

Abstract
The purpose of the present investigation was to prepare matrix tablets of naproxen using a hydrophobic polymer, i.e., Eudragit RLPO, RSPO, and combination of both, by wet granulation method. The tablets were further coated with different concentrations of Eudragit S-100, a pH-sensitive polymer, by dip immerse method. In vitro drug release studies of tablets were carried out in different dissolution media, i.e., 0.1 N HCl (pH 1.2), phosphate buffers pH 6.8 and 7.4, with or without rat cecal content. The swelling studies of the optimized formulation were carried out. The physicochemical parameters of all the formulations were found to be in compliance with the pharmacopoeial standards. The effect of dissolution medium on the surface of matrix tablet was determined by using Scanning Electron Microscopy technique. The stability studies of all formulations were performed as per ICH guidelines. The results demonstrated that the tablets coated with Eudragit S-100 (2% w/v) showed a sustained release of 94.67% for 24 h, but drug release increased to about 98.60% for 24 h in the presence of rat cecal content while the uncoated tablets released the drug within 5 h. With regard to release kinetics, the data were best fitted with the Higuchi model with non-Fickian drug release kinetics mechanism. The stability studies of tablets showed less degradation during accelerated and room temperature storage conditions for 6 months. The enteric-coated Eudragit S-100 coated matrix tablets of naproxen showed promising site-specific drug delivery in the colon region.
Keywords: Eudragit S-100, matrix tablets, naproxen, non-Fickian, rat cecal
INTRODUCTION
Mainly the colon specific drug delivery system has provided the importance for drugs, which are especially absorbed from colon region by preventing the degradation in upper gastrointestinal tract (GIT). Drug release at this site will ensure maximum therapeutic benefits.[1–3] Colon-targeted delivery systems are convenient for treating localized colonic diseases, i.e., Crohn's disease, ulcerative colitis, and constipation, which can be treated most efficiently by local delivery of drugs.[4] The colon-specific delivery system should protect the drug from absorption in the stomach and small intestine, thus prevent a sudden onset of drug release upon entry into less aggressive ambience of the colon. Various drug delivery approaches have been developed for colon-specific drug delivery, which include pH-sensitive system, time-dependent system, pro-drugs, and microflora-activated system to deliver anti-inflammatory agents to the sites of inflammation, and hence systemic drug absorption should be reduced as this leads to unwanted systemic side effects.[5–7]
Of all the systems formulated for colon-specific drug delivery, pH-sensitive system and time-dependent system are mostly used.[8–10] The pH variation along the GIT is based on the strategy of using pH as a trigger to achieve drug release in the colon. The high individual variability together with similarity in pH between the small intestine and the colon make the site specificity of pH-dependent system not very reliable.[11,12] Most of the strategies in time-dependent drug delivery are dependent on the principle to delay the drug release until approximate influx in the colon region. Although the relative consistency of transit times in the small intestine is because of the potentially large variation in gastric emptying time, the colon influx time cannot be exactly predicted.[12] Therefore, by suppressing drug release in the stomach and thus reducing the effect of variations in gastric residence time, appropriate integration of pH-sensitive and time-dependent systems in a single dosage form should improve colon drug delivery.
The release of water-soluble drug from a water-soluble polymeric platform is often rapid, and therefore hydrophobic polymer may be included within the matrix formulation to offer a greater control drug release.[13] Among the various polymers, Eudragit RSPO and Eudragit RLPO are the hydrophobic polymers which have been used successfully to formulate appropriate sustained release matrix formulations.[14] Matrix tablets have been developed by direct compression based on combination of hydrophobic polymers (RSPO and RLPO) and a gelling hydrophilic polymer, HPMC 60SH, to achieve a 20-h sustained release formulation of diltiazem hydrochloride. Film coated matrix tablets using Eudragit NE30D produce a delivery system in which the release of diltiazem hydrochloride is pH independent (from 1.2 to 7.4). Eudragit RSPO was employed to delay the penetration of dissolution medium into the matrix, thereby decreasing drug release rate.[15]
The pH-dependent coating polymers are methacrylic acid polymers, e.g., Eudragit S-100 is used for coating solid dosage forms because it solubilizes at pH 7. So, the main purpose was to develop a single coating layer to prevent the drug release in stomach or small intestine and to slow down the drug release in the target site (colon).[16,17] Hence, appropriate combinations of a time- and pH-dependent systems are suitable for adequate sustained release and assure more reproducible drug release behavior.
Naproxen is a member of the arylacetic acid group of nonsteroidal anti-inflammatory drugs (NSAIDs). It has analgesic and antipyretic properties. However, its ability to inhibit prostaglandin synthesis may be involved in the anti-inflammatory effect. The mechanism of action of naproxen, like that of other NSAIDs, is believed to be associated with the inhibition of cyclooxygenase (COX) activity. Inhibition of COX-1 is thought to be associated with gastrointestinal and renal toxicity, while inhibition of COX-2 provides anti-inflammatory activity.[18,19]
Taking above information in view, the purpose of present investigation was to formulate the enteric coated time release matrix tablets of naproxen using Eudragit S-100 as a pH dependent polymer.
MATERIALS AND METHODS
Materials
Naproxen was obtained as a gift sample from Micro labs, Bangalore, India. Eudragit RLPO, Eudragit RSPO, and Eudragit S-100 were obtained as gift samples from Evonik Laboratory, Mumbai, India. Polyvinyl pyrrolidone (PVP K-30) was obtained as gift samples from Helios Pharma, Baddi, India. All other chemicals and reagents used in study were of analytical grade.
Methods
Preparation of the matrix tablets
Matrix tablets containing naproxen were dry blended with appropriate quantity of polymers (Eudragit RLPO or RSPO) and granulated using 5% w/v ethanolic solution of PVP K-30 as a binder and prepared by wet granulation method. The wet mass was passed through a sieve No. 22. The wet granules were dried at 50°C for 15 min and then sieved again with No. 22 sieve. The oversized granules retained on sieve No. 22 were kept aside. This granule mixture was blended with magnesium stearate (2% w/w) and compressed using single punch tablet machine, equipped with flat-faced punches of 12 mm diameter (A. K. Industries, Nakodar, India).
Enteric coating of the matrix tablets
The naproxen matrix tablets were further coated with Eudragit S-100 solution. A different concentration (1, 1.5 and 2.5 % w/v) of coating solution of Eudragit S-100 was prepared in a mixture of Isopropyl alcohol: acetone (1:1). The coating of the matrix tablets was performed by immersion in the coating solution followed by dip coating technique.
Characterization of granules
Angle of repose
The angle of repose of granules was determined by the funnel method. The accurately weighed granules were taken in a funnel. The height of the funnel was adjusted in such a way that the tip of the funnel just touched the apex of the heap of the granules. The granules were allowed to flow through the funnel freely onto the surface. The diameter of the powder cone was measured and angle of repose was calculated using the following equation:
tanθ = h/r,
where θ = angle of repose, h = height, and r = radius.
Bulk density
Bulk density is defined as ratio of total mass of powder to the bulk volume of powder. Bulk volume is the volume occupied by a certain mass of powder when gently poured into a measuring cylinder. It was measured by pouring initially weighed powder into a measuring cylinder and the volume (bulk volume) was noted. From this, the bulk density was calculated according to the formula mentioned below. It is expressed in g/ml and is given by
Db = M/Vb,
where M is the mass of powder and Vb is the bulk volume of the powder.
