

Abstract
Intense lung inflammation characterizes respiratory failure associated with Pneumocystis pneumonia. Our laboratory has previously demonstrated that alveolar epithelial cells (AECs) elaborate inflammatory cytokines and chemokines in response to the P. carinii cell wall constituent β-1,3-glucan (PCBG), and that these responses require lactosylceramide, a prominent glycosphingolipid constituent of certain cell membrane microdomains. The relevance of membrane microdomains, also termed plasma membrane lipid rafts, in cell signaling and macromolecule handling has been increasingly recognized in many biologic systems, but their role in P. carinii-induced inflammation is unknown. To investigate the mechanisms of microdomain-dependent P. carinii-induced inflammation, we challenged primary rat AECs with PCBG with or without preincubation with inhibitors of microdomain function. Glycosphingolipid and cholesterol rich microdomain inhibition resulted in significant attenuation of P. carinii-induced expression of TNF-α and the rodent C-X-C chemokine MIP-2, as well as their known inflammatory secondary signaling pathways. We have previously shown that protein kinase C (PKC) is activated by PCBG challenge, and herein show that PKC localizes to AEC microdomains. We also demonstrate by conventional microscopy, fluorescence microscopy, confocal microscopy, and spectrophotofluorimetry that AECs internalize fluorescently labeled PCBG by microdomain-mediated mechanisms, and that anti-microdomain pretreatments prevent internalization. Taken together, these data suggest an important role for AEC microdomain function in PCBG-induced inflammatory responses. This offers a potential novel target for therapeutics for a condition that continues to exert unacceptable morbidity and mortality among immunocompromised populations.
Keywords: Pneumocystis, beta-glucan, epithelial cells, membrane microdomains
INTRODUCTION
Pneumocystis pneumonia remains an important problem in the management of immunocompromised patients.1–3 Despite medical advances, the case mortality for Pneumocystis pneumonia ranges between 15% and 40%, with significantly worse outcomes among non-AIDS immunocompromised persons.1–3 An intense lung inflammation characterized by excess neutrophils and CD8+ lymphocytes is characteristic of severe Pneumocystis pneumonia and results in diffuse alveolar damage, gas exchange impairment, and respiratory failure.1,4,5 Notably, the degree of lung inflammation has been shown to be a stronger predictor of respiratory failure and death rather than the fungal organism burden.2–4,6–9 The mechanism by which Pneumocystis organisms induce lung inflammation remains incompletely elucidated, but recent studies implicate the Pneumocystis cell wall constituent β-1,3-D-glucans in lung inflammation.9,10 Pneumocystis jirovecii is the species of this genus that infects humans, whereas Pneumocystis carinii represents the species infecting rodents.11 A great deal of our understanding of Pneumocystis pathogenesis and all current drugs known to be effective in human Pneumocystis pneumonia have been developed by studies of P. carinii.11
Alveolar epithelial cells (AECs) appear to play an important role in Pneumocystis pneumonia. Ultrastructural studies identify Pneumocystis organisms closely associated with the alveolar epithelium of infected human and animal lungs, supporting the hypothesis that the binding of Pneumocystis to AECs is an integral part of the establishment of infection.12 Evidence also strongly supports that AECs, once considered simply passive gas exchange barriers, actively participate in host defenses against this organism. Our laboratory has demonstrated that rat AECs produce macrophage inflammatory protein-2 (MIP-2) in response to P. carinii β-glucan challenge.13,14 MIP-2, the rodent ortholog of the human C-X-C chemokine interleukin-8, is a potent neutrophil chemoattractant. We have also shown that AECs produce tumor necrosis factor-α (TNF-α) following PCBG challenge, a cytokine known to have numerous pro-inflammatory effects.13–15 We have further demonstrated that, on a cell-by-cell basis, AECs produce more of these inflammation mediators than do identically challenged alveolar macrophages.14 Thus, we have postulated that the MIP-2 and TNF-α produced by AECs contribute to the lung inflammation observed during severe Pneumocystis pneumonia.11
The mechanisms by which Pneumocystis β-glucans stimulate inflammatory cytokine and chemokine production remain incompletely elucidated.16 Previous investigations from our laboratory indicate that lactosylceramide, a prominent cell membrane glycosphingolipid, functions as a major β-glucan receptor on AECs, facilitating the subsequent inflammatory responses.17 Microdomains (lipid rafts) are known to facilitate cellular internalization and transcytosis of a variety of macromolecules by AECs.18,19 Further, microdomain function is required for the establishment and propagation of several known infectious processes among both respiratory and non-respiratory epithelia.19–23 The current investigations explore the relevance of functional AEC membrane lipid microdomains in Pneumocystis β-glucan-induced inflammatory cytokine expression and AEC handling of PCBG.
