more affected than V34 [12, 37].
While the single IIa mutants containing W215A or E217A could bind and hydrolyze FXIII V34P AP, the double mutant WE caused far greater difficulties hence individual kinetic parameters could not be calculated. These results further emphasize the critical nature of W215 and the important interplay of E217 in accommodating a P34-containing FXIII AP segment in the thrombin active site. A FXIII V34P mutant could not be readily activated by the anticoagulant thrombin WE. A combination of WE thrombin and FXIII V34P could help assure that fibrin levels are greatly reduced along with a major loss in cross-linking capability.
In contrast to V34P, FXIII V34F AP could be hydrolyzed by both the single and the double mutants of thrombin (W215A, E217A, and WE). Similar kcat/Km values were observed in the presence of the thrombin mutants W215A and E217A. The double mutant WE then exhibited modest improvement in Km but a critical drop in kcat. Interestingly, the kcat/Km value for FXIII V34F AP was comparable to that of FXIII V34L AP. FXIII V34F may be a promising candidate for creating a therapeutic system in which there is reduced fibrin formation but FXIII based transglutaminase activity can be utilized. Of the FXIII V34X AP mutants, the V34F segment could best handle the loss of the W215 platform and thus could likely still be activated by thrombin. Pairing FXIII V34F with a thrombin W215X mutant may help preserve the kcat value that was diminished with the double mutant WE.
4.8. Conclusions
The individual kinetic parameters determined in this project clearly demonstrate that each FXIII V34X AP exhibits distinct features. For WT-IIa, FXIII V34P AP contributed the strongest Km whereas V34L AP succeeded the greatest in enhancing the kcat. All the FXIII APs could be accommodated by thrombin W60dA. By contrast, loss of Y60a was detrimental for FXIII V34F AP. Thrombin L99A generated a larger S2 pocket that could best be tolerated by FXIII V34F and V34P AP.
Thrombin W215A removed an important substrate platform often compromising Km values. FXIII V34P AP had great difficulties accommodating this loss whereas FXIII V34F AP provided the first example of an FXIII AP segment whose Km underwent little change in this environment. Typically, the thrombin E217A mutation was less detrimental for the kinetic properties of the FXIII V34X AP series. FXIII V34, V34L, and V34F AP each exhibited improved Km values and drastically hindered kcat values with the double thrombin mutant WE relative to the single mutant W215A. By contrast, FXIII V34P AP could not tolerate the loss of both W215A and E217A.
Significant progress is being made to design thrombin species that may be directed to procoagulant or anticoagulant functions. As these studies continue, there will be a need to know how well the FXIII AP segment can be accommodated by these changes and how the interaction can be manipulated to regulate FXIII activation. FXIII V34P AP provides an opportunity to introduce PAR4 character into a FXIII AP segment. The greatest benefit of the V34P substitution is that the pyrrolidine ring helps promote binding interactions with the wild-type thrombin active site surface. The V34P is however highly reliant on the thrombin W215 platform. Utilizing FXIII V34P in conjunction with an anticoagulant such as WE may help diminish both fibrin formation and transglutaminase based crosslinking. FXIII V34F provides an intriguing alternative candidate within the clotting environment. Similar to the common polymorphism V34L, V34F exhibits the beneficial P4 to P2 interaction with the thrombin active site surface. The F provides the added benefit that its binding interactions are well maintained with such thrombin mutants as W60dA, W215A, E217A, and WE. FXIII V34F may be a promising candidate for generating a therapeutic system that possesses some transglutaminase cross-linking ability in the presence of much reduced clotting of fibrinogen. From these different studies, new knowledge is being gained on how to design novel FXIII species whose activation may be selectively controlled by wild-type and mutant thrombins.
Highlights
FXIII V34F and V34P AP show promise for controlling thrombin activation of FXIII.
FXIII V34F AP is highly reliant on thrombin Y60a and FXIII V34P on thrombin W215A.
FXIII V34P AP with its PAR4 character promotes best binding to wild-type thrombin.