Tapped density
It is the ratio of total mass of the powder to the tapped volume of the powder. Volume was measured by tapping the powder for 100 times and then the tapped volume was noted. It is expressed in g/ml and is given by
Dt = M/Vt,
where M is the mass of powder and Vt is the tapped volume of the powder.
Compressibility index
It indicates powder flow properties. It is expressed in percentage and is given by
I = (Dt – Db) × 100,
where Dt is the tapped density of the powder and Db is the bulk density of the powder.
Hausner ratio
The Hausner ratio is a number that is correlated to the flowability of powder. It is calculated by the following formula:
Hausner ratio = Dt/Db,
where Dt is the tapped density and Db is the bulk density.
A Hausner ratio greater than 1.25 is considered to be an indicator of poor flowability.
FTIR study
The Fourier transform infrared (FTIR) spectra of pure drug (naproxen), Eudragit RLPO, Eudragit S-100, physical mixture of naproxen–Eudragit RLPO, and physical mixture of naproxen–Eudragit RLPO–Eudragit S-100 were recorded using a FTIR spectrophotometer (Perkin Elmer Spectrum 400, England) according to the KBr disk technique. The smoothing of the spectra and the baseline correlation procedures were applied. The FTIR measurements were performed in the scanning range of 4000-400 cm–1 at ambient temperature.
Physicochemical evaluation of naproxen matrix tablets
Weight variation
For estimating weight variation, 20 tablets of each formulation were weighed using an electronic balance (Denver Instrument, Gottingen, Germany) and the test was performed according to the official test.
Thickness
The thickness of the tablet was measured using a Digital Vernier Calliper (Mitutoya Digimatic Calliper, Kanagawa Japan).
Hardness
The crushing strength of ten tablets was measured using Monsanto tablet hardness tester (Interlabs, Ambala, India). A tablet hardness of about 5-7 kg/cm2 is considered adequate for mechanical stability.
Friability
The friability of the tablets was determined in Roche Friabilator (Model 902, EI product, Panchkula, India). Six tablets were weighed accurately from each batch of tablets and placed in the tumbling chamber and rotated at 25 rpm for a period of 4 min. Tablets were taken and again weighed. The percentage loss was determined by using the formula:
![]()
Drug content
Tablets were finely powdered and quantity of the powder equivalent to 250 mg was accurately weighed and transferred to a volumetric flask containing 50 ml of phosphate buffer, pH 7.4. The flask was shaken to solubilize the drug and the volume was made up to 100 ml with phosphate buffer 7.4. The solution was filtered through a membrane filter (0.22 μm) and analyzed for drug content using UV/Visible spectrophotometer (AU-2701, Systronics, Mumbai, India) at 271 nm.
In vitro drug release studies
The in vitro dissolution studies were performed for the naproxen matrix tablet using USP II dissolution apparatus (Lab India, DS 8000, Mumbai, India) in 900 ml dissolution medium at 100 rpm, 37°C ± 0.5°C. The dissolution media with pH 1.2, phosphate buffer pH 6.8, and phosphate buffer pH 7.4 were used in order to simulate the pH change along the GIT. The in vitro drug release experiments were performed at pH 1.2 for 2 h. Then pH 1.2 buffer was replaced with pH 6.8 phosphate buffer dissolution media for 3 h. After performing the experiments for 3 h with pH 6.8 phosphate buffer, the dissolution media were replaced finally with phosphate buffer pH 7.4 for the next 19 h. At regular time intervals, samples were withdrawn from the dissolution media and filtered with Whatman filter paper (0.22 μm). The absorbance was measured using UV/Visible spectrophotometer (AU-2701, Systronics) at 271 nm. The graph was plotted against cumulative percentage drug release versus time. The experiment was done in triplicate and data were expressed in mean ± SD.[20]
Preparation of rat cecal content for dissolution studies
The in vitro drug release study was also performed in presence of rat cecal content medium to simulate the human intestinal microflora. Male Wistar rats weighing 100-150 g maintained on a normal diet were used for the study. Using carbon dioxide (CO2), the rats were asphyxiated. To stimulate enzymes which specifically hydrolyze Eudragit RLPO, enzyme induction was done. For enzyme induction, 2 ml of a 1% w/v dispersion of Eudragit RLPO in water was administered to the rats daily for 7 days. The abdomens were cut, the ceci were isolated, ligated at both ends, and dissected. About 8 g of the cecal content was immediately weighed and transferred into 200 ml of pH 7.4 phosphate buffer to provide 4% (w/v) dilution. Carbon dioxide (CO2) was continuously passed through pooled content so as to keep the environment in anaerobic condition. All the experimental protocols were approved by Institutional Animal Ethical Committee constituted as per the Committee for the Purpose of Control and Supervision on Experiments on Animals (IAEC proposal no. 06/10, CPCSEA registration no. IAEC/CCP/11/PR-11).[21]
In vitro drug release studies in the presence of rat cecal content
The drug release studies were carried out using USP dissolution rate test apparatus in 900 ml dissolution medium at 100 rpm, 37°C ± 0.5°C, with slight modification, as previously discussed by Mundargi et al.[21] After completing the dissolution studies in pH 1.2 phosphate buffer for 2 h, the matrix tablet was transferred to pH 6.8 phosphate buffer for 3 h. After placing matrix tablet in pH 6.8 phosphate buffer, it was transferred to 200 ml phosphate buffer pH 7.4 diluted with 4% (w/v) rat cecal medium for 19 h. As cecum is naturally anaerobic, the experiment was carried out with continuous CO2 supply into the beakers. At regular time intervals, 2 ml of the dissolution samples was withdrawn and replaced with 2 ml of 4% (w/v) rat cecal medium. The absorbance was measured using UV/Visible spectrophotometer (AU-2701, Systronics) at 271 nm.[21]
Kinetic analysis of dissolution data
To study the mechanism of drug release from the matrix tablets, the release data were fitted to zero-order, first-order, and Higuchi equation. The dissolution data were also fitted to the well-known exponential equation (Korsmeyer–Peppas equation), which is often used to describe the drug release behavior from polymeric systems:
log Mt/M∞ = log k + n log t,
where Mt is the amount of drug released at time t, M ∞ is the amount of drug released after infinite time, k is a release rate constant incorporating structural and geometric characteristics of the tablet, and n is the diffusional exponent indicative of the mechanism of drug release.