MATERIALS AND METHODS
Reagents and Organisms
Unless otherwise noted, all general reagents were from Sigma Chemical Co. (St. Louis, MO). All animal experiments were reviewed and approved by the Mayo Institutional Animal Care and Usage Committee prior to initiation of these studies. Pneumocystis carinii (P. carinii) was originally obtained through the American Type Culture Collection (ATCC, Bethesda, MD) and maintained in our colony of dexamethasone-treated immunosuppressed Long Evans rats (HSD, Inc., Indianapolis, IN), as we previously reported.24
A P. carinii β-D-glucan enriched cell wall isolate (PCBG) was prepared, as we have previously described.10 Briefly, P. carinii organisms were isolated from lungs of heavily infected rats, autoclaved and disrupted by ultrasonication. Glucan was isolated by NaOH digestion and lipid extraction, washed first with 0.1% SDS, and then vigorously washed with distilled physiological saline to remove the detergent. This isolate was confirmed to contain a predominantly glucose-rich complex carbohydrate complex, which was largely degraded by β-1,3-glucanases. Only those β-glucan preparations that consistently displayed <0.125 units of endotoxin by the Limulus amebocyte lysate method were utilized in these studies.
Alveolar Epithelial Cell Isolation
In our hands, primary alveolar epithelial cells provide most robust chemokine responses to P. carinii β-glucans, and provide the advantage of studying rat host specific epithelial cells challenged with rat-derived PCBG. Accordingly, rat alveolar epithelial cells (AECs) were isolated as described by Dobbs and colleagues.25 In short, pentobarbital-anesthetized rats (~250 g) were sacrificed by transection of the inferior vena cava. The pulmonary vasculature was perfused with saline. The trachea was isolated and the lungs depleted of alveolar macrophages by multiple lavages. AECs were separated from the basement membrane by incubation with porcine elastase, and the lungs were minced, filtered, and centrifuged. Recovered cells were suspended in serum-free DMEM and incubated for 1 hour in Petri dishes coated with rat IgG to remove residual macrophages. The supernatants ware collected and centrifuged prior to suspension of the epithelial cells in DMEM containing 10% bovine calf serum, penicillin (50,000 units/liter) and streptomycin (50 mg/liter). AECs were counted using a standard haemocytometer. The AECs were incubated (37°C, 5% CO2) and allowed to adhere to culture plates for at least 48 hours. The media was changed after the initial 24 hours. After 48 hours, the cells had largely lost lamellar inclusion bodies, and were flattened and well spread, as well as firmly attached, displaying morphology more reminiscent of Type I cells than the Type II cells. Prior studies have demonstrated the Pneumocystis interacts predominantly with Type I cells, but also interact to a lesser degree with Type II cells.11
PCBG Challenge of AECs and MIP-2 Responses
As we previously reported, primary AECs were maintained in monolayer culture for approximately 48 hours prior to challenge with PCBG (100 μg/mL, ~5 × 106 particles/ml) for 6 hours. Culture media was then collected and submitted to MIP-2 sandwich ELISA (Biosource, Camarillo, CA) to characterize the inflammatory response. To define the effects of inhibitors of internalization on eventual PCBG-induced MIP-2 expression, we challenged AECs with or without various pretreatments. Endocytotic function was nonspecifically impaired by maintaining the cell culture at 4°C for 30 minutes prior to challenge and throughout the challenge. Microdomain formation and function was disrupted by 30 minutes pretreatment with concentrations of agents known to inhibit plasma membrane lipid microdomains including; genistein 5 μM, methyl-β-cyclodextrin 5mM or nystatin 25 μg/mL (Sigma Nystatin N1638) prior to challenge of the AECs with PCBG.26,27 In contrast, clathrin-mediated internalization was impaired by 30 minutes pretreatment with chlorpromazine 8 μg/mL. 26,27
Microdomain-localized protein kinase C activation
We have previously demonstrated that PCBG challenge induces MIP-2 production by NF-κB and PKC-dependent mechanisms.13 Because PKC has been reported to be associated with certain membrane microdomains, we isolated microdomains from both PCBG challenged and PCBG naïve AECs by Triton X-100 fractionation.28 After homogenization of the samples, the microdomains were separated by SDS-PAGE, and then submitted to immunoblotting using a rabbit monoclonal phospho-PKC (pan) antibody (Antibody 9371, Cell Signaling, Danvers, MA). The samples were next exposed to HRP-conjugated anti-rabbit secondary antibody and ECL chemiluminescent detection (Amersham, Piscataway, NJ).
