

Abstract
Arabidopsis thaliana (Arabidopsis) is ideally suited for studies of natural phenotypic variation. This species has also provided an unparalleled experimental system to explore the mechanistic link between genetic and epigenetic variation, especially with regard to cytosine methylation. Using high-throughput sequencing methods, genotype to epigenotype to phenotype observations can now be extended to plant populations. We review the evidence for induced and spontaneous epigenetic variants that have been identified in Arabidopsis in the laboratory and discuss how these experimental observations could explain existing variation in the wild.
Natural genetic variation
Diversity within and between species is primarily driven by genetic variation and interactions with the environment. Arabidopsis thaliana has adapted to numerous environments, as it exists naturally and broadly throughout the northern hemisphere [1]. It is a selfing species that contains a largely isogenic genome, although residual heterozygosity does exist due to infrequent admixture [2]. The genome is compact and depleted of transposons and is relatively small (~150 Mb) compared to other plant species [3,4]. Recently, the Arabidopsis 1001 Genomes Project (see 1001genomes.org and signal.salk.edu/atg1001) was initiated with the goal of identifying the total genetic variation present within this species and to enable scientists around the globe to study their favorite quantitative trait of interest using association or linkage mapping techniques [1,5,6]. One challenge for adaptation of Arabidopsis to novel environments may be a limited set of mutations [7,8]. Therefore, it is essential to identify not only genetic variation but also natural epigenetic (see Glossary) variation, for which Arabidopsis is ideally suited [9,10]. In this regard, in addition to sequencing genomes [11–15], efforts are underway by the Arabidopsis 1001 Epigenomes Project (http://www.nsf.gov/awardsearch/showAward.do?AwardNumber=1122246) to sequence transcriptomes and DNA methylomes, which will be useful for identifying natural epigenetic variants.
Epigenetic alleles (Epialleles)
The extent to which epigenetic variation contributes to phenotypes remains to be determined [16]. However, several examples of phenotypic variation that can arise through epigenetic variation are known, including the peloric (Linaria vulgaris) [17], colorless non-ripening (cnr – Solanum lycopersicum) [18] and B′ (Zea mays) [19] epialleles. Studies in Arabidopsis thaliana have provided the richest source of known epialleles [20–32]. Many of the Arabidopsis epialleles require the activities of the RNA-directed DNA methylation (RdDM) machinery [33] for their formation and stability. RdDM functions to silence repetitive and transposon sequences and in some cases genes by a complex set of machinery that initiates transcription, generates 24nt RNAs and methylates targeted DNA in all cytosine contexts (for review see [33]). In fact, a number of these epialleles were used in genetic screens or as markers [21,22,28,29] to uncover novel components and mechanisms of the RdDM pathway [34–48].
Epigenetics is the study of mitotically and/or meiotically heritable changes in phenotype that arise independent of genetic variation. This definition is especially difficult to reconcile in studies of natural epigenetic variation, as by definition the populations under study exhibit extensive genetic variation. Recognizing this conundrum and the complexity of epiallele formation, a classification system that grouped epialleles into one of three classes based on their dependence on genotype has been proposed [49]. The first class called obligate epialleles display a complete dependency on a genetic variant, whereas the second class called facilitated epialleles form from the presence of a genetic variant such as a nearby transposon insertion, but their maintenance is not necessarily dependent on this variant. The third class, pure epialleles, form independent of any genetic variation. The epialleles thus far identified in Arabidopsis are either obligate epialleles [21,24] or are facilitated epialleles, as their formation has been induced using mutations [23,27,28,50] in machinery required for maintenance of DNA methylation or they were recovered from mutagenesis experiments [22,29]. Mitotically pure epialleles exist [51], but until recently [52–54] the identification of naturally occurring meiotically pure epialleles has remained elusive.