To determine the release exponent for different batches of matrix tablet, the log value of percentage drug dissolved was plotted against log time for each batch. A value of n = 0.45 indicates > 0.45 Fickian (case I) release, <0.89 indicates non-Fickian (anomalous) release, and n > 0.89 indicates super case II type of release. Case II generally refers to the erosion of the polymeric chain, and anomalous transport (non-Fickian) refers to a combination of both diffusion and erosion controlled drug release.[21]
Swelling studies
The swelling studies were carried out of optimized formulations, i.e., from F7 to F9. These tablets were glued to a piece of glass slide. The glass slide and tablets were weighed initially (W1). The tablets were placed in different dissolution media (pH 1.2, 6.8, and 7.4) using USP type II dissolution apparatus (Lab India, DS 8000, Mumbai, India) in 900 ml dissolution medium at 100 rpm, 37°C ± 0.5°C. The swollen tablets were taken out at predetermined time intervals, and using tissue paper, excess of water on their surface was carefully removed and tablets were reweighed (W2). The study was carried out over a period of 12 h and the tablets were observed for rupturing. The % swelling index was calculated using the following formula:[21]
![]()
Scanning electron microscopy
The morphology and surface appearance of naproxen matrix tablets before and after the release test were examined using a scanning electron microscope (JSM 6100, JEOL, Tokyo, Japan) equipped with an image analysis system. Prior to examination, samples were gold sputter-coated to render them electrically conductive.
Stability Study
The stability study was conducted according to International Conference on Harmonisation (ICH) guidelines. All the formulations were stored in aluminum packaging laminated with polyethylene (cellophane packets) and kept in humidity chamber (Oswal Scientific, Chandigarh, India) at 30°C ± 2°C/65% ±5% RH (room temperature studies) and 40°C ± 2°C/75% ±5% RH (accelerated temperature studies) for 6 months. The tablets were analyzed after 0 day, 1 month, 2 months, 3 months, and 6 months. At the end of the study period, the tablets were observed for the change in physical appearance, color, and drug content.[21]
RESULTS AND DISCUSSION
Characterization of Granules
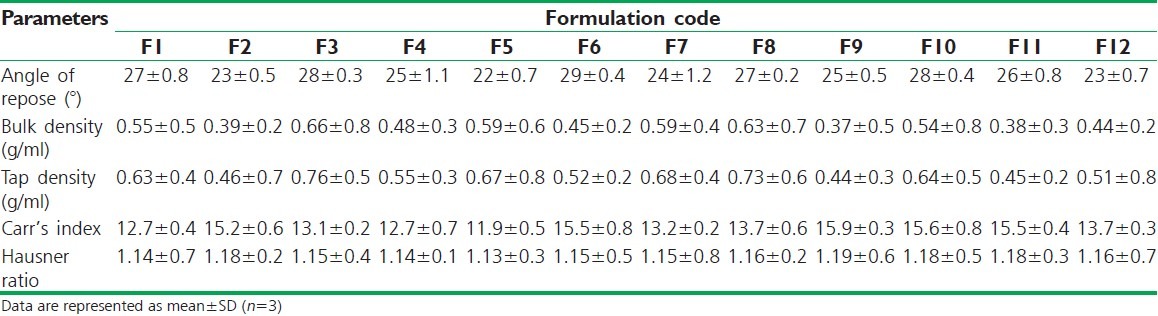
The granules for naproxen matrix tablets were prepared by wet granulation method according to the formula shown in Table 1. The granules were characterized with respect to angle of repose, bulk density, tapped density, Carr's index, and Hausner ratio. The parameters for evaluation of granules are depicted in Table 2. The angle of repose of different formulation batches from F1 to F12 was found to be from 22° ± 0.7° to 29° ± 0.4°. The angle of repose was less than 30° for all the formulation batches of granules, indicating good flow behavior. Similarly, bulk density and tap density of all the formulation batches from F1 to F12 were found to be from 0.37 ± 0.5 to 0.66 ± 0.8 g/ml and from 0.44 ± 0.3 to 0.76 ± 0.5 g/ml, depicting good flow properties of the granules. The Carr's index of all formulation batches was in the acceptable range from 11.9 ± 0.5 to 15.9 ± 0.3. The Hausner ratio of all formulation batches from F1 to F12 was found to be from 1.13 ± 0.3 to 1.19 ± 0.6. The Hausner ratio less than 1.25 indicates good flowability.[26]
Table 1.
Formulation composition of matrix tablets of naproxen
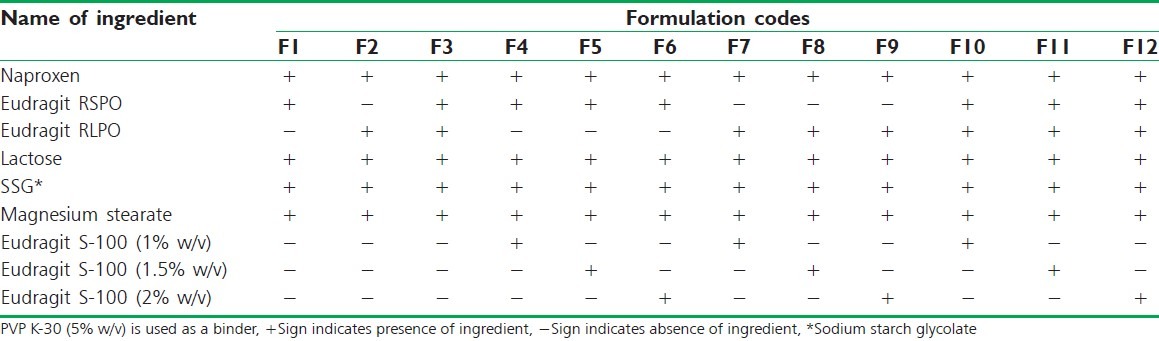
Table 2.
Characterization of naproxen granules
FTIR Study
The FTIR spectra of naproxen, Eudragit RLPO, Eudragit S-100, and physical mixture are shown in Figure 1. FTIR spectrum of naproxen showed characteristic peaks at 1725.6 cm–1 due to the carboxyl group, at 1176.5 cm-1 due to presence of C–O stretch, the C = C (aromatic stretch) at 1602.9 cm–1, –CH3 bend at 1457.2 cm–1, and due to the presence of O–H (carboxylic acid), the peak was found at 2968.1 cm–1. FTIR spectrum of Eudragit S-100 showed the peak at 2953.9 cm–1 due to presence of O–H (carboxylic acid), at 1450.7 cm–1 due to -CH3 bend, and at 1731.2 cm–1 due to the presence of C = O (ester). FTIR spectrum of Eudragit RLPO showed the peak at 3432.1 cm–1 due to the presence of tertiary amine, at 1731.4 cm–1 due to the presence of C = O (ester), and at 1450.2 cm–1 due to –CH3 bend. FTIR spectrum of the physical mixture of drug and polymer showed the peak at 1728.6 cm–1 due to the presence of C = O (ester), at 1174.8 cm–1 due to the presence of C-O stretch, the C = C (aromatic stretch) at 1604.1 cm–1, –CH3 bend at 1453.1 cm–1, at 1728.6 cm–1 due to the carboxyl group, and at 2960.9 cm–1 due to presence of O–H (carboxylic acid). Therefore, FTIR study concluded that no interaction occurred between the drug and polymer.
Figure 1.