Conventional, fluorescence, and confocal microscopy detection of PCBG internalization
Dichlorotriazinylaminofluorescein (DTAF, 10 mg in 0.1M Borax solution) was incubated overnight with 30 mg PCBG, then repeatedly centrifuged and rinsed with sterile 0.9% normal saline until all unbound DTAF was removed. AECs grown in monolayers on LabTek II slides were exposed to DTAF-labeled PCBG for one hour, with or without pretreatment to impede internalization. Cultures were maintained at 4°C for 30 minutes prior to challenge and throughout the subsequent challenge to non-specifically impair internalization. Glycosphingolipid-cholesterol rich microdomain internalization was impaired using nystatin (25 μg/mL) applied 30 minutes prior to challenge.29 In addition we also pretreated the cells with anti-lactosylceramide antibody (anti-CDw17, 200 μg/mL), or anti-lactosylceramide antibody in the presence of added exogenous lactosylceramide (1 mg/mL).13
Samples were then challenged for two hours with DTAF-labeled PCBG. The media were removed and the cells submitted to two rapid washes with PBS before repeated acid stripping with DMEM at pH 2.2 to remove all non-internalized PCBG. The cells were next aggressively washed with cold PBS, fixed with 4% paraformaldehyde, and quenched with glycine. The media chambers were removed from the slides and coverslips applied with Slow Fade (Molecular Probes, Carlsbad, CA). The cells were examined by fluorescence (excitation 490 nm, emission 520 nm) and phase contrast microscopy using an Olympus IX70 microscope (Melville, NY).
To confirm the internalization of the DTAF-labeled PCBG, AECs were grown and challenged as described above (no pretreatments were performed). The cells were acid-stripped and fixed as described above. To define the plasma membrane borders, the challenged cells were incubated with Vybrant DiI cell labeling solution (Molecular Probes, Eugene, OR; 1:200 dilution; absorption 549 nm, emission 565 nm) for approximately eight minutes. The fixed, labeled cells were then submitted to confocal microscopy using a Zeiss model 510 microscope equipped with a 100× (1.4 numerical aperture) objective. Sequential laser acquisition was performed for the two dyes, and all images were examined in multiple z-planes to confirm cellular location of the DTAF-labeled PCBG particles.
Spectophotofluorimetric Assessment of PCBG internalization
To quantify the effects on AEC internalization of PCBG, we pretreated cells with several different inhibitors of internalization for 30 minutes prior to challenge. We utilized pretreatment with anti-CDw17 and nystatin to inhibit lactosylceramide rich microdomains. We also pretreated with antibodies against an arbitrary membrane glycosphingolipid, anti-GM1 (200 μg/ml), as well as cytochalasin D (5 μg/ml) to generally inhibit phagocytic function. Four hours after PCBG challenge, the cells were acid stripped, then lysed with 2N NaOH. The lysates were submitted to conventional spectrophotofluorimetry using an Aminco SLM 8000C spectrophotofluorimeter (SLM Instruments, Inc., Urbana, IL), optimized for DTAF as noted above. Samples were maximally shielded from light prior to analysis and agitated to ensure even distribution of particles immediately before analysis. At least three trials for each condition and at least four aliquots from each sample were analyzed.
Data Analysis
All data analysis was preformed using JMP 5 software (SAS Institute, Cary, NC). The data are expressed as mean ± standard error of the mean. Differences between groups were defined using a two-tailed Student’s t-test, with p<0.05 being considered a significant difference.
RESULTS
MIP-2 production following PBCG challenge is inhibited by low temperature and by inhibitors of plasma membrane microdomains
We have previously demonstrated that challenge AEC with PCBG induced robust chemokine production.13 Consistent with our previous reports, by six hours after PCBG challenge, the AECs had generated significant concentrations of MIP-2 in the culture media (Figure 1). Furthermore, production of MIP-2 was significantly inhibited by culturing the cells at 4°C or by treating the cells with all three tested inhibitors of membrane microdomain function: genistein, methyl-β-cyclodextrin, or nystatin.30 In contrast, treatment of AECs with chlorpromazine at concentrations known to inhibit clathrin-coated pit-mediated internalization had no effect on MIP-2 production. These results suggest that AEC stimulation to release chemokines following PCBG stimulation required intact cell membrane microdomain function, but not clathrin mediated function.
Figure 1. Disruption of microdomain function impairs P. carinii β-glucan-induced inflammatory cytokine expression.

In order to determine the effect of various internalization mechanisms, primary AECs were challenged with PCBG with or without inhibitory pretreatment. To impair general internalization, cells were maintained at 4°C for 30 minutes prior to challenge. Microdomain function was disrupted by several mechanisms, including 30 minutes pretreatment with genistein, methyl-β-cyclodextran or nystatin. In contrast, internalization via clathrin-mediated mechanisms was inhibited by 30 minutes pretreatment with chlorpromazine. ELISA detection of MIP-2 was performed on the supernatant after four hours of challenge. These experiments showed significant suppression of PCBG-induced MIP-2 expression following both cold and anti-microdomain pretreatments, while anti-clathrin pretreatment had little effect. (* Denotes P<0.05).
