

Abstract
The objective of the study was to prepare semisolid capsules (SSCs) of poorly water-soluble drug amlodipine besilate (AB) using a combination of technologies involving solid dispersion (SD) preparation and converting it into semisolid matrix filled in hard gelatin capsules (termed as SSCs) with the aim of reducing lag time in drug release and to improve the dissolution rate. AB is used for its anti-arrhythmic, anti-anginal, and anti-hypertensive activity. These are the emergency activities which should be treated as fast as possible like in the case of angina attack (heart attack). Any lag time that is generated due to its poor dissolution can add on in this emergency and that can be avoided by developing a readily dissolvable formulation: SDs of AB. SD of AB was prepared by fusion method using varying combinations of Poloxamer 407 and Plasdone S630. A total of nine batches (SD1–SD9) were characterized for the in vitro dissolution behavior in phosphate buffer pH7.4. SD8 with 95.8% cumulative drug release in 60 min, t50% = 4.1 min and DE30 Min = 84.2% were selected for the development of the semisolid matrix. Differential scanning calorimetry of SD8 revealed molecular dispersion of AB and Plasdone S630 in Poloxamer 407. SD8 was then formulated as SSCs using gelucire 44/14 and PEG 400 as semisolid components and PEG 6000 as a suspending agent to achieve the reduction in lag time for drug release. A total of seven SSC formulations were prepared and evaluated for drug release. Formulation of SSC4 showed maximum cumulative drug release (CDR) of 98.9% within 20 min that was almost a threefold reduction in the time required to achieve similar CDR by SD of AB. Thus, SSCs present an excellent approach to enhance the dissolution as well as to reduce the lag time of dissolution for poor water-soluble drugs especially to those therapeutic classes that are intended for faster onset of action.
Keywords: Amlodipine besilate, dissolution, semisolid capsules, solid dispersions
INTRODUCTION
Amlodipine besilate (AB) is a dihydropyridine calcium antagonist (calcium ion antagonist or slow channel blocker) and is used for its anti-arrhythmic, anti-anginal, and anti-hypertensive activity. These are the emergency activities which should be treated as fast as possible like in the case of angina attack (heart attack). Any lag time that is generated due to its poor dissolution can add on in this emergency and that can be avoided by developing a readily dissolutable formulation: solid dispersions (SDs) of AB. SDs of AB, when combined with the semisolid matrix and filled in the capsule, are able to release the drug readily and improve dissolution.
AB inhibits calcium ion influx across cell membranes selectively, with a greater effect on vascular smooth muscle cells than on cardiac muscle cells.[1] Chemically it is 3-ethyl-5-methyl (±)-2-[(2-aminoethoxy) methyl] -4-(2-chlorophenyl)-1, 4-dihydro -6-methyl-3, 5-pyridinedicarboxylate, mono benzenesulfonate monohydrate.[2] It is a poorly water-soluble drug.
Various techniques like SDs, salt formation, solubilization, and particle size reduction have commonly been used to increase the dissolution rate, oral absorption, and bioavailability of such drugs. Of these techniques, the SD technique is frequently used for solubility enhancement of poor water-soluble drugs. The term ‘solid dispersion’ has been utilized to describe a family of dosage forms where the drug is dispersed in a biologically inert matrix, usually with a view to enhance oral bioavailability.[3] The number of techniques had been reported for the preparation of SDs, such as spray drying, co-evaporation, co-precipitation, and freeze-drying. All these methods require expensive equipment involving complex procedures based on use of solvents. On the other hand, the hot melting method is a non-solvent technology; therefore, it is environment friendly and is cost-effective from commercial point of view.[4,5]
The carriers employed for preparing SDs by the hot melting technology should be water soluble with low melting point. Poloxamer 407 and Plasdone S630 were selected as carriers for SD preparation. Poloxamers are block co-polymers that consist of hydrophilic corona (ethylene oxide) and hydrophobic core (polypropylene oxide) blocks arranged in a triblock structure resulting in an amphiphilic structure, characterized by hydrophilic-lipophilic balance values. Plasdone S630 is used for solubility enhancement of poorly water-soluble drugs.[6] SD is proposed to be formulated as semisolid capsules (SSCs) using Gelucire 44/14 and PEG 400. Selection of Gelucire 44/14 for the formulation of SSCs was based on the following properties: an inert semisolid waxy material, amphiphilic in nature, unique emulsifying properties that make it ideal for prompt release formulations. As a well-documented fact, Gelucire with high HLB (hydrophilic–lipophilic balance) values is preferred for use in faster release of drugs.
Thus, the outline of the present project is the preparation of SDs of AB by solvent-free fusion method using Plasdone S630 and Poloxamer 407. The SDs will then be formulated as a semisolid system with PEG 400, Gelucire 44/14 as vehicles, and PEG 6000 as the suspending agent that will be filled into hard gelatin capsules and assessed for rapid dissolution capability.
MATERIALS AND METHODS
Materials
AB was obtained as a gift sample from Cadila Pharmaceuticals (Pvt) Ltd, Ankleshwar, India. Poloxamer 407 was gifted by BASF Corporation, NJ, USA. Plasdone S630 was gifted by ISP technologies, USA. PEG 400 and PEG 6000 were purchased from CDH, India, and Gelucire 44/14 was obtained from Colorcon Asia Pvt Ltd, India. All other chemicals were of analytical grade.