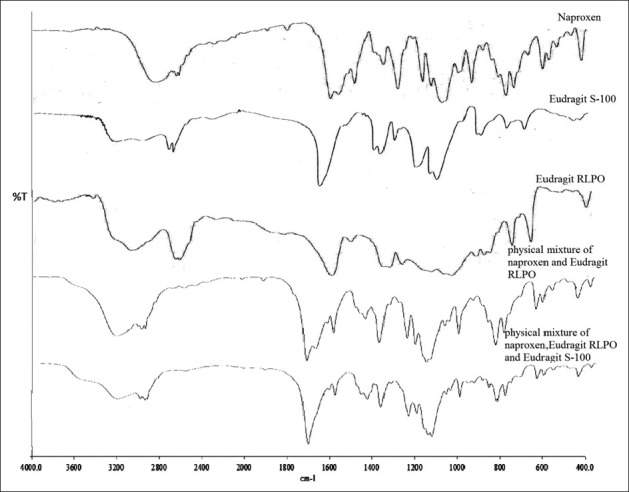
FTIR spectra of naproxen, Eudragit S-100, Eudragit RLPO, physical mixture of naproxen and Eudragit RLPO, and physical mixture of naproxen, Eudragit RLPO, and Eudragit S-100
Physicochemical Evaluation of Naproxen Matrix Tablets
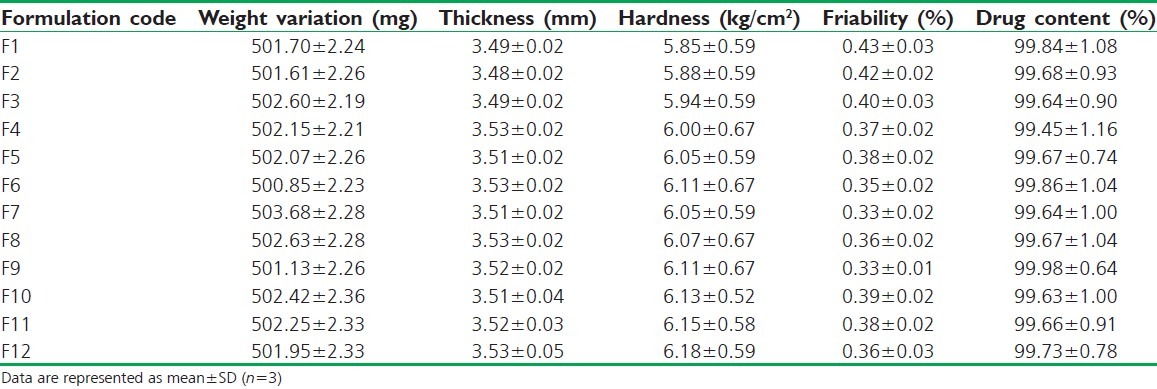
The tablets of different formulations with and without coating were subjected to various evaluation tests of weight variation, thickness, hardness, friability, and drug content. In Table 3 is presented the results of physicochemical evaluation of all batches of naproxen matrix tablets. The weight variation of all formulated batches, i.e., from F1 to F12, was found to be from 500.85 ± 2.23 to 503.68 ± 2.28 mg. The weight variation of all formulation batches was less than 4% which was in acceptable range. The thickness of formulation F1-F3 was found to be between 3.48 ± 0.02 and 3.49 ± 0.02 mm and F4-F12 was between 3.51 ± 0.02 and 3.53 ± 0.05 mm. The hardness of formulation F1-F3 was found to be from 5.85 ± 0.59 to 5.94 ± 0.59 kg/cm2 and F4-F12 was found to be from 6.00 ± 0.67 to 6.18 ± 0.59 kg/cm2. The thickness and hardness of tablets in formulation F4-F12 increased due to coating of the Eudragit S-100 over the entire surface of the tablet than the uncoated tablets from formulation from F1 to F3. The same effect of Eudragit S-100 was reported in previous literature.[27] The hardness was found to be in acceptable range. The friability of all tablets in formulation was in acceptable range of less than 1%, ranging from 0.33% ±0.01% to 0.43% ±0.03%. The drug content of all formulation batches was found to be between 99.45% ±1.16% and 99.98% ±0.64%. The maximum drug content was obtained for formulation F9 which consists of Eudragit RLPO matrix coated with Eudragit S-100 (2% w/v). The Eudragit RLPO is more permeable to the dissolution media which might result in increased drug content.
Table 3.
Physicochemical evaluation of naproxen matrix tablets
In vitro Drug Release Studies
The in vitro drug release studies of the of core tablets, i.e., F1-F3, showed 95.48% ±0.95%, 99.39% ±0.82%, and 97.37% ±0.70% drug release within 5 h in pH 7.4 phosphate buffer. Therefore, different dissolution media (pH 1.2, 6.8, 7.4) were used for the remaining formulation batches of matrix tablets to perform in vitro drug release studies. The formulation containing Eudragit RSPO showed decreased drug release at the target site (colon) at the end of 24 h due to less permeability in media.[14] The formulation containing Eudragit RLPO showed maximum drug release before reaching at the target site (colon), due to more permeability in media. But the formulations containing Eudragit RSPO and RLPO in combination showed maximum drug release in stomach and small intestine, and decreased drug release at the target site (colon) at the end of 24 h due to the presence of Eudragit RSPO as a retardant [Figure 2].[15] Therefore, it is observed that for sustained drug release, there should be coating of Eudragit S-100 with different coating concentrations (1% w/v, 1.5% w/v, and 2% w/v) over the entire surface area of the tablets [Table 1].
Figure 2.
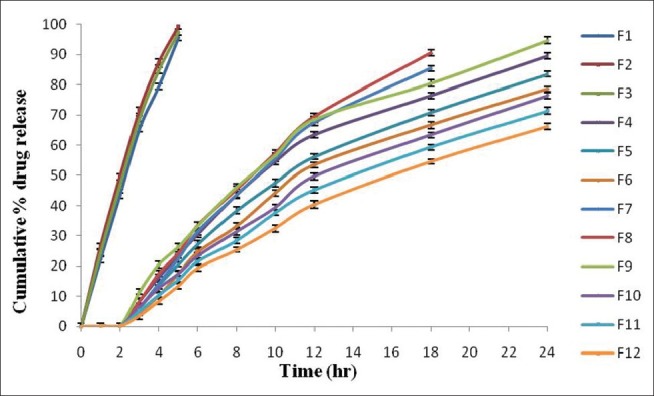
Comparative in vitro percent drug release (%) of all formulations of naproxen matrix tablets with or without Eudragit S-100 coating. Data are represented as mean ± SD (n = 3)
The in vitro drug release of formulations F1 and F2 was found to be 95.48% ±0.95% and 99.39% ±0.82%, respectively, within 5 h in pH 7.4 phosphate buffer [Figure 3]. The drug release from formulation F1 was less due to the presence of Eudragit RSPO, which has low permeability as compared with Eudragit RLPO, which has higher permeability in the dissolution media. Among the formulations F1-F3, the formulation containing the combination of Eudragit RSPO and RLPO (F3) showed less drug release, i.e., 97.37% ±0.70%, within 5 h in pH 7.4 phosphate buffer due to the presence of Eudragit RSPO, which may act as a release retardant polymer, as compared to formulations F1 and F2. The coating of the tablet using different coating concentrations (1% w/v, 1.5% w/v, and 2% w/v) of Eudragit S-100, pH-dependent methacrylic acid copolymer, increased the lag time and provided entire coat that allowed the tablet to pass intact through the stomach and small intestine to target in the colon. Therefore, different formulation batches were coated, as it was not possible to control uncoated tablet for longer duration in the dissolution media.
