

Abstract
Takayasu's arteritis is a rare, systemic vasculitis with varied presentations across multiple medical specialities. Here, we present a young woman who had recurrent episodes of erythema nodosum on the background of a low-grade fever and no vascular manifestations. The presence of a high erythrocyte sedimentation rate generated a high index of suspicion for underlying vasculitis, and radioimaging confirmed the suspicion of Takayasu's arteritis. The patient was found to have type III diseases in the vasculitic stage and was managed with systemic corticosteroids.
Background
The clinical symptoms and signs of Takayasu's arteritis may often be so trivial that the disease may go unrecognised, and at times may present with catastrophic and life-threatening stroke or cardiac failure. The clinical heterogeneity, together with inadequate specific laboratory markers and complex/expensive investigative procedures, makes the clinician apprehensive of diagnosing this condition, especially with limited resources. With the wider availability of conventional and non-invasive angiographic techniques, the diagnosis of Takayasu's arteritis has become feasible, provided the imaging findings are interpreted in the clinical context.
Case presentation
A 20-year-old woman presented to our medicine outpatient department with the complaints of malaise and low-grade fever of 1 year. Since 1 week prior to this visit, she had painful nodular skin lesions on bilateral lower limbs. She had similar episodes of skin lesions twice for the past 1 year which had resolved with oral antibiotics and analgesics. She had no history to suggest trauma or insect bite, tuberculosis or diabetes.
On general examination, the patient was hypertensive with blood pressure in the right upper limb as 144/94 mm Hg and left upper limb 140/90 sitting position. The blood pressure in the lower limbs was normal. The radial pulse was normal and equal in both upper limbs and all peripheral pulses were felt normally. There was no carotid, subclavian, abdominal or renal bruit. Lower limb examination showed multiple tender erythematous nodular skin swellings over bilateral shins (figure 1). Rest of the systemic examination was unremarkable.
Figure 1.
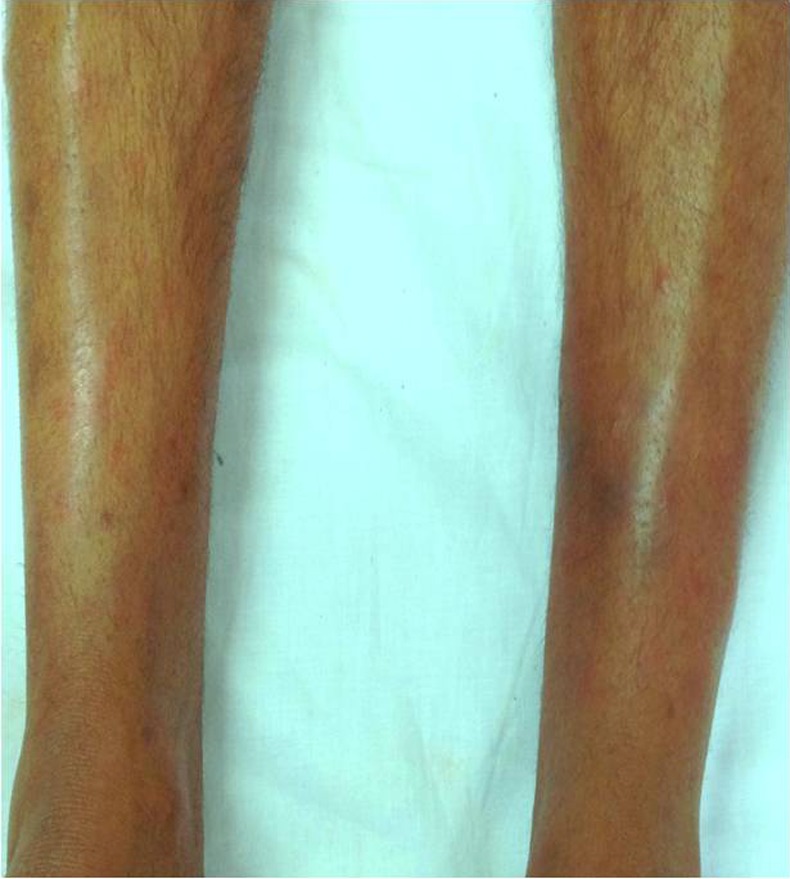
Clinical photograph showing multiple erythematous nodular swelling on bilateral shins.