Determination of Equilibrium and Phase Solubility
To determine equilibrium solubility, an excess amount of AB was added to three different 25 mL conical flasks, containing 10 mL of double distilled water, pH 5.86, 0.1 Mol L-1 HCl buffer, pH 1.2, and phosphate buffer, pH 7.4, respectively. Flasks were placed in a mechanical shaker maintained at 37 ± 5°C for 72 h. At the end of 72 h, samples were filtered through the Whatman filter paper No. 2 (Grade 1, with a pore size of 11 μm). 1 mL of the filtered sample was suitably diluted and analyzed at 239 nm using UV spectrophotometer (Shimadzu, PharmaSpec1700, Tokyo, Japan). For phase solubility determination, excess amount of drug was added to five different 25 mL conical flasks containing varying concentrations of Poloxamer 407 (0.02%, 0.04%, 0.06%, 0.08%, and 0.1% by weight) and Plasdone S630 (0.2%, 0.4%, 0.6%, 0.8%, and 1.0% by weight) along with 10 mL of double distilled water. Further steps were similar to that in equilibrium solubility study.
Preparation of Formulations
Preparation of SDs
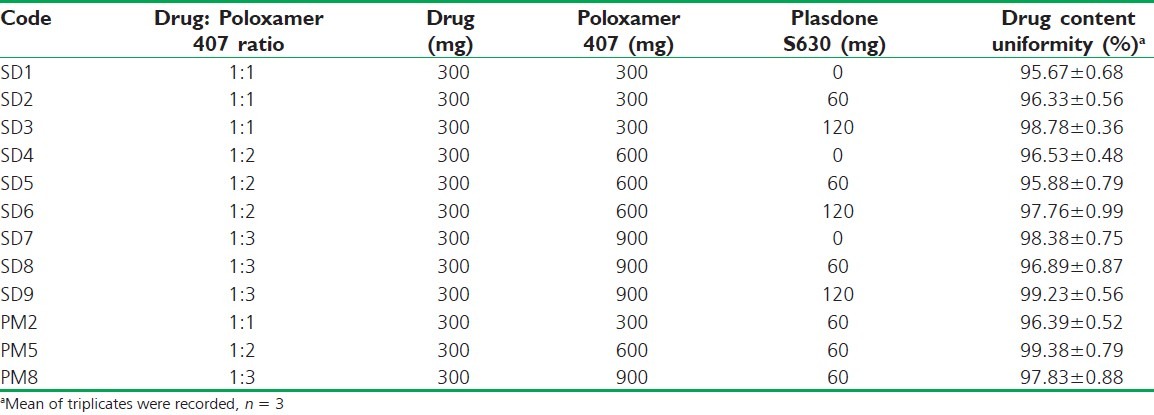
The carriers Plasdone S630 and Poloxamer 407 were heated on a water bath to a temperature 100 ± 0.5°C, above their melting points and the weighed amount of the drug was gradually incorporated into this molten mass. The mixture was cooled with constant stirring to homogenously disperse the drug throughout the matrix, without melting the drug into it. The cooled mass was crushed in glass mortar, passed through a sieve of 355 μm, and stored in a desiccator over anhydrous calcium chloride, until use. A total of nine SDs were made wherein the drug Poloxamer 407 was varied in 1:1, 1:2, 1:3 ratios to get binary SDs and Plasdone S630 was incorporated as 20% and 40% by mass to getternary SD within each ratio [Table 1].
Table 1.
Design for solid dispersions and selected physical mixtures of AB using Poloxamer 407 and Plasdone 630 as carriers along with the drug content
Preparation of physical mixture
The physical mixtures of AB, Poloxamer 407 and Plasdone S630, were prepared by mixing all components in a mortar for 5 min followed by sieving through a sieve of 355 μm.
Preparation of SSCs
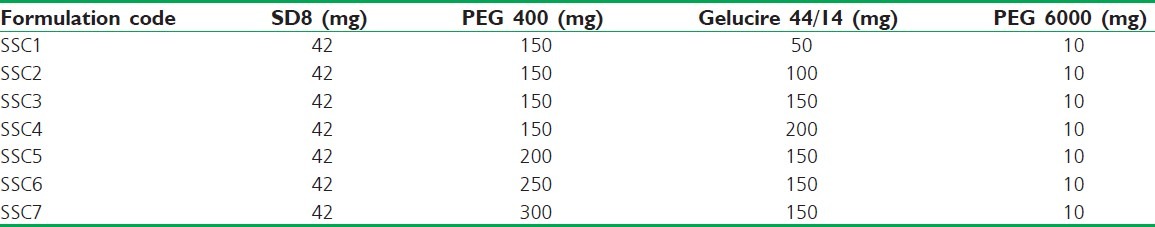
The semisolid matrix was prepared by the melting method. Calculated amounts of PEG 400, Gelucire 44/14, and PEG 6000 were weighed and mixed together in a beaker. The mix was heated on a water bath at 80°C with continuous stirring to obtain a homogeneous melt to which SD8 (optimized SD), equivalent to 10 mg, was gradually incorporated. The heated mixture was poured into a preheated (by immersing into the hot water at 80°C) plastic injector and transferred in hard gelatin capsules. A total of seven batches of SSCs containing 10 mg AB per capsule were prepared [Table 2].