PKC activation by PCBG is localized to membrane microdomains
We have also previously demonstrated that PCBG-induced cytokine and chemokine production by AECs is PKC-dependent.13 We next sought to determine whether active phospho-PKC was localized to the microdomains of AECs following PCBG stimulation (Figure 2). Indeed, PKC was found to localize to isolated microdomains as early as 15 minutes following PCBG stimulation. By 30 minutes the PKC signal was decreased. Our studies further revealed that this PKC effect was best appreciated when using antibodies against pan-phospho-PKC (shown), suggesting that non-α/non-β PKC isoforms may play a prominent role in this function.
Figure 2. P. carinii β-glucan induces microdomain-localized protein kinase C activation.
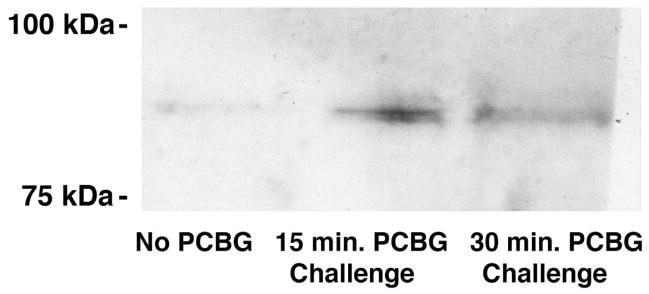
Primary AECs were harvested from monolayer with or without PCBG challenge. Lipid microdomains (rafts) were then isolated from the samples by Triton X-100 fractionation.28 Following homogenization, the samples were separated by SDS-PAGE then submitted to Western blotting using a pan-phospho-PKC antibody. Samples were exposed to HPR-conjugated secondary antibody and ECL chemiluminescent detection system, prior to autoradiography. While the unchallenged cell samples showed little activated (phosporylated) PKC, the PCBG-challenged cells demonstrated a significant increase in the autoradiographic signal. Double bands are noted, due to multiple PKC isoforms. This effect appeared maximal at 15 minutes and began to decline around 30 minutes.
PCBG is internalized by AECs, which can be suppressed by inhibitors of membrane microdomain function
Confocal microscopy was further utilized to determine whether PCBG is internalized by AECs (Figure 3). Fluorescent DTAF-labeled PCBG was fully internalized by AECs within one hour of challenge. The internalized location was confirmed by analysis in multiple z-planes, using lipophilic labeling to delineate the cell extent. We then demonstrated that PCBG internalization was interrupted by disrupting microdomain function; either by directing antibodies against lactosylceramide, culturing the cells at 4°C at the time of challenge, or pretreatment with nystatin, an agent that inhibits glycosphingolipid-cholesterol rich microdomains, including caveoli (Figure 4).
Figure 3. Alveolar epithelial cells internalize fluorescently labeled P. carinii β-glucan.
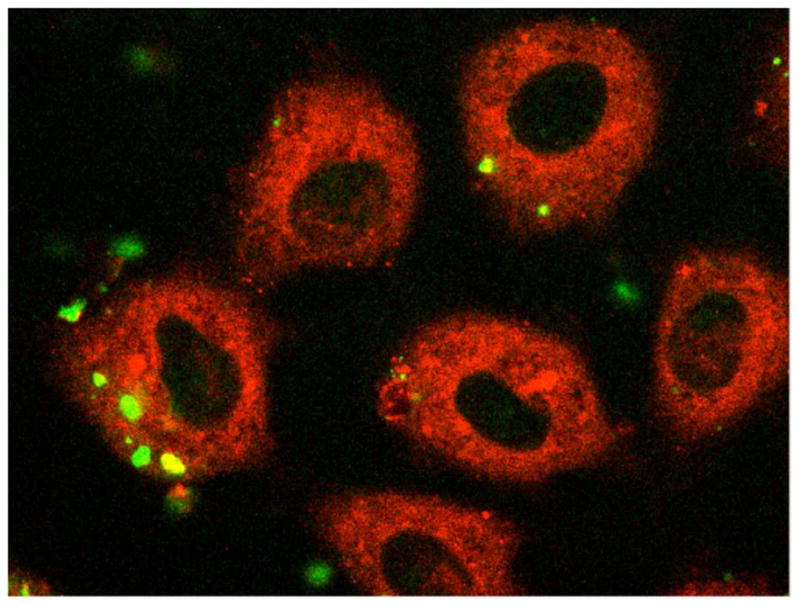
After approximately 48 hours culture, primary AECs were exposed to DTAF-labeled PCBG for one hour. The samples were then acid stripped, formalin fixed, and treated with a lipophilic counterstain prior to being submitted to confocal fluorescent microscopy. Multiple images in the z-plane were obtained to confirm complete internalization of PCBG particle. This photomicrograph demonstrates DTAF-labeled PCBG (green) in various stages of internalization. The internalized PCBG displayed yellow coloration when imaged in this manner.