Spontaneous epigenetic variation
Epialleles have the potential for reversibility and often exist in metastable states, as has been observed for both the peloric and cnr natural epialleles [17,18]. With the exception of transposon-induced events, this feature is unique to epialleles as genetic mutations are typically considered unidirectional over evolutionary time. The molecular spectrum and rate of spontaneous mutations are relatively well understood especially when compared to our understanding of spontaneous epigenetic variation. Genome resequencing of a collection of Arabidopsis thaliana mutation accumulation (MA) lines, which were created from a lone founder by single seed descent growth for 30 generations [55], revealed just under one mutation per line per generation [56]. Recently, base pair resolution DNA methylomes of this same population were determined, which uncovered a rate of base level spontaneous variation in DNA methylation (single methylation polymorphisms – SMPs) that was at least four orders of magnitude greater than genetic mutations [52,53]. Furthermore, these studies captured the formation of pure epialleles, some of which resulted in significant transcriptional variation (~10- to 1000-fold change) of the affected locus [52,53]. One study proposed that these pure epialleles may have arisen through incomplete reinforcement of methylation states that occurs postfertilization [52] (Figure 1), because the sites of epiallelic variation overlapped significantly with loci that are also affected in the maintenance methyltransferase met1 and in the triple mutant of DNA demethylases rdd (REPRESSOROFSILENCING1; DEMETER-LIKE2; DEMETER-LIKE 3) [57–59]. The involvement of epigenetic mechanisms that require the activities of small RNAs and/or DNA methylation machinery are widespread in plant reproductive development. In fact, both the maternally and the paternally inherited genomes in Arabidopsis require intricate mechanisms for ensuring their identity and for inheritance of epigenomic information from generation to generation [60,61]. One feature of these mechanisms is to correct errors (loss of DNA methylation) through the use of smRNA (small RNA) guides [62], but inefficiencies in transferring this information across generations may result in the formation of a pure epiallele.
Figure 1.

Incorrect maintenance of methylation states over generational time at certain loci may lead to the formation of pure (nongenetic) epialleles. It has been proposed that the activities of MET1 and RDD are required to maintain methylation states in the germ line during postfertilization [52]. A failure to maintain the appropriate methylation state by plants wild type for either MET1 [(a) – loss of DNA methylation] or RDD [(b) – gain of methylation] or additional enzymes (X) may result in the formation of pure epialleles, unlinked to any cis or trans genetic variation. Gold lines, CG methylation; blue lines, CHG methylation; pink lines, CHH methylation.
The variation in methylation observed in the MA lines was not uniformly distributed throughout the genome. This was especially the case for CG gene body methylation [9,63,64], which still has an unknown role and requires MET1 for its maintenance. In fact, there was a depletion of variation associated with transposon sequences, exons and the pericentromeres [52,53]. These genomic regions all are associated with nucleosome occupancy [65] and may reflect that methyltransferase enzymes such as MET1 maintain nucleosome-associated DNA methylation at a higher fidelity than nucleosome-free DNA methylation.
In general, the rates of spontaneous variation in DNA methylation were similar between all of the MA lines except for one (line 69). This hypervariability is unlikely due to the environment because the entire population was derived in a common greenhouse in synchrony, which would be expected to affect numerous lines if there were such pressures. Additionally, line 69 was identified as being hypervariable in two independent laboratories [52,53]. The basis for this hypervariability has been examined by resequencing the genome of line 69 [53]. Intriguingly, an additional nonsynonymous SNP (G–T, Ala–Ser) not previously reported in this line [56] was identified in MEE57 [53], a homolog of the maintenance methyltransferase MET1 [66]. It is conceivable that the variation in DNA methylation observed in line 69 may be due to this mutation in MEE57, which could potentially represent one route to the formation of genome-wide epigenetic variation.
Mutation-induced epigenetic variation
As described above, spontaneous genetic variation may provide a source of natural epigenetic variation. Proof-of-concept for this type of epiallele formation was originally demonstrated by using mutants in DDM1 (DECREASE IN DNA METHYLATION 1 [67]), which encodes a SWI–SNF2-like protein that is related to chromatin remodelers in mouse and humans [68]. ddm1 mutants result in a genome-wide depletion of DNA methylation, especially in repeated regions of the genome [67]. Remarkably, remethylation of ddm1 chromosomes does not occur immediately upon introduction to wild-type chromosomes, instead remethylation occurs gradually over generational time [67]. Genome-wide depletion of DNA methylation [57,63,69] and epiallele formation [25] can also be induced in met1 mutants, which can result in a variety of developmental phenotypes [70,71]. It is noteworthy that ddm1 mutants do not display major developmental abnormalities until successive generations removed from the originally mutated plant [30,72]. However, much of this mutation-induced epigenetic variation results in the stable transmission of morphological phenotypes across generations, even when outcrossed from the original mutant backgrounds [23,27,50,73,74]. Although the major molecular phenotype of ddm1 or met1 mutants is a depletion of DNA methylation, examples of genetic variation in the form of genomic rearrangements, copy number variants (CNVs) and frequent DNA transposition have also been observed and may account for a considerable amount of phenotypic variability [21,50,73,75–77].