Table 2.
Formulation design for SSCs of AB solid dispersion (SD8) prepared using PEG 6000 as the suspending agent
Evaluation of Formulations
Drug content
Accurately weighed (10mg) SDs were dissolved in 10 ml of volumetric flask using phosphate buffer pH 7.4. Samples were prepared by suitable dilutions after filtering the stock solution and the drug content was assayed spectrophotometrically in triplicate.
In vitro dissolution
In vitro dissolution of pure drug, its physical mixture, and SDs was carried out in USP XXIII[7] apparatus II containing 900 mL of phosphate buffer (pH7.4) at an agitation rate of 100 rpm. The temperature of the medium was maintained at 37 ± 0.5°C. A 5 mL sample was withdrawn at regular intervals, 0, 5, 10, 20, 30, 40, 50, and 60 min and replaced with equal volume of fresh dissolution medium, filtered, and analyzed spectrophotometrically.
In vitro release from SSCs
In vitro release test carried conditions were similar to the in vitro dissolution test except for the sampling time points t: 0, 5, 10, 15, and 20 min. The study was done in triplicate and statistically operated for standard deviation determinations. The release profiles were analyzed by model-independent parameters calculated by PCP Disso 2.0 v software, Bharti Vidyapeeth, Pune, India. Percent dissolution efficiency (%DE) was computed to compare the relative performance polymers in SDs. The magnitude of %DE for SDs was computed as the percent ratio of area under the dissolution curve up to time t, to that of the area of the rectangle described by 100% dissolution at the same time.[8]
Interaction Studies
Differential scanning calorimetry
The samples were sealed in aluminum pans and analyzed using a DSC 60, Shimadzu, Japan. 5–10 mg of sample was sealed in aluminum pan and analyzed at a scanning rate of 10°/min between 30 and 300°C. A nitrogen purge (20 mL/min) was maintained throughout the runs, using an empty sealed pan as reference. Temperature and heat flow calibrations were performed using indium as a standard.
Scanning electron microscopy
The surface morphology of pure drug, carriers, and SD8 was examined using FEI Quanta™ 200 scanning electron microscope, USA. The samples were fixed on a brass stub using a double-sided tape and then gold coated in a vacuum by a sputter coater and examined at a magnification of 100×.
Diffuse reflectance spectroscopy
Diffuse reflectance spectrographs of pure drug, carriers, SD8, and their physical mixture (PM) were obtained on Shimadzu, FTIR-8400, with DRS attachment, Tokyo, Japan in the scanning range of 500-4000 cm-1. The resolution was maintained at 1 cm-1.
Dispersion Homogeneity Test of Semisolid Mix
This test was performed to ensure the homogeneity of a semisolid mix (in turn, homogeneity of drug within the system) before filling it into the capsules. A weighed quantity of the semisolid mix prepared by SD8 was transferred to test tubes, mixed with 10 mL of distilled water, and centrifuged (4000 rpm) for 10 min. The homogeneity of the material was examined visually for any sediment at the bottom of the tube and floating residue on the top layer of the sample. Additionally, 1 mL volume was sampled from the top and bottom of the tubes and the drug content was determined spectrophotometrically.
Stability Studies
Stability studies were carried out for SSCs at 40 ± 2°C/75 ± 5% RH for 3 months. The physical appearance, drug content, and in vitro release of SSCs were determined at the end of 1, 2, and 3 months, and compared with that of freshly prepared SDs. The profiles were compared by calculating the similarity factor (f2).[9]
RESULTS AND DISCUSSION
Solubility
The equilibrium solubility of AB was found to be 80.31 ± 0.19, 75.92 ± 0.23, and 151.01 ± 0.29 μg/mL in double distilled water, phosphate buffer pH 7.4, and 0.1 M HCl, respectively. Phase solubility studies of AB in the presence of Plasdone S630 showed a linear increase (AL curve, R2 = 0.979) in the solubility [Figure 1] as a function of increasing concentration of carrier indicating formation of soluble 1:1 molar complex of AB and Plasdone S 630. The stability constant (Ks) of the complex was calculated from slope of AL-type solubility curve and was found to be 73.43 Mol/L. The low value of KS indicates poor stability of the complex but is adequate for its formation that can contribute in improving the bioavailability of poorly water-soluble drug.[10]
Figure 1.