FXIII V34F AP is first activation peptide that accommodates loss of thrombin W215.
Kinetics of thrombin W215A/E217A with FXIII V34F AP is comparable to cardioprotective FXIII V34L
Acknowledgements
The authors thank Dr. Enrico Di Cera and Ms. Leslie Pelc from Saint Louis University for generously supplying the recombinant thrombins. RCL and WNG acknowledge undergraduate research fellowships from both the University of Louisville Summer Research Opportunity Program (SROP) and the Institute of Molecular Diversity and Drug Design (IMD3). Critical evaluation of the results by R. Woofter, M. Malovichko, and P. Doiphode are greatly appreciated. This work was supported by a grant from the National Institutes of Health (R01 HL68440).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The abbreviations used are: FXIII (28–41) AP, blood clotting Factor XIII Activation Peptide; FXIII V34 AP, wild type Factor XIII Activation Peptide; FXIII V34L AP, Val to Leu polymorphism of Factor XIII Activation Peptide; FXIII V34F AP, Val to Phe substitution; FXIII V34P AP, Val to Pro substitution; IIa, thrombin; ABE-I, anion-binding exosite-I; ABE-II, anion-binding exosite-II; Fbg, fibrinogen; PC, protein C; PAR1, protease activated receptor 1; PAR4, protease activated receptor 4.
Amino acids of thrombin are designated by single letter abbreviations and chymotrypsin numbering is employed. This numbering scheme requires accommodations for the thrombin insertion loops. As a result, Y60a corresponds to the tyrosine in the first residue position of the 60- or β-insertion loop. W60d corresponds to the tryptophan at the fourth or “d” position of this same loop. WT is used to define wild type thrombin. Y60aA, W60dA, L99A, W215A, and E217A correspond to mutants in which the wild-type residue is replaced with an alanine. The thrombin double mutant W215A/E217A may be abbreviated as WE thrombin.
The P nomenclature system is used to assign the individual amino acid positions on the substrate peptides. The scissile bond is designated by P1-P1′. The substrate amino acids to the left of the hydrolysis site are labeled P2, P3, P4, etc. whereas those to the right are labeled P2′, P3′, and so on. Likewise, the S nomenclature is used to assign positions on the enzyme. S1 accepts the P1 residue from the peptide substrate and so on.
References
- [1].Di Cera E. Thrombin. Mol Aspects Med. 2008;29:203–254. doi: 10.1016/j.mam.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Weisel JW. Fibrinogen and fibrin. Adv Protein Chem. 2005;70:247–299. doi: 10.1016/S0065-3233(05)70008-5. [DOI] [PubMed] [Google Scholar]
- [3].Lord ST. Molecular mechanisms affecting fibrin structure and stability. Arterioscler Thromb Vasc Biol. 2011;31:494–499. doi: 10.1161/ATVBAHA.110.213389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Muszbek L, Bagoly Z, Bereczky Z, Katona E. The involvement of blood coagulation factor XIII in fibrinolysis and thrombosis. Cardiovasc Hematol Agents Med Chem. 2008;6:190–205. doi: 10.2174/187152508784871990. [DOI] [PubMed] [Google Scholar]
- [5].Esmon CT. The protein C pathway. Chest. 2003;124:26S–32S. doi: 10.1378/chest.124.3_suppl.26s. [DOI] [PubMed] [Google Scholar]
- [6].Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3:1800–1814. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- [7].Wells CM, Di Cera E. Thrombin is a Na(+)-activated enzyme. Biochemistry. 1992;31:11721–11730. doi: 10.1021/bi00162a008. [DOI] [PubMed] [Google Scholar]