Barbara McClintock proposed the concept of gene regulation by transposon insertion within or near genes [78]. It is now commonly accepted that variation in gene expression can be regulated by the position of nearby transposons [79–82]. For example, transposon insertions in certain accessions of Arabidopsis thaliana can result in a rapid cycling flowering behavior [24]. Most fascinating is the recent identification of the ONSEN class of transposons that are heat responsive [83]. The transcriptional activation of ONSEN can persist for at least three days after the heat stress has been removed and can result in transposition in the germline of RNAi mutants [83]. Moreover, genome reinsertion of ONSEN in the RNAi mutants in the nonstressed next generation confers heat responsiveness to nearby genes [83]. Even though plant species have developed a wide range of mechanisms to ensure faithful transposon silencing, transpositions can occur over generational time either in the wild or in cell culture as well as in mutant backgrounds [62,76,83,84] and can provide a substrate for epigenetic variation to nearby genes (Figure 2).
Figure 2.
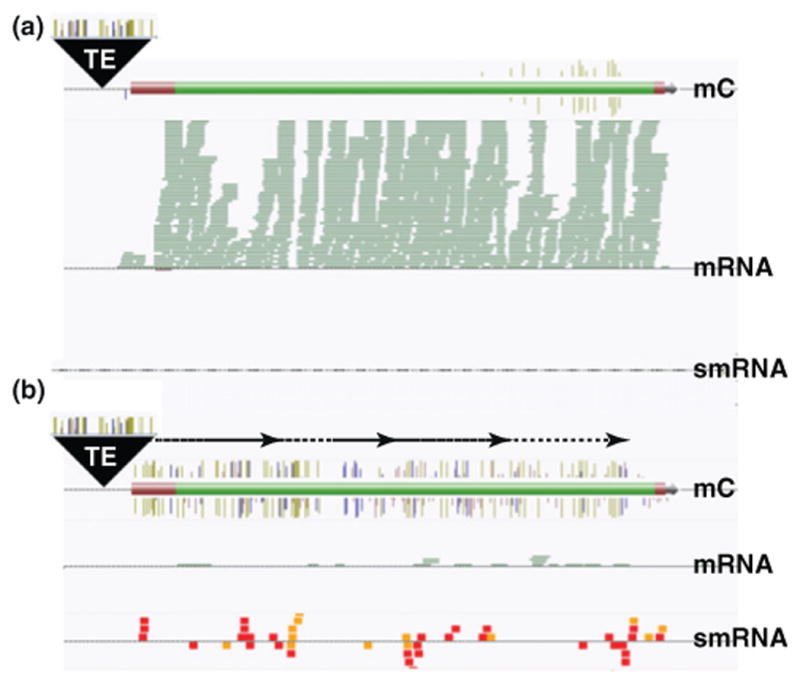
Transposon-derived epiallele formation. The presence of transposons and repetitive sequences can provide a nucleation point for spreading of DNA methylation and smRNAs into nearby genes, which can result in a decrease in gene expression. (a) Methylation of a transposon insertion upstream of the transcriptional start site of a gene may have no affect on downstream gene expression. (b) By a poorly understood mechanism(s), spreading of transposon methylation and smRNAs to a nearby gene results in reduced expression. Gold lines, CG methylation; blue lines, CHG methylation; pink lines, CHH methylation; green rectangles, mRNA sequencing reads; red boxes, 24 nucleotide smRNAs; orange boxes, 23 nucleotide smRNAs. Abbreviations: mC, methylation of the 5-position of cytosine.
Epigenetic recombinant inbred lines (epiRILs)
The relatively stable transmission of epigenetic variation observed in ddm1 and met1 mutants provided the basis for creation of epigenetic recombinant inbred lines or epiRILs [23,27]. RIL populations are commonly constructed in plants and are used for mapping causal variants for traits that are variable between two naturally occurring parental lineages. The epiRIL populations provide a unique opportunity to reduce genetic variability and increase epigenetic variability. Two independent epiRIL populations were created using either ddm1 [23] or met1 [27] mutants that were crossed to wild-type Col-0 plants to generate F1 hybrids. Segregating populations of plants were screened for the presence of wild-type alleles of either DDM1 or MET1 and propagated by single seed descent for multiple generations to increase homozygosity. Each epiRIL within the population essentially contains a mosaic epigenome derived from either wild type and ddm1 or wild type and met1. Using microarray-based epigenomic approaches, it was determined that although some of the epialleles remained stable throughout construction of the epiRIL population, many slowly reverted back to the wild-type status over generational time [23,27]. This type of reversion is not uncommon and probably results from de novo methylation through smRNA guides [62]. In addition to epigenetic variation, there was clear evidence of genetic variation in the form of transpositions in the epiRILs [23,27,76].