Investigations
The haematological investigations revealed a normocytic normochromic anaemia with a haemoglobin 10 g/dl, total leucocyte count 8200/mm3, with a lymphocyte predominance of 40% on differential count. The erythrocyte sedimentation rate (ESR) was high, 70 mm in the first hour by the Westergren method, and the C reactive protein was elevated at 15.2 mg/dl. The serum electrolytes and renal functions were within normal limits. The serum ACE levels were in the normal range. The tuberculin test showed an induration of 15×15 mm, indicating that it was reactive. The chest radiograph did not reveal any pulmonary lesion, mediastinal widening or cardiomegaly. A contrast-enhanced CT (CECT) chest was also performed; however, it was also normal. Doppler ultrasonography of the abdomen demonstrated a dilated (2.7 cm) upper abdominal aorta with calcification of its wall, and aneurysmal dilation of superior mesenteric artery at its origin, with tubular flow. Biopsy from the skin lesions showed septal panniculitis with perivascular inflammatory lymphocytic infiltrate suggestive of erythema nodosum (figure 2).
Figure 2.
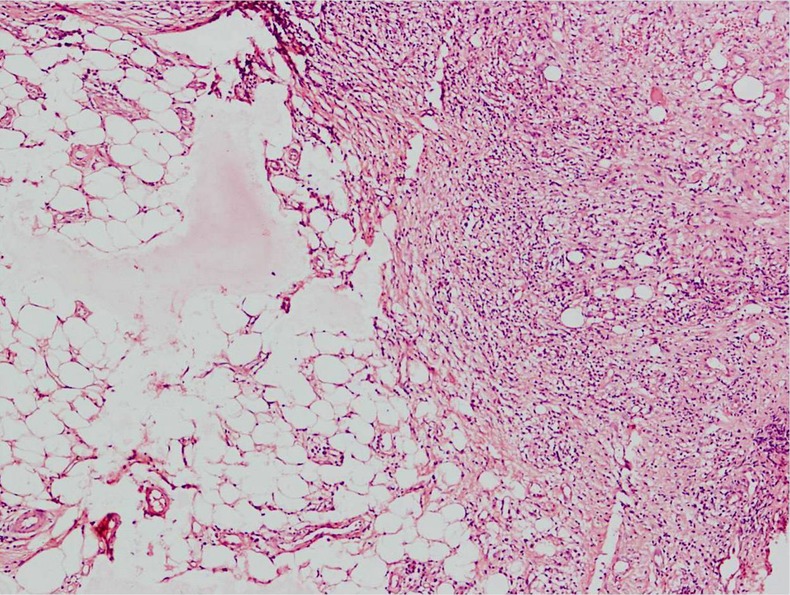
Skin histopathology showing inflammatory exudate with septal panniculitis.
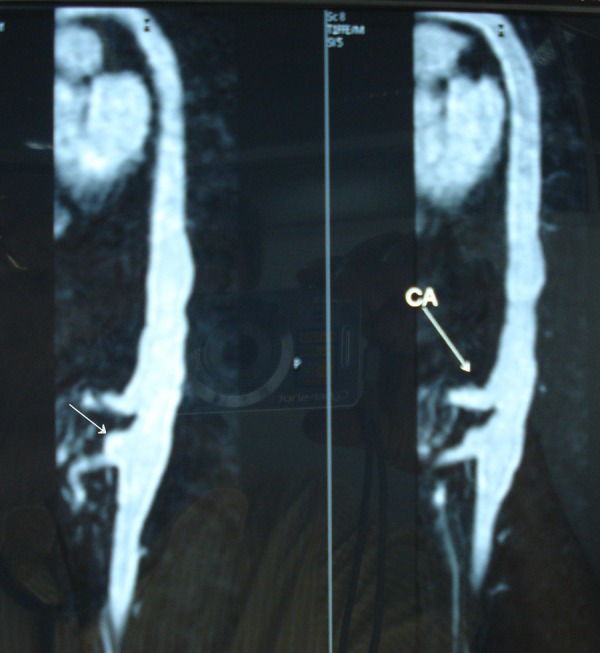
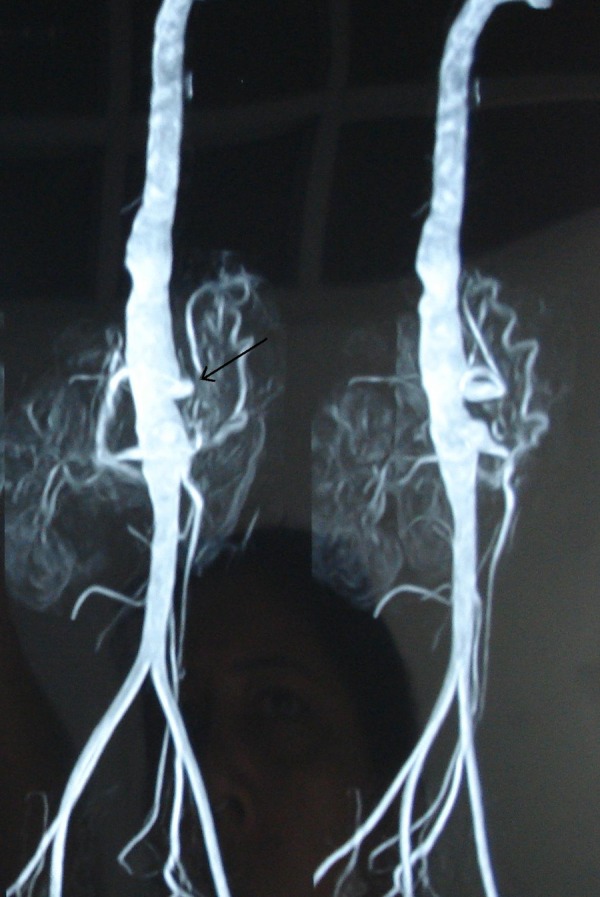
MR angiogram of aorta and its branches was done to confirm and evaluate the extent of disease involvement. Thoracoabdominal aorta shows irregular dilation (maximum diameter 25 mm) with lobulated outline extending from D10 level up to the infrarenal part just below the origin of the renal arteries involving approximately 9 cm long statement. There was evidence of a small fusiform aneurysm (13.6 × 10.1 mm) seen arising from the anterior aspect of the abdominal aorta just above the origin of the superior mesenteric artery (figure 3). The celiac axis was also irregular in outline. Right renal artery was normal, but left renal artery was irregular and had a short narrowed segment just distal to its origin with no post-stenotic dilation (figure 4). An accessory renal artery was visualised supplying the lower pole of the left kidney. These findings were suggestive of type III disease (involvement of thoracic descending aorta, abdominal aorta, renal arteries or a combination).
Figure 3.
MR angiogram depicting lobulated irregular outline of aorta with small fusiform aneurysm (thin white arrow) arising from the anterior aspect of the abdominal aorta just above the origin of the superior mesenteric artery.