- [8].Gianni S, Ivarsson Y, Bah A, Bush-Pelc LA, Di Cera E. Mechanism of Na(+) binding to thrombin resolved by ultra-rapid kinetics. Biophys Chem. 2007;131:111–114. doi: 10.1016/j.bpc.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bode W, Turk D, Karshikov A. The refined 1.9-A X-ray crystal structure of D-Phe-Pro-Arg chloromethylketone-inhibited human alpha-thrombin: structure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure-function relationships. Protein Sci. 1992;1:426–471. doi: 10.1002/pro.5560010402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bode W. The structure of thrombin: a janus-headed proteinase. Semin Thromb Hemost. 2006;32(Suppl 1):16–31. doi: 10.1055/s-2006-939551. [DOI] [PubMed] [Google Scholar]
- [11].Pineda AO, Carrell CJ, Bush LA, Prasad S, Caccia S, Chen ZW, Mathews FS, Di Cera E. Molecular dissection of Na+ binding to thrombin. J Biol Chem. 2004;279:31842–31853. doi: 10.1074/jbc.M401756200. [DOI] [PubMed] [Google Scholar]
- [12].Cantwell AM, Di Cera E. Rational design of a potent anticoagulant thrombin. J Biol Chem. 2000;275:39827–39830. doi: 10.1074/jbc.C000751200. [DOI] [PubMed] [Google Scholar]
- [13].Arosio D, Ayala YM, Di Cera E. Mutation of W215 compromises thrombin cleavage of fibrinogen, but not of PAR-1 or protein C. Biochemistry. 2000;39:8095–8101. doi: 10.1021/bi0006215. [DOI] [PubMed] [Google Scholar]
- [14].Tsiang M, Jain AK, Dunn KE, Rojas ME, Leung LL, Gibbs CS. Functional mapping of the surface residues of human thrombin. J Biol Chem. 1995;270:16854–16863. doi: 10.1074/jbc.270.28.16854. [DOI] [PubMed] [Google Scholar]
- [15].Tsiang M, Paborsky LR, Li WX, Jain AK, Mao CT, Dunn KE, Lee DW, Matsumura SY, Matteucci MD, Coutre SE, Leung LL, Gibbs CS. Protein engineering thrombin for optimal specificity and potency of anticoagulant activity in vivo. Biochemistry. 1996;35:16449–16457. doi: 10.1021/bi9616108. [DOI] [PubMed] [Google Scholar]
- [16].Leung LL, Gibbs CS. Modulation of thrombin's procoagulant and anticoagulant properties. Thromb Haemost. 1997;78:577–580. [PubMed] [Google Scholar]
- [17].Pineda AO, Chen ZW, Caccia S, Cantwell AM, Savvides SN, Waksman G, Mathews FS, Di Cera E. The anticoagulant thrombin mutant W215A/E217A has a collapsed primary specificity pocket. J Biol Chem. 2004;279:39824–39828. doi: 10.1074/jbc.M407272200. [DOI] [PubMed] [Google Scholar]
- [18].Gandhi PS, Page MJ, Chen Z, Bush-Pelc L, Di Cera E. Mechanism of the anticoagulant activity of thrombin mutant W215A/E217A. J Biol Chem. 2009;284:24098–24105. doi: 10.1074/jbc.M109.025403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kamath P, Huntington JA, Krishnaswamy S. Ligand Binding Shuttles Thrombin along a Continuum of Zymogen-and Proteinase-like States. Journal of Biological Chemistry. 2010;285:28651–28658. doi: 10.1074/jbc.M110.154914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Trumbo TA, Maurer MC. Examining thrombin hydrolysis of the factor XIII activation peptide segment leads to a proposal for explaining the cardioprotective effects observed with the factor XIII V34L mutation. J Biol Chem. 2000;275:20627–20631. doi: 10.1074/jbc.M000209200. [DOI] [PubMed] [Google Scholar]