Figure 3.
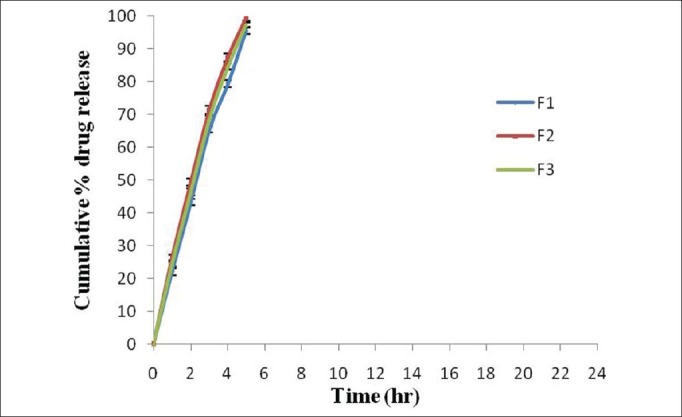
Comparative in vitro drug release (%) of naproxen matrix tablet (core tablet). Data are represented as mean ± SD (n = 3)
The in vitro drug release studies of different formulations F4-F6 containing Eudragit RSPO in matrix tablets with different coating concentrations (1% w/v, 1.5% w/v, and 2% w/v) of Eudragit S-100 were carried out in different dissolution media (pH 1.2, 6.8, and 7.4). The formulations F4-F6 showed 89.44% ± 0.93%, 83.54% ±0.97%, and 78.52% ±0.98% drug release within 24 h [Figure 4]. The lag time for drug release increased with increase in the coating concentration of Eudragit S-100. With increase in the Eudragit S-100 coating concentration, the coat became more impermeable which resulted in decrease in drug release in the dissolution medium. The other reason may be the presence of Eudragit RSPO which is less swellable due to the presence of least amount of quaternary ammonium groups in the structure and less impermeable to the dissolution media due to which the drug release decreased.[28,29]
Figure 4.
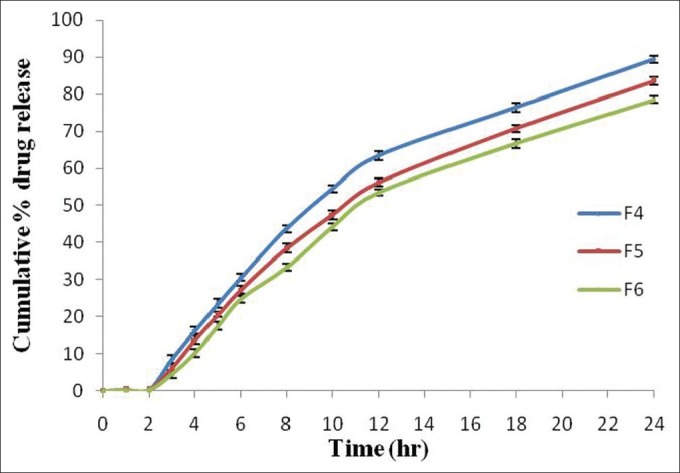
Comparative in vitro drug release profile (%) of Eudragit RSPO based naproxen matrix tablet (Eudragit S-100 coated). Data are represented as mean ± SD (n = 3)
The in vitro drug release studies of formulations F7-F9 were carried out in pH 1.2, pH 6.8, and pH 7.4 dissolution media containing Eudragit RLPO in matrix tablets with different coating concentrations (1% w/v, 1.5% w/v, and 2% w/v) of Eudragit S-100. The formulations F7 and F8 showed 85.34% ±0.97% and 90.45% ±1.15% drug release within 18 h and formulation F9 showed 94.67% ±1.17% drug release within 24 h [Figure 5]. The dissolution studies of formulation batches F7 and F8 showed maximum drug release in 18 h, and after that the drug release started decreasing, therefore the study was carried out till 18 h. There was no effect of Eudragit S-100 coating, and lag time also decreased as most of the drug was released in stomach and small intestine and less amount of drug was released in the colon site. But formulation F9 showed sustained drug release till 24 h due to increase in the coating concentration of Eudragit S-100. The coating concentration was optimized to provide suitable coating over the surface of the tablet to pass thorough the stomach and small intestine without any drug release and to target to colon site. The Eudragit RLPO was more sellable due to presence of higher number of quaternary ammonium groups that could be contribute to increased drug release from the tablets.[30]
Figure 5.
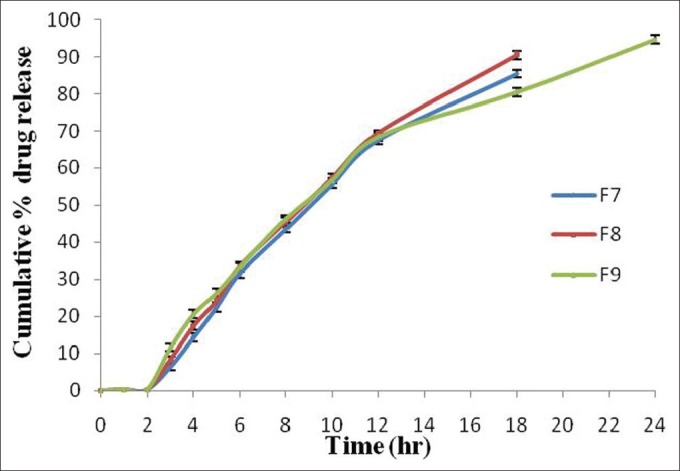
Comparative in vitro drug release profile (%) of Eudragit RLPO based naproxen matrix tablets (Eudragit S-100 coated). Data are represented as mean ± SD (n = 3)
In vitro drug release studies of optimized formulations containing both Eudragit RLPO and RSPO with coating of different concentration Eudragit S-100 (F10 to F12) were performed in different dissolution media (pH 1.2, 6.8 and 7.4) in the presence of rat cecal content. The formulations F10-F12 showed 76.37% ±1.04%, 71.33% ±1.11%, and 66.22% ±0.95% drug release within 24 h [Figure 6]. The lag time for drug release increased with increase in the coating concentration of Eudragit S-100 as the coat became more impermeable which resulted in decreased drug release. The other reason may be the presence of less quaternary ammonium groups in the polymer structure and retardant property of Eudragit RSPO in the dissolution media.[31]
Figure 6.