Both epiRIL populations contained extensive phenotypic variation that was stable throughout each population for numerous traits suchastime to flowering, plant height and fruit size [23,27]. Phenotypes present within the met1 epiRIL population resembled variation observed in natural Arabidopsis accessions [27]. Moreover, significant heritability estimates and continuous distribution of phenotypes were observed in the ddm1 epiRILs, indicating that multiple loci contribute to the observed phenotypic variation and these loci can potentially be identified through linkage mapping approaches [85]. Recently, the ddm1 epiRIL population was subjected to extensive phenotyping alongside natural Arabidopsis accessions in a common garden to test the contribution of epigenetic variation to phenotypic diversity [86]. The findings demonstrate that phenotypic variation in the epiRIL population displays heritability estimates similar to natural (wild) accessions grown in parallel [86]. Although epiRILs represent a set of artificially induced epialleles, these results provide compelling evidence that if spontaneous mutations do occur in components of the methylation machinery then an explosion of largely heritable epigenetic variation may quickly arise, providing a substrate for natural selection to act upon. As described above, a spontaneous mutation occurring in line 69 of the MA population was found in a component of methylation machinery (MEE57) [53], which could account for the increased hypervariability of methylation polymorphisms in these plants [52,53].
Natural epigenetic variation
Although natural epigenetic variation exists in Arabidopsis [9,10], understanding the basis for this variation presents a complex challenge due to the inherent genetic variation also present in these same accessions [11–15]. The experimental systems mentioned above provide alternative routes by which epigenetic variants can arise and spread within populations. There is, however, evidence for other events occurring in the wild [21,24,87,88]. A survey of Arabidopsis accessions for variation in methylation of the 178-bp centromeric satellite repeat identified hypomethylation of this region in the Bor-4 accession [88]. The causal variant for the hypomethylation was identified through linkage mapping as it segregated as a single recessive locus and encodes a methylcytosine binding protein named VIM1 (VARIANT IN METHYLATION 1)[88].Because the methylation status of the centromeric repeat was entirely dependent on the presence of the VIM1 variant, methylation variation of centromeric repeats in Bor-4 represents an example of ‘obligate’ epigenetic variation [49]. This case nicely illustrates the ability of genetic variants to affect epigenetic variation in trans similar to what is observed in the epiRIL population. Another example of natural epigenetic variation that uncovers the complexity of epiallele formation exists at the PAI (PHOSPHORIBOSYLANTHRANILATE ISOMERASE) loci in certain Arabidopsis accessions [21,89]. In the Col-0 genome the PAI loci are present as a three-member gene family with high nucleotide sequence identity. In some accessions, the PAI1 locus exists as a duplicated inverted repeat as well as in other rearranged forms depending on the accession [89]. This inverted repeat leads to silencing by the RdDM pathway both in cis at PAI1 as well as in trans at PAI2 and PAI3[45,90]. The methylated PAI loci provides an additional example of naturally occurring ‘obligate’ epialleles as the silencing of these loci dissipates when either PAI2 or PAI3 are segregated away by outcrossing from the inverted repeat at PAI1[21].
Epigenomic approaches for studying natural epigenetic variation
The type of epiallelic variation present at the PAI gene family in some accessions of Arabidopsis is prevalent within this species. Using a comparative epigenomic approach, extensive methylation variation was identified between the Col-0 and Ler accessions; some of which overlapped with copy number variants identified within these same accessions [9]. Two major groups of methylation variants were identified in this study; one group contained hypervariable genic methylation patterns although a second group included transposons, which were invariably methylated. This second group was strongly associated with smRNAs typical of the RdDM pathway, whereas the genic methylation showed no such relationship [9].
An array-based epigenomic approach [10] was used to examine the genome-wide patterns and heritability of methylation polymorphisms between two different Arabidopsis accessions. This study revealed that inheritance of methylation was largely additive, as intermediate methylation levels were observed in F1 hybrids. They also determined that methylation variation immediately upstream of genes was strongly correlated with transcriptional variation, whereas variation of gene body methylation had no significant impact on gene expression levels [10].