- [21].Trumbo TA, Maurer MC. Thrombin hydrolysis of V29F and V34L mutants of factor XIII (28-41) reveals roles of the P(9) and P(4) positions in factor XIII activation. Biochemistry. 2002;41:2859–2868. doi: 10.1021/bi0157823. [DOI] [PubMed] [Google Scholar]
- [22].Ayala YM, Cantwell AM, Rose T, Bush LA, Arosio D, Di Cera E. Molecular mapping of thrombin-receptor interactions. Proteins. 2001;45:107–116. doi: 10.1002/prot.1130. [DOI] [PubMed] [Google Scholar]
- [23].Schechter I, Berger A. On the active site of proteases. 3. Mapping the active site of papain; specific peptide inhibitors of papain. Biochem Biophys Res Commun. 1968;32:898–902. doi: 10.1016/0006-291x(68)90326-4. [DOI] [PubMed] [Google Scholar]
- [24].Scheraga HA. Chemical basis of thrombin interactions with fibrinogen. Ann N Y Acad Sci. 1986;485:124–133. doi: 10.1111/j.1749-6632.1986.tb34574.x. [DOI] [PubMed] [Google Scholar]
- [25].Kohler HP, Grant PJ. The role of factor XIIIVal34Leu in cardiovascular disease. QJM. 1999;92:67–72. doi: 10.1093/qjmed/92.2.67. [DOI] [PubMed] [Google Scholar]
- [26].Hancer VS, Diz-Kucukkaya R, Bilge AK, Ozben B, Oncul A, Ergen G, Nalcaci M. The association between factor XIII Val34Leu polymorphism and early myocardial infarction. Circ J. 2006;70:239–242. doi: 10.1253/circj.70.239. [DOI] [PubMed] [Google Scholar]
- [27].Ariens RA, Lai TS, Weisel JW, Greenberg CS, Grant PJ. Role of factor XIII in fibrin clot formation and effects of genetic polymorphisms. Blood. 2002;100:743–754. doi: 10.1182/blood.v100.3.743. [DOI] [PubMed] [Google Scholar]
- [28].Kohler HP, Stickland MH, Ossei-Gerning N, Carter A, Mikkola H, Grant PJ. Association of a common polymorphism in the factor XIII gene with myocardial infarction. Thromb Haemost. 1998;79:8–13. [PubMed] [Google Scholar]
- [29].Wartiovaara U, Perola M, Mikkola H, Totterman K, Savolainen V, Penttila A, Grant PJ, Tikkanen MJ, Vartiainen E, Karhunen PJ, Peltonen L, Palotie A. Association of FXIII Val34Leu with decreased risk of myocardial infarction in Finnish males. Atherosclerosis. 1999;142:295–300. doi: 10.1016/s0021-9150(98)00241-x. [DOI] [PubMed] [Google Scholar]
- [30].Ariens RA, Philippou H, Nagaswami C, Weisel JW, Lane DA, Grant PJ. The factor XIII V34L polymorphism accelerates thrombin activation of factor XIII and affects cross-linked fibrin structure. Blood. 2000;96:988–995. [PubMed] [Google Scholar]
- [31].Balogh I, Szoke G, Karpati L, Wartiovaara U, Katona E, Komaromi I, Haramura G, Pfliegler G, Mikkola H, Muszbek L. Val34Leu polymorphism of plasma factor XIII: biochemistry and epidemiology in familial thrombophilia. Blood. 2000;96:2479–2486. [PubMed] [Google Scholar]
- [32].Wartiovaara U, Mikkola H, Szoke G, Haramura G, Karpati L, Balogh I, Lassila R, Muszbek L, Palotie A. Effect of Val34Leu polymorphism on the activation of the coagulation factor XIII-A. Thromb Haemost. 2000;84:595–600. [PubMed] [Google Scholar]
- [33].Lim BC, Ariens RA, Carter AM, Weisel JW, Grant PJ. Genetic regulation of fibrin structure and function: complex gene-environment interactions may modulate vascular risk. Lancet. 2003;361:1424–1431. doi: 10.1016/S0140-6736(03)13135-2. [DOI] [PubMed] [Google Scholar]
- [34].Trumbo TA, Maurer MC. V34I and V34A substitutions within the factor XIII activation peptide segment (28-41) affect interactions with the thrombin active site. Thromb Haemost. 2003;89:647–653. [PubMed] [Google Scholar]