Figure 4.
MR angiogram showing irregular left renal artery with a short narrowed segment just distal to its origin (black arrow). An accessory renal artery is also visualised.
Differential diagnosis
Various conditions that mimic Takayasu's arteritis include fibromuscular dysplasia (FMD), temporal arteritis, syphilitic aortitis, coarctation of aorta and atherosclerotic disease. FMD has a beaded appearance, usually does not affect the aorta, and is rare in the subclavian artery. Temporal arteritis is seen in the elderly and usually does not involve the common carotid arteries. Syphilitic aortitis is extremely uncommon now and typically causes calcification in the ascending aorta. Erythema nodosum in the setting of vasculitis may be commonly seen in polyarteritis nodosa, sarcoidosis, fungal infections, drug reactions and Behçet's disease.
Treatment
She was started on oral prednisolone (1 mg/kg/day) and antipyretics. Inflammatory parameters such as ESR normalised within 3 weeks. Prednisolone was continued for initial 2 months following which it was gradually reduced to a maintenance dosage of 10 mg/day for 1 year. She was also prescribed 2.5 mg of amlodipine for hypertension control.
Outcome and follow-up
She is off steroids now for the last 1.5 years and has remained free of symptoms.
Discussion
Takayasu's arteritis is a chronic granulomatous inflammation of large elastic arteries usually affecting the aorta, its larger branches and pulmonary arteries. Initially, in 1827, R Adams termed this pulseless disease as non-specific aorto-arteritis (NSAA), and in 1908, Mikito Takayasu named it Takayasu's arteritis. Takayasu's arteritis occurs worldwide, but more so in the Asian countries and is predominantly a disease of young women of child-bearing age. The disease has protean manifestations which may differ with the age of the patient, geographical location and the stage and severity of disease. Because of the heterogeneity in its presentation, it is often called as ‘the great imitator’ and the diagnosis may be delayed or go unrecognised. Although traditionally the disease has been classified as having early or late stage, or occasionally a triphasic course (prevasculitic, vascular inflammation and burnt-out stage), the categorisation is often blurred and overlapping symptoms predominate. Advanced lesions can be stenotic, occlusive or aneurysmal and display a panarteritis with intimal proliferation on histopathology.