There are currently a relatively small number of natural epialleles known in Arabidopsis, but with the use of epigenomic approaches this situation is likely to rapidly change [91]. Early attempts at profiling genome-wide patterns of histone modifications and DNA methylation resulted in low-resolution maps (~50–1000 bp) and were unable to provide information regarding specific (base resolution) DNA methylation contexts [9,10,64,92,93,63,94]. However, it is now possible to survey complete genomes for DNA methylation at base pair resolution by combining the gold standard for methylation detection sodium bisulfite conversion [95–101], with high-throughput DNA sequencing [57,69]. The cytosine DNA methylome maps produced using these new methodologies make it possible to distinguish between the commonly observed gene body CG methylation and the rarely observed gene body methylation that occurs in all three sequence contexts (CG, CHG, CHH), which is typically targeted by the RdDM pathway. As described above in the spontaneous epigenetic variation section, the ability to survey base resolution methylomes will rapidly accelerate the identification of natural epigenetic variants in any plant species that contains a reference genome.
Concluding remarks and future outlook
There exists natural epigenetic variation within Arabidopsis, but it is becoming increasingly clear that many forms of cis and trans genetic variation can contribute to the formation of epialleles. In the near future, it will be possible to use association mapping techniques that combine both nucleotide (CNV or SNP) and methylation variation [differentially methylated region (DMR) or SMP] present within this species to map causal variants for much of this epigenetic variation. It is also clear that perturbations to the machinery required for RNAi and/or DNA methylation probably result in a release of transcriptional silencing, promoting both genetic variation as well as the formation of novel epialleles. Methods now exist for detecting base resolution DNA methylomes, shifting the need for improvement to input sample preparation. Essentially all published studies of plant epigenomes have utilized complex tissues, comprising numerous cell types, for DNA methylome profiling. Recent developments now enable enriching samples for specific cell types [102–104]. Future improvement may even allow interrogation of the epigenomes of single cells, which would provide unprecedented resolution for detection of epigenomic variation. These higher resolution views will be essential to unravel the intricate relationship between genotype, epigenotype and phenotype, allowing this knowledge to be applied to a wide range of species, including plants and people.
Acknowledgments
We would like to thank Milos Tanurdzic and anonymous reviewers for their helpful suggestions to improve the clarity and thoroughness of this review. R.J.S. was supported by an National Institutes of Health National Research Service Award (NIH NRSA) postdoctoral fellowship (F32-HG004830). This work was supported by the National Science Foundation (MCB-0929402 and MCB1122246), the Howard Hughes Medical Institute and the Gordon and Betty Moore Foundation (J.R.E.). J.R.E. is a Howard Hughes Medical Institute–Gordon and Betty Moore Foundation (HHMI–GBMF) Investigator.
Glossary
- Accession
a natural (wild) isolate from a specific geographic location
- CNV
copy number variant
- DMR
differentially methylated region
- Epiallele
a heritable variant of an allele that is not reflected by a change in its DNA sequence, but does have an effect on gene expression
- Epigenetics
the study of mitotically and/or meiotically heritable changes in phenotype that arise independent of genetic variation
- Epigenomics
the genome-wide study of modifications to DNA and associated proteins such as a variety of modified and variant histones, nucleosome positioning, transcription factors as well as RNAs
- epiRIL population
recombinant inbred lines that have wild-type genotypes, but contain mosaic epigenomes based on the parental lines used to establish the population
- MethylC-Seq
a genome-wide technique to survey base resolution maps of DNA methylation using high-throughput sequencing
- RNA-directed DNA methylation (RdDM)
a mechanism that is involved in silencing through a feedback loop consisting of small RNAs guiding DNA methylation and histone modifications
- Small RNAs (smRNAs)
21–24 nucleotides (nt) RNA species that can guide machinery to silence loci post-transcriptionally (21 nt smRNAs) and/or transcriptionally (24 nt smRNAs)
- SMP
single methylation polymorphism
References
- 1.Nordborg M, Weigel D. Next-generation genetics in plants. Nature. 2008;456:720–723. doi: 10.1038/nature07629. [DOI] [PubMed] [Google Scholar]
- 2.Tang C, et al. The evolution of selfing in Arabidopsis thaliana. Science. 2007;317:1070–1072. doi: 10.1126/science.1143153. [DOI] [PubMed] [Google Scholar]