- [35].Isetti G, Maurer MC. Probing thrombin's ability to accommodate a V34F substitution within the factor XIII activation peptide segment (28-41) J Pept Res. 2004;63:241–252. doi: 10.1111/j.1399-3011.2004.00132.x. [DOI] [PubMed] [Google Scholar]
- [36].Isetti G, Maurer MC. Thrombin activity is unaltered by N-terminal truncation of factor XIII activation peptides. Biochemistry. 2004;43:4150–4159. doi: 10.1021/bi049796v. [DOI] [PubMed] [Google Scholar]
- [37].Isetti G, Maurer MC. Employing mutants to study thrombin residues responsible for factor XIII activation peptide recognition: a kinetic study. Biochemistry. 2007;46:2444–2452. doi: 10.1021/bi0622120. [DOI] [PubMed] [Google Scholar]
- [38].Philippou H, Rance J, Myles T, Hall SW, Ariens RA, Grant PJ, Leung L, Lane DA. Roles of low specificity and cofactor interaction sites on thrombin during factor XIII activation. Competition for cofactor sites on thrombin determines its fate. J Biol Chem. 2003;278:32020–32026. doi: 10.1074/jbc.M305364200. [DOI] [PubMed] [Google Scholar]
- [39].Ni F, Ripoll DR, Martin PD, Edwards BF. Solution structure of a platelet receptor peptide bound to bovine alpha-thrombin. Biochemistry. 1992;31:11551–11557. doi: 10.1021/bi00161a037. [DOI] [PubMed] [Google Scholar]
- [40].Mathews, Padmanabhan KP, Ganesh V, Tulinsky A, Ishii M, Chen J, Turck CW, Coughlin SR, Fenton JW., 2nd Crystallographic structures of thrombin complexed with thrombin receptor peptides: existence of expected and novel binding modes. Biochemistry. 1994;33:3266–3279. doi: 10.1021/bi00177a018. [DOI] [PubMed] [Google Scholar]
- [41].Gandhi PS, Chen Z, Di Cera E. Crystal structure of thrombin bound to the uncleaved extracellular fragment of PAR1. J Biol Chem. 2010;285:15393–15398. doi: 10.1074/jbc.M110.115337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nair DG, Sunilkumar PN, Sadasivan C. Modeling of factor XIII activation peptide (28-41) V34L mutant bound to thrombin. J Biomol Struct Dyn. 2008;26:387–394. doi: 10.1080/07391102.2008.10507253. [DOI] [PubMed] [Google Scholar]
- [43].Cleary DB, Trumbo TA, Maurer MC. Protease-activated receptor 4-like peptides bind to thrombin through an optimized interaction with the enzyme active site surface. Arch Biochem Biophys. 2002;403:179–188. doi: 10.1016/s0003-9861(02)00220-5. [DOI] [PubMed] [Google Scholar]
- [44].Jacques SL, Kuliopulos A. Protease-activated receptor-4 uses dual prolines and an anionic retention motif for thrombin recognition and cleavage. Biochem J. 2003;376:733–740. doi: 10.1042/BJ20030954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Nieman MT, Schmaier AH. Interaction of thrombin with PAR1 and PAR4 at the thrombin cleavage site. Biochemistry. 2007;46:8603–8610. doi: 10.1021/bi700597p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bah A, Chen Z, Bush-Pelc LA, Mathews FS, Di Cera E. Crystal structures of murine thrombin in complex with the extracellular fragments of murine protease-activated receptors PAR3 and PAR4. Proc Natl Acad Sci U S A. 2007;104:11603–11608. doi: 10.1073/pnas.0704409104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Marino F, Pelc LA, Vogt A, Gandhi PS, Di Cera E. Engineering thrombin for selective specificity toward protein C and PAR1. J Biol Chem. 2010;285:19145–19152. doi: 10.1074/jbc.M110.119875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Dang QD, Sabetta M, Di Cera E. Selective loss of fibrinogen clotting in a loop-less thrombin. J Biol Chem. 1997;272:19649–19651. doi: 10.1074/jbc.272.32.19649. [DOI] [PubMed] [Google Scholar]