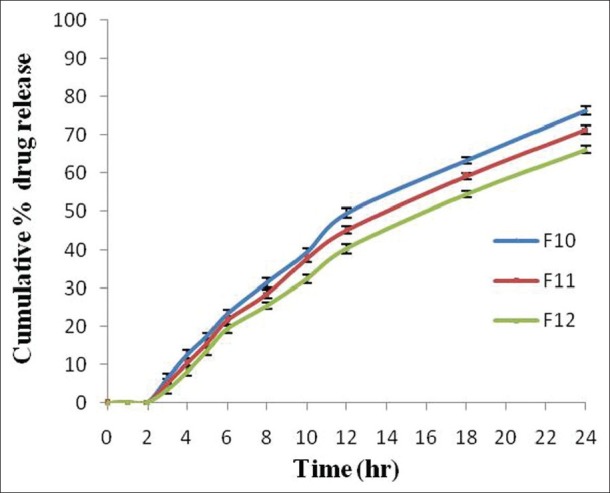
Comparative in vitro drug release profile of both Eudragit RSPO and RLPO (1:1 ratio) based naproxen (Eudragit S-100 coated). Data are represented as mean ± SD (n = 3)
In vitro Drug Release Studies in the presence of Rat Cecal Content
In vitro drug release studies of optimized formulations containing Eudragit RLPO with coating of different concentration Eudragit S-100 (F7 to F9) were performed in different dissolution media (pH 1.2, 6.8 and 7.4) in the presence of rat cecal content. The formulations F7 and F8 showed 90.31% ±1.31% and 95.49% ±1.12% drug release within 18 h and formulation F9 showed 98.60% ±1.17% drug release within 24 h [Figure 7]. After placing the swollen tablet in pH 1.2 for 2 h and in pH 6.8 for 3 h, it was transferred to 4% w/v rat cecal medium present in phosphate buffer pH 7.4 for 19 h to carry out dissolution studies. The formulation F9 showed maximum drug release of 98.60% ±1.17% in rat cecal medium, as compared to lower drug release of about 94.67% ±1.17% without rat cecal medium. This shows that due to the presence of bacteria in it, coating material was eroded from the surface of tablet quickly and maximum drug release was observed.[32]
Figure 7.
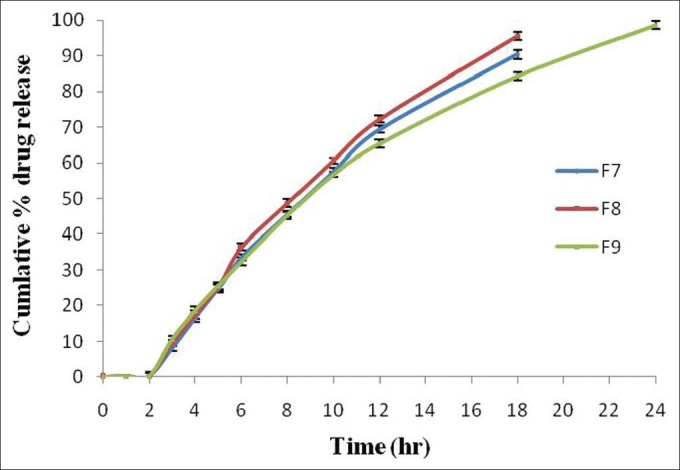
Comparative in vitro drug release profile of optimized formulations (F7, F8, and F9) in the presence of rat cecal content (4% w/v). Data are represented as mean ± SD (n = 3)
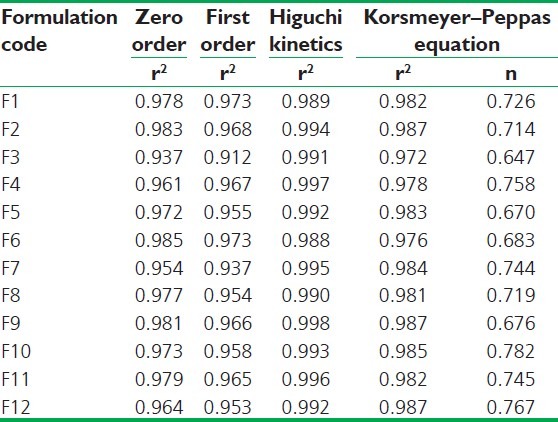
Kinetic Analysis of Dissolution Data
The mechanism and kinetics of drug release of naproxen was determined by the application of Korsmeyer-Peppas model, Higuchi model, zero-order kinetics, and first-order kinetics, as shown in Table 4. The different formulations followed Higuchi model as their r2 values were between 0.988 and 0.998. The mechanism of drug release is non-Fickian diffusion (anomalous transport), since t hey fitted well with Korsmeyer–Peppas models (r2 in the range of 0.972-0.987 with n value less than 1). This indicates the drug release depends on swelling and diffusion mechanism of release.[33]
Table 4.
Regression coefficient (r2) values of drug release data obtained from various kinetic models and “n” value (diffusional exponent) according to Korsmeyer–Peppas model
Swelling Studies
The swelling studies of optimized formulations F7-F9 containing Eudragit RLPO in the same proportion with different coating concentrations (1% w/v, 1.5% w/v, and 2% w/v) of Eudragit S-100 were carried out in different dissolution media (pH 1.2, 6.8, and 7.4). The swelling study of formulation F7 of the matrix tablets with a coating concentration of about 1% w/v showed that coating layer was removed in an earlier stage during the transfer to pH 7.4 dissolution media, with low swelling index as well as drug release, and the tablet got burst. But formulation F8 with increase in coating concentration of Eudragit S-100 (1.5% w/v) increased the lag time and showed high swelling index as well as drug release, compared to formulation 7, but at the end of 12 h, the tablet got ruptured. At last, in formulation F9, with increase in coating concentration of Eudragit S-100 (2% w/v), there was increase in lag time and drug release with maximum swelling index as the tablet got ruptured at the end of 12 h. From this, it is concluded that with maximum coating level of Eudragit S-100 (2% w/v) was considered as an optimized formulation as it showed maximum swelling index as well as drug release due to the presence of RLPO polymer which is more swellable to the dissolution media. The swelling studies of optimized formulation are shown in Figure 8.[34,35]
Figure 8.
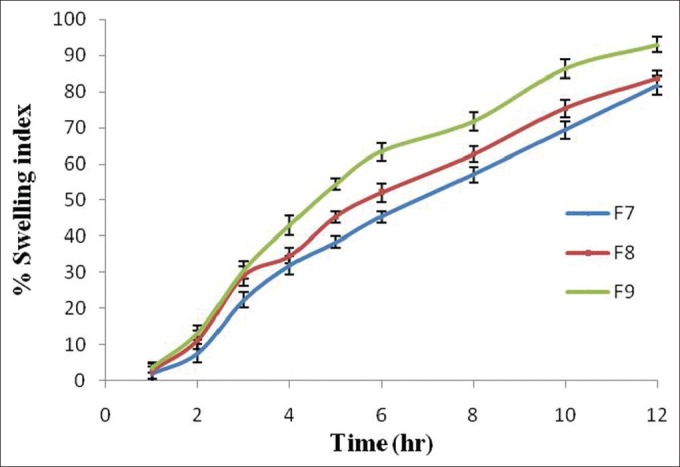
Swelling index (%) of optimized formulation of naproxen tablets. Data are represented as mean ± SD (n = 3)
Scanning Electron Microscopy of Matrix Tablets
Scanning Electron Microscopy (SEM) analyses were performed on the optimized formulation (F9) with different dissolution media (pH 1.2, 6.8, and 7.4). SEM photomicrographs of the outer surface of the coated matrix tablet showed a homogenous and quite compact structure without any pore before dissolution, as depicted in Figure 9a. The coated matrix tablet placed in pH 1.2 for 2 h showed the presence of numerous pores and channels indicating limited swelling of the matrix tablet; drug diffusion could occur thorough a porous network filled by the solvent that penetrated into the matrix [Figure 9b]. Then, the coated matrix tablet was transferred to pH 6.8 for 3 h in which the medium penetrated through channels and pores during the release test, mainly due to Eudragit RLPO which was more permeable to the dissolution media. This caused the coating to break, producing deep fracture and irregular cavities which appeared evident on the tablet surface [Figure 9c]. Then, at last, the coated matrix tablets transferred in pH 7.4 for 19 h showed increase in diameter of the pores and swelling of the tablets through the matrix at different time intervals. The presence of both swelling and diffusion mechanism indicates formation of pores, and gelling structure on the tablet surface showed sustained drug release from coated naproxen matrix tablets [Figure 9d].[36]
Figure 9.