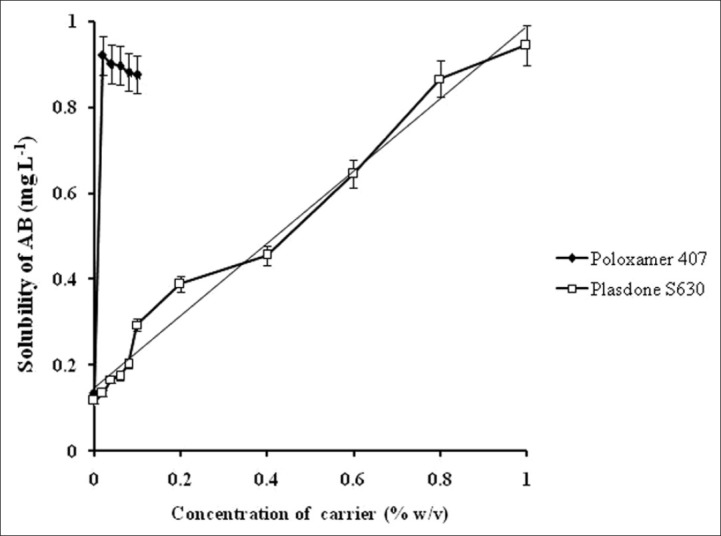
Phase solubility profile of AB in the presence of different concentrations of carriers at 37 ± 0.5°C
On the other hand, the solubility of AB in the presence of Poloxamer 407 showed a sharp increase of 0.921 mg/?mL at 0.02% w/v of Poloxamer 407 and thereafter the solubility decreased. The solubility enhancement with increasing Poloxamer 407 concentration may be due to its surfactant properties that resulted in micellar structure and consequently increased the solubility. Beyond 0.02% w/v, the formation of complex micellar structures probably led to decrease in its availability for drug solubilization. mL at 0.02% w/v of Poloxamer 407 and thereafter the solubility decreased. The solubility enhancement with increasing Poloxamer 407 concentration may be due to its surfactant properties that resulted in micellar structure and consequently increased the solubility. Beyond 0.02% w/v, the formation of complex micellar structures probably led to decrease in its availability for drug solubilization.[11] Based on the phase solubility results, SDs were made in ratios of 1:1, 1:2, and 1:3 (drug: Poloxamer 407) and within each ratio the synergistic effect of Plasdone S630 was analyzed in reference to the binary SDs. Table 1 shows the design of SDs along with the drug content determination results that might indicate uniformity in drug content.
In vitro Dissolution of SDs
Dissolution of pure AB in phosphate buffer, pH 7.4, documented 19.4% cumulative drug release (CDR) in 60 min that was lower as compared to physical mixtures and SDs [Figure 2]. Enhanced dissolution rate of AB from physical mixtures can be correlated with the hydrophilic nature of the carriers that were in close contact or adhered to the drug particles as a result of mixing. The mixture when came in contact with aqueous media resulted in rapid hydration of the carrier particles that contributed to the increase in wettability and dissolution of the drug particles.[12] The SDs, however, showed a %CDR twice as high as the corresponding physical mixtures at the end of 60 min. The SDs also displayed an ascending order of CDR increments on increasing drug: Poloxamer 407 ratios (1:1 < 1:2 < 1:3) were attributable to the association of monomolecular micelles to form aggregates of varying size, have the ability to increase drug dissolution,[13] and is consistent with the results of phase solubility studies.
Figure 2.
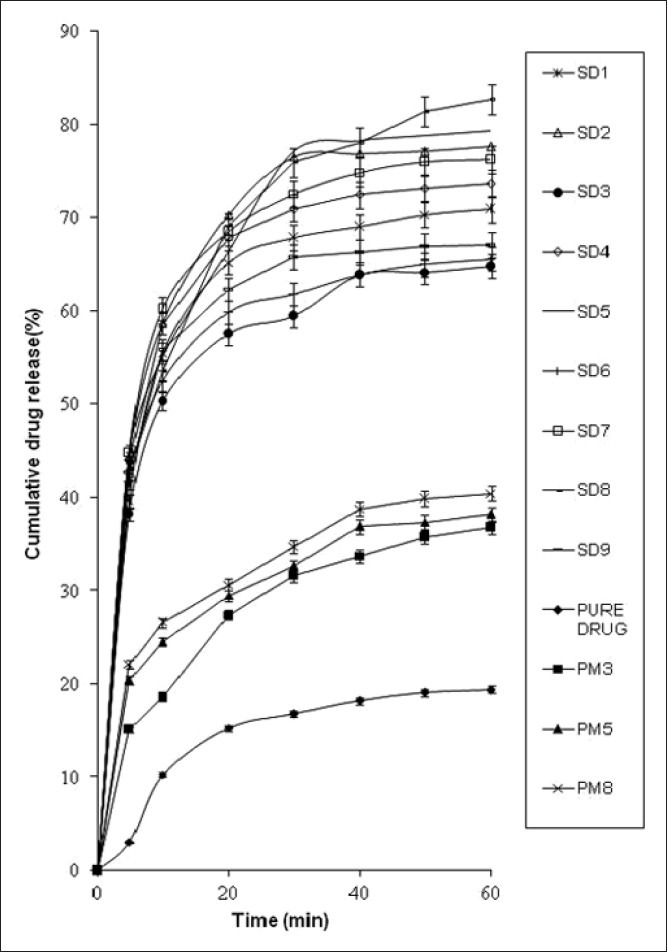
In vitro drug dissolution profiles of solid dispersions of AB in phosphate buffer pH 7.4, n = 3
Effect of second polymer, Plasdone S630, on the dissolution profile was found to be higher when used at 20% concentration. A slight decrement at high concentration of Plasdone S630 (40%) can be due to the formation of a grid-like structure around drug molecules that hindered the release medium to reach the drug molecules. Thus, it can be concluded that Plasdone S630 exhibited self-inhibitory effect above certain concentrations.[14] And thus, an optimum blend of the selected polymers was required.