Figure 4. Microdomain disruption impairs internalization of DTAF-labeled P. carinii β-glucan by alveolar epithelial cells.

Primary AECs were challenged with DTAF-labeled PCBG with or without pretreatment, and then submitted to conventional fluorescence microscopy. A. DTAF-labeled PCBG was internalized by alveolar epithelial cells 2 hours after challenge at 37°C. B. In contrast, culture of the AECs at 4°C prevented PCBG uptake. C. Monoclonal antibodies targeting the microdomain glycosphingolipid constituent lactosylceramide also prevented PCBG uptake. D. However, preincubation of the anti-lactosylceramide antibody with free lactosylceramide abrogated this effect. E. Finally, disruption of lipid microdomains with nystatin also inhibited PCBG uptake.
To further quantify the amount of labeled PCBG internalized under various conditions, spectophotofluorimetry of the cultured AEC lysates was performed following various AEC membrane treatments and PCBG challenge (Figure 5). Once again, treatment with either nystatin or anti-lactosylceramide antibodies that impaired microdomain function each significantly suppressed PCBG internalization. In contrast, antibodies against an arbitrary glycosphingolipid (GM1) had no such effect. As anticipated, generalized impairment of actin function with cytochalasin D impaired internalization approximately as much as anti-lactosylceramide antibody pretreatment, but less than nystatin inhibition of caveolae. Overall, these data support that membrane microdomains rich in glycosphingolipids and cholesterol particularly lactosylceramide participate in epithelial cell activation to release cytokines in response to the pro-inflammatory Pneumocystis β-glucan cell wall components.
Figure 5. Impaired microdomain function decreases internalized DTAF-labeled P. carinii β-glucan signal.

Primary rat AECs were challenged with DTAF-labeled PCBG with or without pretreatment by a variety of inhibitors of internalization. After four hours of challenge, the samples were acid stripped, then lysed with sodium hydroxide. Homogenized equal volumes from each sample were then assayed by conventional photospectrofluorimetry. Anti-lactosylceramide antibodies and nystatin both significantly impaired the internalization of DTAF-labeled PCBG, as measured by this method. Cytochalasin D had a lesser, but still significant, effect. Antibodies against an arbitrary glycosphingolipid (GM-1) had no effect on PCBG uptake. (* Denotes P<0.01).
DISCUSSION
Our prior investigations have convincingly demonstrated that PCBG induces inflammatory responses from AECs.13,14 Further, we have shown that these responses are dependent upon PCBG interaction with lactosylceramide at the cell surface and that secondary signaling by NF-κB and PKC occur following challenge.13,14 The current investigations further emphasize the importance of plasma membrane microdomain function in PCBG-induced inflammation.
Handling of macromolecules in the airspaces is highly dependent upon the AECs and alveolar macrophages that internalize the particles that deposit peripherally. AECs are essential for the recycling of surfactant proteins, and are adept at bidirectional transcytosis of proteins and carbohydrates.18,31–36 Given the mass of particles inhaled as humans ventilate around 10 L ambient air per minute, this macromolecule processing function of epithelial cells is critical as it would overwhelm the capacity of professional phagocytes, such as the infrequently encountered alveolar macrophage. AECs are also known to internalize cellular elements and toxins from respiratory pathogens,23 as well as whole organisms including Aspergillus fumigatus,21 Streptococcus pneumoniae22, and Pseudomonas aeruginosa.20 Membrane microdomains appear necessary for internalization of several pathogens, including atypical caveolar endocytosis of SV-40 virus and FimH-associated endocytosis of Escherichia coli.19 Pathogen internalization functions of microdomains have been demonstrated in several organs and cell types, but we are unaware of any data regarding microdomain-mediated internalization of Pneumocystis or any of its components.
Microdomains are localized regions of the plasma membrane enriched in glycosphingolipids and cholesterol, and measuring 10–200 nm in size.19,31,37 Microdomains have been implicated in cell signaling and uptake of particulate material.29 Thus, it is intriguing that 1–2 μm PCBG particles require microdomain function for internalization. This may be possibly explained by the notion that internalization is initiated by microdomain-localized cell signaling events (perhaps PKC activation), while the process of mechanical internalization occurs by otherwise microdomain-independent phagocytosis. This would be consistent with our observation that both microdomain disruption and cytochalasin D pretreatment inhibit internalization. It is also notable that Pneumocystis interaction with AECs during lung infection induces plasmalemmal activation, resulting in dramatic alterations of the surface membrane structure of the type 1 and type 2 pneumocytes observed by electron microscopy.38 It has been speculated that this allows the pathogen to more efficiently extract nutrients from the host.