Scanning electron microscopy of optimized formulation (F9) after (a) 0 h, (b) 2 h, (c) 3 h, and (d) 19 h of dissolution study in pH 1.2, 6.8 and 7.4 media
Stability Study
The stability studies of all formulations for targeting naproxen to the colon were carried out at 30°C ± 2°C/65% ±5% RH (room temperature studies) and at 40°C ± 2°C/75% ±5% RH (accelerated temperature studies) for 6 months as per ICH guidelines. After storage for 0 day, 1 month, 2 months, 3 months, and 6 months, the tablets were observed for physical appearance and color change and were subjected to drug content study. The insignificant change in the physical appearance, color, and drug content after storage, when compared to the tablets of the same batch before storage indicated that formulation batch F9 is stable and considered to be optimized which could provide a good shelf life for 6 months. The graphical representation of drug content of all formulation batches of naproxen matrix tablets at room temperature and accelerated temperature is shown in Figure 10.[37]
Figure 10.
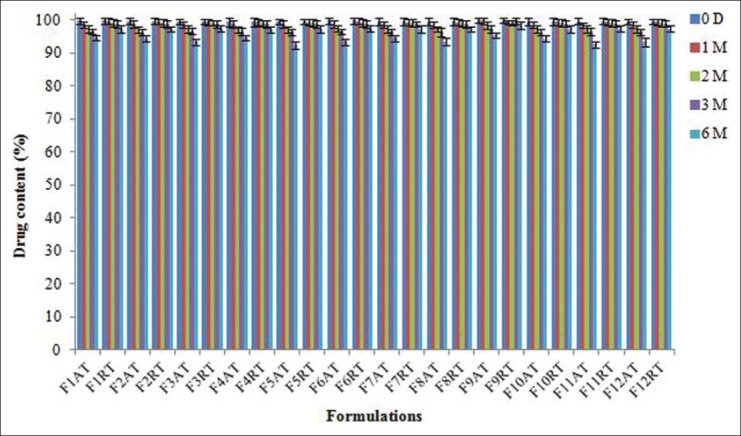
Stability studies of all formulated naproxen matrix tablets under accelerated and room temperature storage conditions (AT = accelerated temperature, RT = room temperature). Data are represented as mean ± SD (n = 3)
CONCLUSION
It is concluded from the present study that appropriate combination of a pH-dependent polymer (Eudragit S-100) with a pH-independent polymer (Eudragit RLPO) was suitable for adequately sustained drug release and to protect naproxen from being released in the upper region of the GI system. The in vitro drug release studies and SEM studies indicate that the optimized formulation was a promising system targeting naproxen to the colon. The drug release pattern from all formulations was best fitted with Higuchi release model and the drug release mechanism was followed Korsmeyer-Pappas equation with non-Fickian diffusion kinetics.
ACKNOWLEDGEMENTS
The authors are thankful to Micro Lab. Pvt. Ltd., Bangalore, India. for providing naproxen as a kind gift sample. The authors are also grateful to Dr. Madhu Chitkara, Vice Chancellor, Chitkara University, Patiala, Punjab, India, and Dr. Ashok Chitkara, Chairman, Chitkara Educational Trust, Chandigarh, India, for support and institutional facilities.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
REFERENCES
- 1.Ashford M, Fell JT. Targeting drugs to the colon: delivery systems for oral administration. J Drug Target. 1994;2:241–57. doi: 10.3109/10611869408996806. [DOI] [PubMed] [Google Scholar]
- 2.Rubinstein A. Approaches and opportunities in colon-specific drug delivery. Crit Rev Ther Drug Carrier Syst. 1995;12:101–49. doi: 10.1615/critrevtherdrugcarriersyst.v12.i2-3.10. [DOI] [PubMed] [Google Scholar]
- 3.Watts PJ, Lllum L. Colonic Drug Delivery. Drug Dev Ind Pharm. 1997;23:893–913. [Google Scholar]
- 4.Kinget R, Kalala W, Vervoort L, Van Der Mooter G. Colonic drug targeting. J Drug Target. 1998;6:129–49. doi: 10.3109/10611869808997888. [DOI] [PubMed] [Google Scholar]
- 5.Niwa K, Takaya T, Morimoto T, Takada K. Preparation and evaluation of a time-controlled release capsule made of ethylcellulose for colon delivery of drugs. J Drug Target. 1995;3:83–9. doi: 10.3109/10611869509059209. [DOI] [PubMed] [Google Scholar]
- 6.Chourasia MK, Jain SK. Pharmaceutical approaches to colon targeted drug delivery systems. J Pharm Pharm Sci. 2003;6:33–66. [PubMed] [Google Scholar]
- 7.Mura P, Maestrelli F, Cirri M, González Rodríguez ML, Rabasco Alvarez AM. Development of enteric-coated pectin-based matrix tablets for colonic delivery of theophylline. J Drug Target. 2003;11:365–71. doi: 10.1080/10611860310001639130. [DOI] [PubMed] [Google Scholar]
- 8.Asghar LF, Azeemuddin M, Jain V, Chandran S. Design and in vitro evaluation of formulations with pH and transit time controlled sigmoidal release profile for colon-specific delivery. Drug Deliv. 2009;16:205–13. doi: 10.1080/10717540902823960. [DOI] [PubMed] [Google Scholar]
- 9.Hu Z, Shimokawa T, Ohno T, Kimura G, Mawatari SS, Kamitsuna M, et al. Characterization of norfloxacine release from tablet coated with a new pH-sensitive polymer, P-4135F. J Drug Target. 1999;7:223–32. doi: 10.3109/10611869909085505. [DOI] [PubMed] [Google Scholar]
- 10.Qi M, Wang P, Wu D. A novel pH- and time-dependent system for colonic drug delivery. Drug Dev Ind Pharm. 2003;29:661–7. doi: 10.1081/ddc-120021315. [DOI] [PubMed] [Google Scholar]
- 11.Ashford M, Fell JT, Attwood D, Woodhead PJ. An in vitro investigation into the suitability of pH-dependent polymers for colonic targeting. Int J Pharm. 1993;91:241–5. [Google Scholar]
- 12.Yang L, Chu JS, Fix JA. Colon-specific drug delivery: new approaches and in vitro/in vivo evaluation. Int J Pharm. 2002;235:1–15. doi: 10.1016/s0378-5173(02)00004-2. [DOI] [PubMed] [Google Scholar]
- 13.Abu-Diak OA, Andrews GP, Jones DS. Siepmann J, Siegel RA, Rathbone MJ, editors. Hydrophobic polymer of pharmaceutical significance. Fundamentals and applications of controlled release drug delivery. 2011:49–50. [Google Scholar]
- 14.Patra CN, Kumar AB, Pandit HK, Singh SP, Devi MV. Design and evaluation of sustained release bilayer tablets of propranolol hydrochloride. Acta Pharm. 2007;57:479–89. doi: 10.2478/v10007-007-0038-0. [DOI] [PubMed] [Google Scholar]
- 15.Boyapally H, Nukala RK, Douroumis D. Development and release mechanism of diltiazem HCl prolonged release matrix tablets. Drug Deliv. 2009;16:67–74. doi: 10.1080/10717540802586220. [DOI] [PubMed] [Google Scholar]