Selection of SD
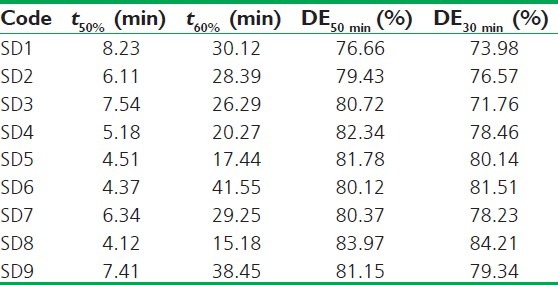
For selection of the best SD, the dissolution profiles were subjected to model-independent dissolution parameter analysis [Table 3]. The model-independent parameters of SD8 favoured its selection as t50% (4.12 min) and t60% (15.18 min) were found to be least and the percent DE at 30 and 50 min highest amongst all.
Table 3.
Model-independent parameters of solid dispersions of AB
Interaction Studies
Differential scanning calorimetry and ultraviolet spectroscopy
The DSC thermograms of AB showed endothermic peak at 207.98°C corresponding to its melting point [Figure 3]. The thermograms of Plasdone S630 and Poloxamer 407 showed a single broad peak at 86.16°C and a sharp peak at 56.85°C, respectively. In SD8, the characteristic peak of AB was completely lost which indicates that the drug might be dispersed in the molecular state in the dispersion whereas the endothermic peak of Poloxamer 407 was retained. The data were supplemented with UV spectroscopy (secondary method) to show that the loss of drug-related melting peak was due to solubilization of drug in carriers rather than thermal degradation. The UV maximum of 239 nm was retained in the case of SD8 confirming the dispersion of intact drug molecule in the carriers. The endothermic peak of Plasdone S 630 was also lost in the thermogram of SD8, suggesting that drug and Plasdone S 630 might have been solubilized into Poloxamer407.[15]
Figure 3.
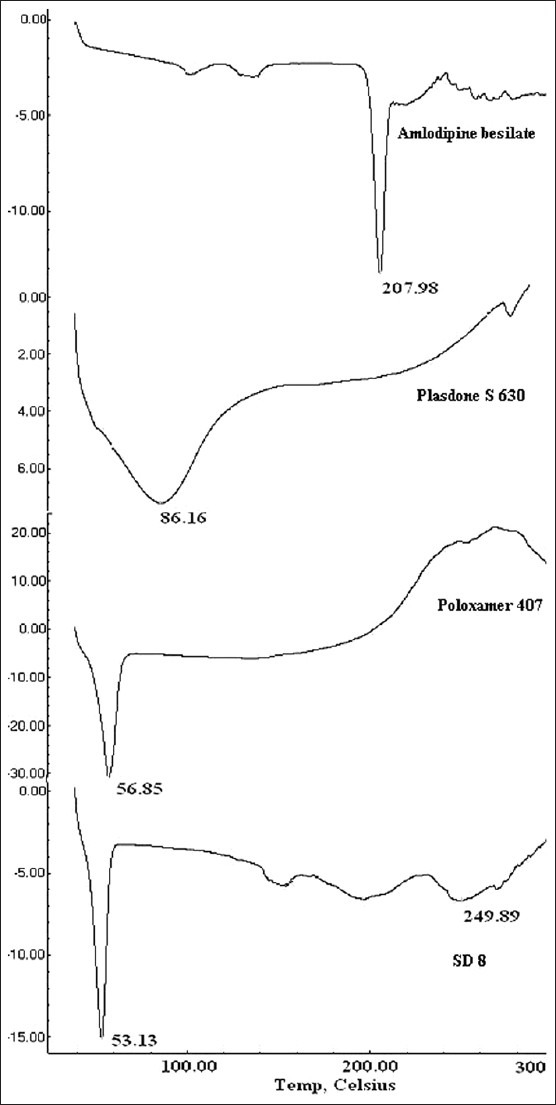
Differential scanning calorimetric thermograms of AB, Poloxamer 407, Plasdone S630, and SD8
Scanning electron microscopy
The scanning electron micrographs of AB [Figure 4] revealed needle-shaped crystals. Poloxamer 407 was characterized by large nearly spherical particles and the micrograph of Plasdone S630 showed molten surface with numerous folds that might have formed due to melting of the sample during its processing. The individuality of all these micrographs was lost in SD8. As a result, the crystalline structure of AB was completely disappeared that is consistent with the DSC results. Similarly, Plasdone S630 was presumably dissolved in poloxamer as indicated by DSC studies and hence the micrograph of SD8 predominantly was closer to Poloxamer 407 with non-uniform surface texture comprising of numerous depressions and striations that can be considered remnants of the processing steps used for SD preparation.
Figure 4.

Scanning electron micrograph of (a) AB, (b) Poloxamer 407, (c) Plasdone S630, and (d) SD8
Diffuse reflectance spectroscopy
The spectrographs of pure drug, carriers, SD8, and its physical mixture did not show any potential interaction and incompatibility between the carriers and drug molecules as shown in Figure 5. Characteristic peaks of pure drug, AB, were analyzed at 3155 cm-1 and 3290 cm-1 for primary anime group - NH2, along with the carboxylic group peak at 1683 cm-1 which were retained in SD8 but with lesser intensity, which might be due to the dilution with polymer.
Figure 5.