The relevance of the current observation lies primarily in the potential therapeutic implications. Pneumocystis pneumonia continues to exert an unacceptably high case-mortality rate among immunocompromised patients, particularly those who are immunosuppressed for reasons other than HIV disease.1–3 The improved mortality observed following the introduction of systemic corticosteroids to the typical antimicrobial regimens for P. carinii pneumonia in the 1980s first indicated the importance of host inflammation in the morbidity associated with the disease.39 Additional studies revealed that, in fact, markers of exaggerated host inflammation were stronger predictors of mortality than were pathogen factors, such as organism burden.6–8
Previous data from our laboratory have suggested that inhibition of NF-κB signaling can reduce the AEC-generated PCBG-induced inflammatory response.13,14 In addition, inflammatory mediator release is also significantly reduced with PKC inhibition. Both of these interventions provide potential therapeutic targets to improve human outcomes. The observation that membrane disruption with nystatin inhibits both internalization of PCBG and dramatically attenuates PCBG-induced chemokine production may offer an additional appealing target to modulate Pneumocystis induced inflammation. Nystatin is a polyene antibiotic with fungistatic and fungicidal properties. The in vitro antifungal activity probably arises due to sterol binding resulting in excessive cell membrane permeability. 40 Binding to mammalian cholesterol appears to also underlie its anti-microdomain effects, as well.
It is further important to note that we observed nystatin to inhibit internalization, cell signaling, and mediator release in response to PCBG. In addition, since Pneumocystis organisms appear to establish infections at the level of type 1 pneumocytes and that such associations may alter activation of microdomains in AECs, we further speculate that treatment with nystatin (or a similar agent) may also impede the establishment or propagation of infection with this pathogen, though that hypothesis remains to be tested. The therapeutic potential for reducing Pneumocystis-induced inflammation by nystatin is particularly attractive. At the present time, clinical data does not exist to support its use, and the current study was not designed to address the anti-Pneumocystis activity of any of the inhibitors in the overall progression of infection and lung injury. Nonetheless, these investigations do suggest novel therapeutic targets that may eventually benefit patients facing this severe infection.
Acknowledgments
These studies are dedicated to the memory of our departed collaborator, Richard E. Pagano, Ph.D., who provided key insights and assistance during our studies of the roles of glycosphingolipids in initiating inflammation. These investigations were funded by the Mayo Foundation and NIH grant R01-HL62150 to AHL. We also thank Zvezdana Vuk-Pavlovic’ and David Marks for many helpful discussions during these studies.
References
- 1.Beck JM, Rosen MJ, Peavy HH. Pulmonary complications of HIV infection. Report of the Fourth NHLBI Workshop. Am J Respir Crit Care Med. 2001;164:2120–6. doi: 10.1164/ajrccm.164.11.2102047. [DOI] [PubMed] [Google Scholar]
- 2.Thomas CF, Jr, Limper AH. Pneumocystis pneumonia: clinical presentation and diagnosis in patients with and without acquired immune deficiency syndrome. Semin Respir Infect. 1998;13:289–95. [PubMed] [Google Scholar]
- 3.Thomas CF, Jr, Limper AH. Pneumocystis pneumonia. N Engl J Med. 2004;350:2487–98. doi: 10.1056/NEJMra032588. [DOI] [PubMed] [Google Scholar]
- 4.Festic E, Gajic O, Limper AH, Aksamit TR. Acute respiratory failure due to pneumocystis pneumonia in patients without human immunodeficiency virus infection: outcome and associated features. Chest. 2005;128:573–9. doi: 10.1378/chest.128.2.573. [DOI] [PubMed] [Google Scholar]
- 5.Wright TW, Gigliotti F, Finkelstein JN, McBride JT, An CL, Harmsen AG. Immune-mediated inflammation directly impairs pulmonary function, contributing to the pathogenesis of Pneumocystis carinii pneumonia. J Clin Invest. 1999;104:1307–17. doi: 10.1172/JCI6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hahn PY, Limper AH. The role of inflammation in respiratory impairment during Pneumocystis carinii pneumonia. Semin Respir Infect. 2003;18:40–7. doi: 10.1053/srin.2003.50004. [DOI] [PubMed] [Google Scholar]