- 16.Khan MZ, Stedul HP, Kurjaković N. A pH-dependent colon.targeted oral drug delivery system using methacrylic acid copolymers. II. Manipulation of drug release using Eudragit L100 and Eudragit S100 combinations. Drug Dev Ind Pharm. 2000;26:549–54. doi: 10.1081/ddc-100101266. [DOI] [PubMed] [Google Scholar]
- 17.Asghar LF, Chandran S. Design and evaluation of matrices of Eudragit with polycarbophil and carbopol for colon-specific delivery. J Drug Target. 2008;16:741–57. doi: 10.1080/10611860802473345. [DOI] [PubMed] [Google Scholar]
- 18.Sweetman SC. The Complete Drug Reference. 36th ed. London, Chicago: Pharmaceutical Press; 2009. Martindale; pp. 92–3. [Google Scholar]
- 19.Piao ZZ, Lee MK, Lee BJ. Colonic release and reduced intestinal tissue damage of coated tablets containing naproxen inclusion complex. Int J Pharm. 2008;350:205–11. doi: 10.1016/j.ijpharm.2007.08.044. [DOI] [PubMed] [Google Scholar]
- 20.Veerareddy PR, Manthri RP. Formulation and evaluation of compression coated piroxicam tablets for colon specific drug delivery. Acta Pharm Sci. 2010;52:281–94. [Google Scholar]
- 21.Mundargi RC, Patil SA, Agnihotri SA, Aminabhavi TM. Development of polysaccharide-based colon targeted drug delivery systems for the treatment of amoebiasis. Drug Dev Ind Pharm. 2007;33:255–64. doi: 10.1080/03639040600897127. [DOI] [PubMed] [Google Scholar]
- 22.Ahmadi F, Varshosaz J, Emami J, Tavakoli N, Minaiyan M, Mahzouni P, et al. Preparation and in vitro/in vivo evaluation of dextran matrix tablets of budesonide in experimental ulcerative colitis in rats. Drug Deliv. 2011;18:122–30. doi: 10.3109/10717544.2010.520352. [DOI] [PubMed] [Google Scholar]
- 23.Lopes CM, Sousa Lobo JM, Costa P, Pinto JF. Directly compressed mini matrix tablets containing ibuprofen: Preparation and evaluation of sustained release. Drug Dev Ind Pharm. 2006;32:95–106. doi: 10.1080/03639040500388482. [DOI] [PubMed] [Google Scholar]
- 24.Wu B, Chen Z, Wei X, Sun N, Lu Y, Wu W. Biphasic release of indomethacin from HPMC/pectin/calcium matrix tablet: I.Characterization and mechanistic study. Eur J Pharm Biopharm. 2007;67:707–14. doi: 10.1016/j.ejpb.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 25.Krishnaiah YS, Satyanarayana V, Dinesh Kumar B, Karthikeyan RS. In vitro drug release studies on guar gum-based colon targeted oral drug delivery systems of 5-fluorouracil. Eur J Pharm Sci. 2002;16:185–92. doi: 10.1016/s0928-0987(02)00081-7. [DOI] [PubMed] [Google Scholar]
- 26.Amrutkar JR, Gattani SG. Chitosan-chondroitin sulfate based matrix tablets for colon specific delivery of indomethacin. AAPS Pharm Sci Tech. 2009;10:670–7. doi: 10.1208/s12249-009-9253-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shah R, Patel S, Patel H, Pandey S, Shah S, Shah D. Formulation development of carvedilol compression coated tablet. Pharm Dev Tech. 2011;16:1–10. doi: 10.3109/10837450.2011.598167. [DOI] [PubMed] [Google Scholar]
- 28.Patel MM, Patel SL, Bhadani MN, Shah TJ, Amin AF. A synchronous colon-specific drug delivery system for orally administered mesalamine. Acta Pharm Sci. 2009;51:251–60. [Google Scholar]
- 29.Wadher KJ, Kakde RB, Umekar MJ. Formulation and evaluation of a sustained –release tablets of metformin hydrochloride using hydrophilic synthetic and hydrophilic natural polymers. Indian J Pharm Sci. 2011;73:208–15. doi: 10.4103/0250-474x.91579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alvarez-Fuentes J, Fernández-Arévalo M, González-Rodríguez ML, Cirri M, Mura P. Development of enteric-coated timed-release matrix tablets for colon targeting. J Drug Target. 2004;12:607–12. doi: 10.1080/10611860400013501. [DOI] [PubMed] [Google Scholar]
- 31.Khan MZ, Prebeg Z, Kurjaković N. A pH-dependent colon targeted oral drug delivery system using methacrylic acid copolymers. I. Manipulation Of drug release using Eudragit L100.55 and Eudragit S100 combinations. J Control Release. 1999;58:215–22. doi: 10.1016/s0168-3659(98)00151-5. [DOI] [PubMed] [Google Scholar]
- 32.Prasad YV, Krishnaiah YS, Satyanarayana S. In vitro evaluation of guar gum as a carrier for colon-specific drug delivery. J Control Release. 1998;51:281–7. doi: 10.1016/s0168-3659(97)00181-8. [DOI] [PubMed] [Google Scholar]
- 33.Chandran S, Asghar LF, Mantha N. Design and Evaluation of Ethyl Cellulose Based Matrix Tablets of Ibuprofen with pH Modulated Release Kinetics. Indian J Pharm Sci. 2008;70:596–602. doi: 10.4103/0250-474X.45397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tuğcu-Demiröz F, Acartürk F, Takka S, Konuş-Boyunağa O. In-vitro and in-vivo evaluation of mesalazine-guar gum matrix tablets for colonic drug delivery. J Drug Target. 2004;12:105–12. doi: 10.1080/10611860410001693751. [DOI] [PubMed] [Google Scholar]
- 35.Patel MM, Amin AF. Formulation and development of release modulated colon targeted system of meloxicam for potential application in the prophylaxis of colorectal cancer. Drug Deliv. 2011;18:281–93. doi: 10.3109/10717544.2010.538447. [DOI] [PubMed] [Google Scholar]
- 36.Kuksal A, Tiwary AK, Jain NK, Jain S. Formulation and in vitro, in vivo evaluation of extended- release matrix tablet of zidovudine: Influence of combination of hydrophilic and hydrophobic matrix formers. AAPS Pharm Sci Tech. 2006;7:E1–9. doi: 10.1208/pt070101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ali Asghar LF, Azeemuddin M, Jain V, Chandran S. Design and in vitro evaluation of formulations with pH and transit time controlled sigmoidal release profile for colon-specific delivery. Drug Deliv. 2009;16:295–303. doi: 10.1080/10717540902989936. [DOI] [PubMed] [Google Scholar]