Approximately 10% of patients with Takayasu's arteritis are asymptomatic, with the disease detected based on abnormal vascular findings on examination.1 The symptoms in Takayasu's arteritis could be vascular or extravascular and might present at any stage of the disease. Even though stenotic vascular symptoms are the hallmark, the clinical harbingers of the disease could very well be the extravascular lesions. Cutaneous manifestations are well documented in various case series on aortoarteritis. They frequently occur as acute, tender erythematous nodules on the legs suggestive of erythema nodosum (6–19%), or subacute ulcerated nodules of the legs (<2.5%), erythema induratum, lupus-like malar rash and pyoderma gangrenosum-like ulcerations (<1%), or urticarial lesions with livedo reticularis.1–3 Other dermatological manifestations include Raynaud's phenomenon, probably owing to direct large vessel involvement.3 Histopathological examination of these lesions shows features of granulomatous or necrotising vasculitis or panniculitis. These skin lesions are often so common in several systemic diseases or as allergic manifestation to drugs that a rare underlying cause could be easily overlooked unless a thorough investigations are carried out.
The underlying pathological process common to both Takayasu's arteritis and erythema nodosum is inflammatory, with several aetiological factors having been proposed. These include infections and antigenic stimulation with spirochetes, Mycobacterium tuberculosis, streptococcal organisms or an idiopathic autoimmune process with reactive circulating antibodies, amplification of inflammatory cytokines (IL-6, interleukin-6 receptor) causing systemic and cutaneous vasculitis.
Studies suggest that non-invasive imaging modalities such as MRI, ultrasonography and 18F-fluorodeoxyglucose positron emission tomography (18F-FDG-PET) allow diagnosis of Takayasu's arteritis earlier in the disease course than that of standard angiography and provide a means for monitoring disease activity.4 The standard treatment of Takayasu's arteritis is with oral corticosteroids which are started at 1 mg/kg daily and tapered over weeks to months once symptoms start to subside. Recently, many new promising biological and cytotoxic agents have shown to improve clinical signs and symptoms of Takayasu's arteritis. They include tocilizumab, a humanised monoclonal antibody causing blockade of the soluble IL-6 receptor, and rituximab, a chimaeric IgG1 antibody that binds to CD20, expressed on the surface of B cells.5 6 Patients who have steroid-resistant or relapsing disease, may benefit from methotrexate, azathioprine, cyclophosphamide, mycophenolate mofetil and tacrolimus hydrate.6 These agents have variable success rates and should be reserved for patients with the most severe and refractory disease states.
Learning points.
Takayasu's arteritis is rare and difficult to diagnose. Initial symptoms may be indistinct and may become noticeable until the disease has progressed considerably.
Skin involvement could be a clinical harbinger of the disease. Knowledge of the skin manifestations associated with Takayasu's arteritis remains important for its diagnosis and prompt institution of life-saving treatment.
Footnotes
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Kerr GS, Hallahan CW, Giordano J, et al. Takayasu arteritis. Ann Intern Med 1994;2013:919–29 [DOI] [PubMed] [Google Scholar]
- 2.Soto ME, Espinola N, Flores-Suarez LF, et al. Takayasu arteritis: clinical features in 110 Mexican Mestizo patients and cardiovascular impact on survival and prognosis. Clin Exp Rheumatol 2008;2013:S9–15 [PubMed] [Google Scholar]
- 3.Francès C, Boisnic S, Blétry O, et al. Cutaneous manifestations of Takayasu arteritis. A retrospective study of 80 cases. Dermatologica 1990;2013:266–72 [DOI] [PubMed] [Google Scholar]
- 4.Andrews J, Mason JC. Takayasu's arteritis—recent advances in imaging offer promise. Rheumatology (Oxford) 2007;2013:6–15 [DOI] [PubMed] [Google Scholar]
- 5.Salvarani C, Magnani L, Catanoso M, et al. Tocilizumab: a novel therapy for patients with large-vessel vasculitis. Rheumatology (Oxford) 2012;2013:151–6 [DOI] [PubMed] [Google Scholar]
- 6.Unizony S, Stone JH, Stone JR. New treatment strategies in large-vessel vasculitis. Curr Opin Rheumatol 2013;2013:3–9 [DOI] [PubMed] [Google Scholar]