- 7.Limper AH, Offord KP, Smith TF, Martin WJ., 2nd Pneumocystis carinii pneumonia. Differences in lung parasite number and inflammation in patients with and without AIDS. Am Rev Respir Dis. 1989;140:1204–9. doi: 10.1164/ajrccm/140.5.1204. [DOI] [PubMed] [Google Scholar]
- 8.Mansharamani NG, Garland R, Delaney D, Koziel H. Management and outcome patterns for adult Pneumocystis carinii pneumonia, 1985 to 1995: comparison of HIV-associated cases to other immunocompromised states. Chest. 2000;118:704–11. doi: 10.1378/chest.118.3.704. [DOI] [PubMed] [Google Scholar]
- 9.Wang J, Gigliotti F, Maggirwar S, Johnston C, Finkelstein JN, Wright TW. Pneumocystis carinii activates the NF-kappaB signaling pathway in alveolar epithelial cells. Infect Immun. 2005;73:2766–77. doi: 10.1128/IAI.73.5.2766-2777.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vassallo R, Standing JE, Limper AH. Isolated Pneumocystis carinii cell wall glucan provokes lower respiratory tract inflammatory responses. J Immunol. 2000;164:3755–63. doi: 10.4049/jimmunol.164.7.3755. [DOI] [PubMed] [Google Scholar]
- 11.Thomas CF, Jr, Limper AH. Current insights into the biology and pathogenesis of Pneumocystis pneumonia. Nat Rev Microbiol. 2007;5:298–308. doi: 10.1038/nrmicro1621. [DOI] [PubMed] [Google Scholar]
- 12.Limper AH, Thomas CF, Jr, Anders RA, Leof EB. Interactions of parasite and host epithelial cell cycle regulation during Pneumocystis carinii pneumonia. J Lab Clin Med. 1997;130:132–8. doi: 10.1016/s0022-2143(97)90089-5. [DOI] [PubMed] [Google Scholar]
- 13.Evans SE, Hahn PY, McCann F, Kottom TJ, Pavlovic ZV, Limper AH. Pneumocystis cell wall beta-glucans stimulate alveolar epithelial cell chemokine generation through nuclear factor-kappaB-dependent mechanisms. Am J Respir Cell Mol Biol. 2005;32:490–7. doi: 10.1165/rcmb.2004-0300OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hahn PY, Evans SE, Kottom TJ, Standing JE, Pagano RE, Limper AH. Pneumocystis carinii cell wall beta-glucan induces release of macrophage inflammatory protein-2 from alveolar epithelial cells via a lactosylceramide–mediated mechanism. J Biol Chem. 2003;278:2043–50. doi: 10.1074/jbc.M209715200. [DOI] [PubMed] [Google Scholar]
- 15.Hoffman OA, Standing JE, Limper AH. Pneumocystis carinii stimulates tumor necrosis factor-alpha release from alveolar macrophages through a beta-glucan-mediated mechanism. J Immunol. 1993;150:3932–40. [PubMed] [Google Scholar]
- 16.Benfield TL, Lundgren B, Shelhamer JH, Lundgren JD. Pneumocystis carinii major surface glycoprotein induces interleukin-8 and monocyte chemoattractant protein-1 release from a human alveolar epithelial cell line. Eur J Clin Invest. 1999;29:717–22. doi: 10.1046/j.1365-2362.1999.00517.x. [DOI] [PubMed] [Google Scholar]
- 17.Hahn PY, Limper AH. Pneumocystis carinii beta-glucan induces release of macrophage inflammatory protein-2 from primary rat alveolar epithelial cells via a receptor distinct from CD11b/CD18. J Eukaryot Microbiol. 2001;(Suppl):157S. doi: 10.1111/j.1550-7408.2001.tb00498.x. [DOI] [PubMed] [Google Scholar]
- 18.Gumbleton M. Caveolae as potential macromolecule trafficking compartments within alveolar epithelium. Adv Drug Deliv Rev. 2001;49:281–300. doi: 10.1016/s0169-409x(01)00142-9. [DOI] [PubMed] [Google Scholar]
- 19.Norkin LC. Caveolae in the uptake and targeting of infectious agents and secreted toxins. Adv Drug Deliv Rev. 2001;49:301–15. doi: 10.1016/s0169-409x(01)00143-0. [DOI] [PubMed] [Google Scholar]
- 20.Kannan S, Audet A, Knittel J, Mullegama S, Gao GF, Wu M. Src kinase Lyn is crucial for Pseudomonas aeruginosa internalization into lung cells. Eur J Immunol. 2006 doi: 10.1002/eji.200635973. [DOI] [PubMed] [Google Scholar]
- 21.Paris S, Boisvieux-Ulrich E, Crestani B, et al. Internalization of Aspergillus fumigatus conidia by epithelial and endothelial cells. Infect Immun. 1997;65:1510–4. doi: 10.1128/iai.65.4.1510-1514.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Talbot UM, Paton AW, Paton JC. Uptake of Streptococcus pneumoniae by respiratory epithelial cells. Infect Immun. 1996;64:3772–7. doi: 10.1128/iai.64.9.3772-3777.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Torgersen ML, Skretting G, van Deurs B, Sandvig K. Internalization of cholera toxin by different endocytic mechanisms. J Cell Sci. 2001;114:3737–47. doi: 10.1242/jcs.114.20.3737. [DOI] [PubMed] [Google Scholar]