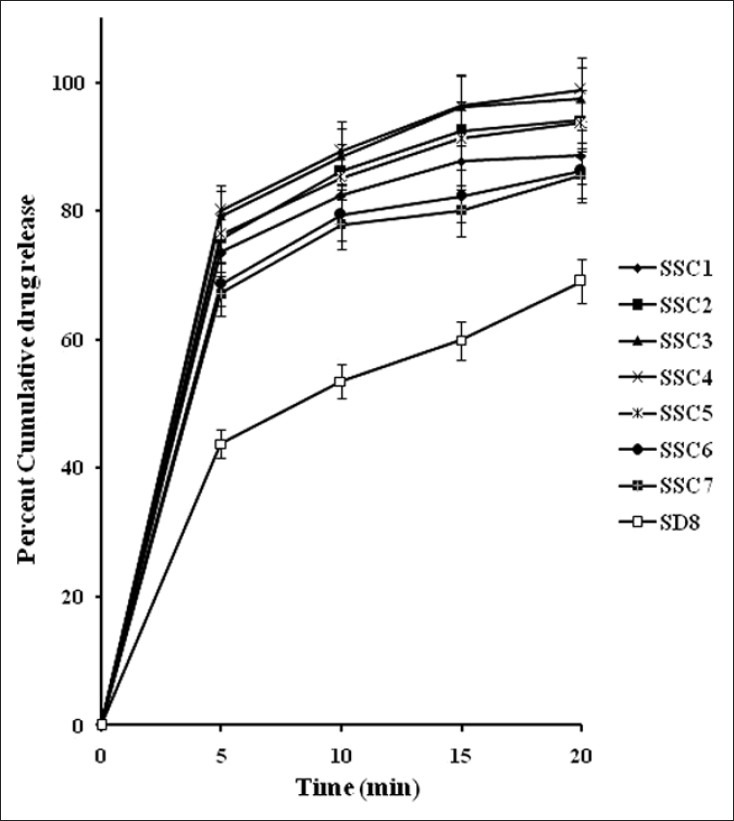
In vitro release profiles of AB from the SSCs in phosphate buffer pH 7.4, n = 3
Dispersion Homogeneity Test of Semisolid Mix
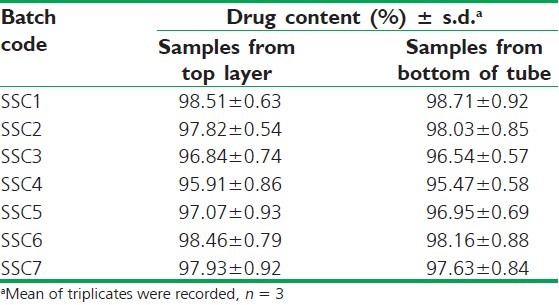
The aqueous dispersion of the semisolid mix when subjected to centrifugation (4000 rpm, 10 min) showed no precipitate/residue at the bottom of the tube or on the surface layer of the test sample. For the further verification, aliquot samples were withdrawn both from the top and bottom end of the samples, and no difference in drug content was evidenced. Results are shown in Table 4, ensuring the uniformity of all the seven semisolid mix formulations.
Table 4.
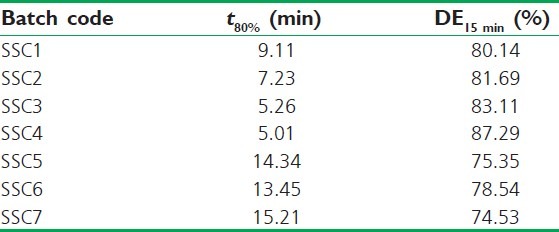
Model-independent parameters of AB released from SSCs
In vitro Drug Release from SSCs Formulations
The in vitro drug release profiles of SSCs [Figure 6] showed a biphasic release: an initial burst phase within 5 min that resulted in CDR ranging from 67.18% to 79.91% that was followed by a slower release of 86.87% to 97.45% CDR up to 20 min. This release pattern is similar to that displayed by SD8 but SD8 showed a biphasic release of lower magnitude (43.08% at 5 min and 69.11% at 20 min) than that of all SSC formulations. This clearly emphasizes the superiority of the SSCs over SDs and which is desirable for antihypertensive drugs like AB, requiring faster onset of action.
Figure 6.
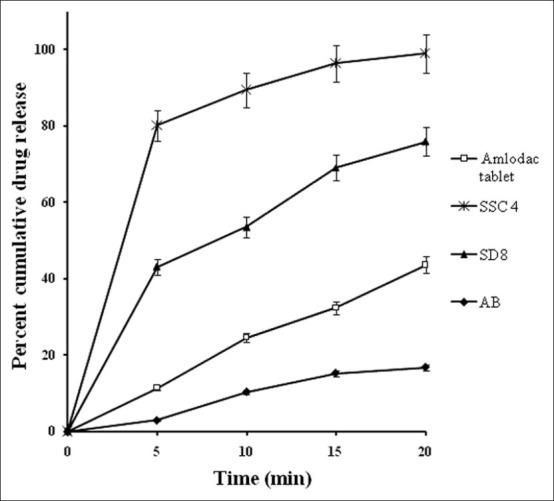
Comparative in vitro dissolution profiles of optimized semisolid capsule (SSC4) and marketed formulation (Amlodac tablet) in phosphate buffer pH 7.4 and reference release profiles of solid dispersion SD8 and pure drug, n = 3
Among the SSCs, SSC4 comprised the lowest level of PEG 400 and highest of Gelucire 44/14 which showed a maximum drug release of 97.55% up to 20 min. On the contrary, SSC7 which consisted of the lowest level of Gelucire 44/14 and the highest amount of PEG 400 showed the least release of 85.56%. Clearly, the levels of both Gelucire 44/14 and PEG 400 played a key role in enhancing the drug release. The model-independent parameters, t80% and DE15 Min were utilized to analyze the release data [Table 5], affirming the similar concept of Gelucire 44/14 and PEG 400 level effect on drug release. The time required for 80% release was found to be least for SSC4. The DE at 15 min was 87.29% and 74.53% for SSC4 and SSC7, respectively. Rest of the formulations displayed intermediate values that were a reflection of the variation in levels of Gelucire 44/14.