- [49].Mengwasser KE, Bush LA, Shih P, Cantwell AM, Di Cera E. Hirudin binding reveals key determinants of thrombin allostery. J Biol Chem. 2005;280:26997–27003. doi: 10.1074/jbc.M502678200. [DOI] [PubMed] [Google Scholar]
- [50].Xu H, Bush LA, Pineda AO, Caccia S, Di Cera E. Thrombomodulin changes the molecular surface of interaction and the rate of complex formation between thrombin and protein C. J Biol Chem. 2005;280:7956–7961. doi: 10.1074/jbc.M412869200. [DOI] [PubMed] [Google Scholar]
- [51].Sadasivan C, Yee VC. Interaction of the factor XIII activation peptide with alpha -thrombin. Crystal structure of its enzyme-substrate analog complex. J Biol Chem. 2000;275:36942–36948. doi: 10.1074/jbc.M006076200. [DOI] [PubMed] [Google Scholar]
- [52].Rezaie AR. Role of Leu99 of thrombin in determining the P2 specificity of serpins. Biochemistry. 1997;36:7437–7446. doi: 10.1021/bi962965u. [DOI] [PubMed] [Google Scholar]
- [53].Lane DA, Philippou H, Huntington JA. Directing thrombin. Blood. 2005;106:2605–2612. doi: 10.1182/blood-2005-04-1710. [DOI] [PubMed] [Google Scholar]
- [54].Martin PD, Robertson W, Turk D, Huber R, Bode W, Edwards BF. The structure of residues 7–16 of the A alpha-chain of human fibrinogen bound to bovine thrombin at 2.3-A resolution. J Biol Chem. 1992;267:7911–7920. [PubMed] [Google Scholar]
- [55].Gruber A, Cantwell AM, Di Cera E, Hanson SR. The thrombin mutant W215A/E217A shows safe and potent anticoagulant and antithrombotic effects in vivo. J Biol Chem. 2002;277:27581–27584. doi: 10.1074/jbc.C200237200. [DOI] [PubMed] [Google Scholar]
- [56].Gruber A, Fernandez JA, Bush L, Marzec U, Griffin JH, Hanson SR, E DIC. Limited generation of activated protein C during infusion of the protein C activator thrombin analog W215A/E217A in primates. J Thromb Haemost. 2006;4:392–397. doi: 10.1111/j.1538-7836.2006.01760.x. [DOI] [PubMed] [Google Scholar]
- [57].Gruber A, Marzec UM, Bush L, Di Cera E, Fernandez JA, Berny MA, Tucker EI, McCarty OJ, Griffin JH, Hanson SR. Relative antithrombotic and antihemostatic effects of protein C activator versus low-molecular-weight heparin in primates. Blood. 2007;109:3733–3740. doi: 10.1182/blood-2006-07-035147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Feistritzer C, Schuepbach RA, Mosnier LO, Bush LA, Di Cera E, Griffin JH, Riewald M. Protective signaling by activated protein C is mechanistically linked to protein C activation on endothelial cells. J Biol Chem. 2006;281:20077–20084. doi: 10.1074/jbc.M600506200. [DOI] [PubMed] [Google Scholar]
- [59].Berny MA, White TC, Tucker EI, Bush-Pelc LA, Di Cera E, Gruber A, McCarty OJ. Thrombin mutant W215A/E217A acts as a platelet GPIb antagonist. Arterioscler Thromb Vasc Biol. 2008;28:329–334. doi: 10.1161/ATVBAHA.107.156273. [DOI] [PubMed] [Google Scholar]
- [60].Flick MJ, Chauhan AK, Frederick M, Talmage KE, Kombrinck KW, Miller W, Mullins ES, Palumbo JS, Zheng XZ, Esmon NL, Esmon CT, Thornton S, Becker A, Pelc LA, Di Cera E, Wagner DD, Degen JL. The development of inflammatory joint disease is attenuated in mice expressing the anticoagulant prothrombin mutant W215A/E217A. Blood. 2011;117:6326–6337. doi: 10.1182/blood-2010-08-304915. [DOI] [PMC free article] [PubMed] [Google Scholar]