- 24.Limper AH, Hoyte JS, Standing JE. The role of alveolar macrophages in Pneumocystis carinii degradation and clearance from the lung. J Clin Invest. 1997;99:2110–7. doi: 10.1172/JCI119384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dobbs LG, Gonzalez R, Williams MC. An improved method for isolating type II cells in high yield and purity. Am Rev Respir Dis. 1986;134:141–5. doi: 10.1164/arrd.1986.134.1.141. [DOI] [PubMed] [Google Scholar]
- 26.Singh RD, Holicky EL, Cheng ZJ, et al. Inhibition of caveolar uptake, SV40 infection, and beta1-integrin signaling by a nonnatural glycosphingolipid stereoisomer. J Cell Biol. 2007;176:895–901. doi: 10.1083/jcb.200609149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh RD, Liu Y, Wheatley CL, et al. Caveolar endocytosis and microdomain association of a glycosphingolipid analog is dependent on its sphingosine stereochemistry. J Biol Chem. 2006;281:30660–8. doi: 10.1074/jbc.M606194200. [DOI] [PubMed] [Google Scholar]
- 28.Mazzone A, Tietz P, Jefferson J, Pagano R, LaRusso NF. Isolation and characterization of lipid microdomains from apical and basolateral plasma membranes of rat hepatocytes. Hepatology. 2006;43:287–96. doi: 10.1002/hep.21039. [DOI] [PubMed] [Google Scholar]
- 29.Singh RD, Holicky EL, Cheng ZJ, et al. Inhibition of caveolar uptake, SV40 infection, and beta1-integrin signaling by a nonnatural glycosphingolipid stereoisomer. J Cell Biol. 2007;176:895–901. doi: 10.1083/jcb.200609149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh RD, Puri V, Valiyaveettil JT, Marks DL, Bittman R, Pagano RE. Selective caveolin-1-dependent endocytosis of glycosphingolipids. Mol Biol Cell. 2003;14:3254–65. doi: 10.1091/mbc.E02-12-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gumbleton M. Caveolae-mediated membrane transport. Adv Drug Deliv Rev. 2001;49:217–21. doi: 10.1016/s0169-409x(01)00137-5. [DOI] [PubMed] [Google Scholar]
- 32.John TA, Vogel SM, Minshall RD, Ridge K, Tiruppathi C, Malik AB. Evidence for the role of alveolar epithelial gp60 in active transalveolar albumin transport in the rat lung. J Physiol. 2001;533:547–59. doi: 10.1111/j.1469-7793.2001.0547a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim KJ, Matsukawa Y, Yamahara H, Kalra VK, Lee VH, Crandall ED. Absorption of intact albumin across rat alveolar epithelial cell monolayers. Am J Physiol Lung Cell Mol Physiol. 2003;284:L458–65. doi: 10.1152/ajplung.00237.2002. [DOI] [PubMed] [Google Scholar]
- 34.Matsukawa Y, Yamahara H, Lee VH, Crandall ED, Kim KJ. Horseradish peroxidase transport across rat alveolar epithelial cell monolayers. Pharm Res. 1996;13:1331–5. doi: 10.1023/a:1016013731237. [DOI] [PubMed] [Google Scholar]
- 35.Matsukawa Y, Yamahara H, Yamashita F, Lee VH, Crandall ED, Kim KJ. Rates of protein transport across rat alveolar epithelial cell monolayers. J Drug Target. 2000;7:335–42. doi: 10.3109/10611869909085516. [DOI] [PubMed] [Google Scholar]
- 36.Widera A, Kim KJ, Crandall ED, Shen WC. Transcytosis of GCSF-transferrin across rat alveolar epithelial cell monolayers. Pharm Res. 2003;20:1231–8. doi: 10.1023/a:1025005232421. [DOI] [PubMed] [Google Scholar]
- 37.Hasegawa H, Zinsser S, Rhee Y, Vik-Mo EO, Davanger S, Hay JC. Mammalian ykt6 is a neuronal SNARE targeted to a specialized compartment by its profilin-like amino terminal domain. Mol Biol Cell. 2003;14:698–720. doi: 10.1091/mbc.E02-09-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Settnes OP, Nielsen MJ. Host-parasite relationship in Pneumocystis carinii infection: activation of the plasmalemmal vesicular system in type I alveolar epithelial cells. J Protozool. 1991;38:174S–6S. [PubMed] [Google Scholar]
- 39.Bozzette SA, Sattler FR, Chiu J, et al. A controlled trial of early adjunctive treatment with corticosteroids for Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome. California Collaborative Treatment Group. N Engl J Med. 1990;323:1451–7. doi: 10.1056/NEJM199011223232104. [DOI] [PubMed] [Google Scholar]
- 40.Fjaervik E, Zotchev SB. Biosynthesis of the polyene macrolide antibiotic nystatin in Streptomyces noursei. Appl Microbiol Biotechnol. 2005;67:436–43. doi: 10.1007/s00253-004-1802-4. [DOI] [PubMed] [Google Scholar]