Table 5.
Drug content determination of centrifuged samples for dispersion homogeneity test of semisolid mix
Gelucire 44/14 (PEG-32 glyceryl laurate) possesses surfactant and self-emulsifying properties and can be used as a binder by the melt granulation technique for poorly water-soluble drugs.[16] In contact with water, it forms a fine emulsion and solubilizes the active substance. The results are in well accordance with numerous research reports as poorly water-soluble drugs have been formulated with Gelucire 44/14, alone or in mixture with other excipients, and have showed enhanced solubility and dissolution. For instance, DMP 323, a protease inhibitor drug,[17] piroxicam,[18] and UC–781 a thiocarboxanilide derivative.[16]
Another hydrophilic component PEG 400 is a low molecular weight molecule that interacts on the surface of the drug to solubilize it and form small units in the medium. These small units get entrapped into the grid-like structures of Plasdone S630 and hinder the release of drug through it.[19] This explains the least CDR value from SSC7 containing highest amount of PEG 400 than the rest of the SSCs.
A comparison was made between marketed formulation (Amlodac®) and the optimized formulation SSC4 [Figure 7]. SSC4 released more than 80% in 5 min and reduced the addition lag time that was taken by marketed formulation to release up to the same level. As stated earlier for an antihypertensive drug, a formulation that has the potential to overcome lag time for the onset of action is desirable that can be better achieved by SSC4 in comparison to the marketed formulation.
Figure 7.
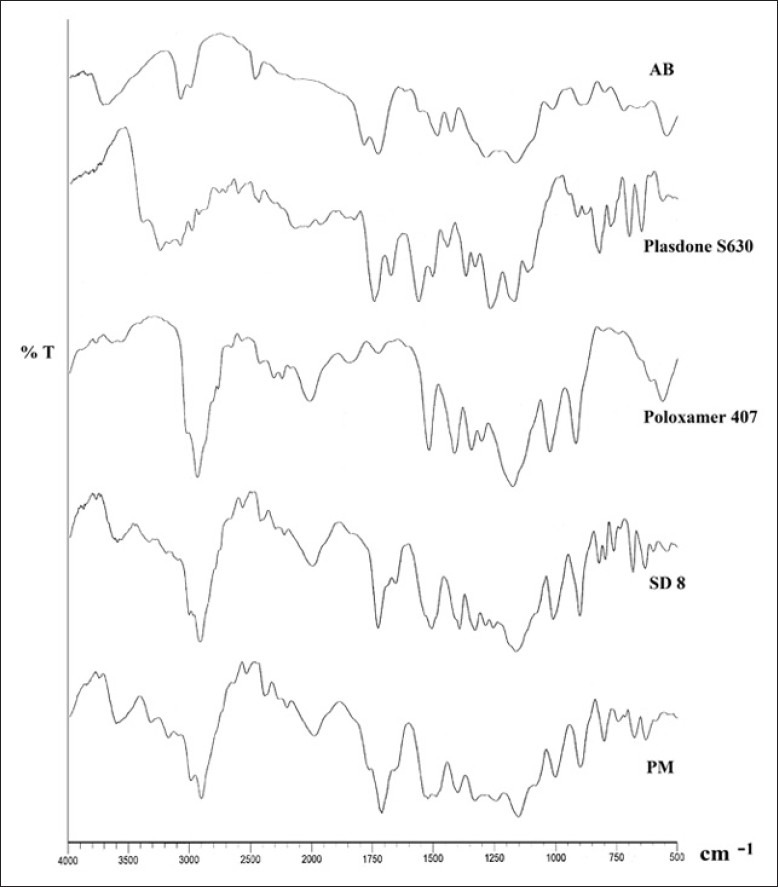
Comparative diffuse reflectance spectrographs amlodipine besilate (AB), Plasdone S630, Poloxamer 407, SD8, and physical mixture of AB and carriers (PM)
Stability
The optimized formulation (SSC4) when stored at 40 ± 2°C and 75 ± 5% RH was devoid of leakage and demonstrated no change in physical appearance. The drug contents were 98.77 ± 0.06%, 98.23 ± 0.05%, and 97.45 ± 0.27% after 1, 2, and 3 months, respectively. These were comparable to the drug content of sample at zero time point, when analyzed statistically using paired t-test, and were found to be significantly similar with P < 0.01. In vitro release profiles (data not shown) after various time points showed a similarity factors (f2) of 83-89 indicating similarity in release profiles of stored and fresh samples of SSCs. Thus, the developed formulation can be considered as a stable dosage form.
CONCLUSION
SSCs of poorly water-soluble drug AB were successfully designed by using a combination of technologies involving SD and semisolid matrix filled in hard gelatin capsules. The drug was molecularly dispersed in SD and hence fast dissolution could be achieved. However, for an antihypertensive drug, an almost zero lag time is desirable. Therefore, SSCs were formulated that were able to reduce the lag time in drug release that was almost three times reduction in the time required by SD to achieve similar CDR. Thus, SSCs present an excellent approach to enhance the dissolution of poorly water-soluble drugs especially those therapeutic classes that are intended for faster onset of action.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
REFERENCES
- 1.Robertson RM, Robertson D, Hardman JG, Limbird LE, Molinoff PE, Ruddon RW, editors. 5th ed. New York: McGraw Hill; 2006. The pharmacological basis of therapeutics. [Google Scholar]
- 2.Sweetman SC. 33rd ed. New York: Pharmaceutical Press; 2002. Martindale's the complete drug reference. [Google Scholar]
- 3.Dhirendra K, Lewis S, Udupa N, Atin K. Solid dispersions: A review. Pak J Pharm Sci. 2009;22:234–46. [PubMed] [Google Scholar]
- 4.Leuner L, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50:47–60. doi: 10.1016/s0939-6411(00)00076-x. [DOI] [PubMed] [Google Scholar]
- 5.Sharma DK, Joshi SB. Solubility enhancement strategies for poorly water soluble drugs in solid dispersions: A review. Asian J Pharm. 2007;1:9–19. [Google Scholar]
- 6.Swarbrick J, Boylan JC. 2nd ed. New York: Marcel Dekker; 2000. Encyclopedia of pharmaceutical technology. [Google Scholar]
- 7.Rockville, MD 20852: United States Pharmacopoeial Convention, Inc; 2004. United States Pharmacopoeia 27/National Formulary 22, Asian Edition; p. 2305. [Google Scholar]
- 8.Costa P, Manuel J, Lobo S. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13:23–133. doi: 10.1016/s0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- 9.Ahuja N, Katare OP, Singh B. Studies on dissolution enhancement and mathematical modeling of drug release of a poorly water-soluble drug using water-soluble carriers. Eur J Pharm Biopharm. 2007;65:26–38. doi: 10.1016/j.ejpb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 10.Newa M, Bhandari KH, Liu XD, Kwon HT, Kim AJ, Yoo KB, et al. Preparation, characterization and in vivo evaluation of ibuprofen binary solid dispersions with poloxamer188. Int J Pharm. 2007;342:228–37. doi: 10.1016/j.ijpharm.2007.05.031. [DOI] [PubMed] [Google Scholar]
- 11.Goddeeris C, Mooter GD. Free flowing solid dispersions of the anti-HIV drug UC 781 with Poloxamer 407 and a maximum amount of TPGS 1000: Investigating the relationship between physicochemical characteristics and dissolution behavior. Eur J Pharm Sci. 2008;35:104–13. doi: 10.1016/j.ejps.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 12.Batra V, Shirolkar VS, Mahaparale PR, Kasture PV, Deshpande AD. Solubility and dissolution enhancement of glipizide by solid dispersion technique. Indian J Pharm Educ Res. 2008;42:372–8. [Google Scholar]
- 13.Ghebremeskel AN, Vemavarapu C, Lodaya M. Use of surfactants as plasticizers in preparing solid dispersions of poorly soluble API: Selection of polymer–surfactant combinations using solubility parameters and testing the processibility. Int J Pharm. 2007;328:119–29. doi: 10.1016/j.ijpharm.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 14.Rowe RC, Sheskey PJ, Owen SC. 5th ed. Great Britain: The Pharmaceutical Press; 2006. Handbook of pharmaceutical excipients. [Google Scholar]
- 15.Dumortier G, Grossiord JL, Agnely F, Chaumeil JC. A review of poloxamer 407 pharmaceutical and pharmacological characteristics. Pharm Res. 2006;23:2709–28. doi: 10.1007/s11095-006-9104-4. [DOI] [PubMed] [Google Scholar]
- 16.Damian F, Blaton N, Kinget R, Mooter RG. Physical stability of solid dispersions of the antiviral agent UC-781 with PEG 6000, Gelucire® 44/14 and PVP K30. Int J Pharm. 2002;244:87–98. doi: 10.1016/s0378-5173(02)00316-2. [DOI] [PubMed] [Google Scholar]
- 17.Aungst BJ, Nguyen NJ, Rogers NJ, Rowe SM, Hussain MA, White SJ, et al. Amphiphilic vehicles improve the oral bioavailability of a poorly soluble HIV protease inhibitor at high doses. Int J Pharm. 1997;156:79–88. [Google Scholar]
- 18.Yuksela N, Karatas A, Ozkanb Y, Savas A. Enhanced bioavailability of piroxicam using Gelucire 44/14 and Labrasol: In vitro and in vivo evaluation. Eur J Pharm Biopharm. 2003;56:453–59. doi: 10.1016/s0939-6411(03)00142-5. [DOI] [PubMed] [Google Scholar]
- 19.Yunzhe S, Rui Y, Wenliang Z, Xing T. Nimodipine semi-solid capsules containing solid dispersion for improving dissolution. Int J Pharm. 2008;359:144–9. doi: 10.1016/j.ijpharm.2008.03.040. [DOI] [PubMed] [Google Scholar]