- 3.Schnable PS, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- 4.Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- 5.Atwell S, et al. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature. 2010;465:627–631. doi: 10.1038/nature08800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weigel D, Mott R. The 1001 genomes project for Arabidopsis thaliana. Genome Biol. 2009;10:107. doi: 10.1186/gb-2009-10-5-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hancock AM, et al. Adaptation to climate across the Arabidopsis thaliana genome. Science. 2011;334:83–86. doi: 10.1126/science.1209244. [DOI] [PubMed] [Google Scholar]
- 8.Fournier-Level A, et al. A map of local adaptation in Arabidopsis thaliana. Science. 2011;334:86–89. doi: 10.1126/science.1209271. [DOI] [PubMed] [Google Scholar]
- 9.Vaughn MW, et al. Epigenetic natural variation in Arabidopsis thaliana. PLoS Biol. 2007;5:e174. doi: 10.1371/journal.pbio.0050174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, et al. Global analysis of genetic, epigenetic and transcriptional polymorphisms in Arabidopsis thaliana using whole genome tiling arrays. PLoS Genet. 2008;4:e1000032. doi: 10.1371/journal.pgen.1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao J, et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet. 2011;43:956–963. doi: 10.1038/ng.911. [DOI] [PubMed] [Google Scholar]
- 12.Gan X, et al. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature. 2011;477:419–423. doi: 10.1038/nature10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ossowski S, et al. Sequencing of natural strains of Arabidopsis thaliana with short reads. Genome Res. 2008;12:2024–2033. doi: 10.1101/gr.080200.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneeberger K, et al. Reference-guided assembly of four diverse Arabidopsis thaliana genomes. Proc Natl Acad Sci USA. 2011;108:10249–10254. doi: 10.1073/pnas.1107739108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santuari L, et al. Substantial deletion overlap among divergent Arabidopsis genomes revealed by intersection of short reads and tiling arrays. Genome Biol. 2010;11:R4. doi: 10.1186/gb-2010-11-1-r4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards EJ. Natural epigenetic variation in plant species: a view from the field. Curr Opin Plant Biol. 2011;14:204–209. doi: 10.1016/j.pbi.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 17.Cubas P, et al. An epigenetic mutation responsible for natural variation in floral symmetry. Nature. 1999;401:157–161. doi: 10.1038/43657. [DOI] [PubMed] [Google Scholar]
- 18.Manning K, et al. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat Genet. 2006;38:948–952. doi: 10.1038/ng1841. [DOI] [PubMed] [Google Scholar]
- 19.Stam M, et al. Differential chromatin structure within a tandem array 100 kb upstream of the maize b1 locus is associated with paramutation. Genes Dev. 2002;16:1906–1918. doi: 10.1101/gad.1006702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foerster AM, et al. Genetic rearrangements can modify chromatin features at epialleles. PLoS Genet. 2011;7:e1002331. doi: 10.1371/journal.pgen.1002331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bender J, Fink GR. Epigenetic control of an endogenous gene family is revealed by a novel blue fluorescent mutant of Arabidopsis. Cell. 1995;83:725–734. doi: 10.1016/0092-8674(95)90185-x. [DOI] [PubMed] [Google Scholar]
- 22.Jacobsen SE, Meyerowitz EM. Hypermethylated SUPERMAN epigenetic alleles in Arabidopsis. Science. 1997;277:1100–1103. doi: 10.1126/science.277.5329.1100. [DOI] [PubMed] [Google Scholar]
- 23.Johannes F, et al. Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet. 2009;5:e1000530. doi: 10.1371/journal.pgen.1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, et al. siRNAs targeting an intronic transposon in the regulation of natural flowering behavior in Arabidopsis. Genes Dev. 2004;18:2873–2878. doi: 10.1101/gad.1217304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mathieu O, et al. Transgenerational stability of the Arabidopsis epigenome is coordinated by CG methylation. Cell. 2007;130:851–862. doi: 10.1016/j.cell.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Mittelsten Scheid O, et al. Formation of stable epialleles and their paramutation-like interaction in tetraploid Arabidopsis thaliana. Nat Genet. 2003;34:450–454. doi: 10.1038/ng1210. [DOI] [PubMed] [Google Scholar]
- 27.Reinders J, et al. Compromised stability of DNA methylation and transposon immobilization in mosaic Arabidopsis epigenomes. Genes Dev. 2009;23:939–950. doi: 10.1101/gad.524609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saze H, Kakutani T. Heritable epigenetic mutation of a transposon-flanked Arabidopsis gene due to lack of the chromatin-remodeling factor DDM1. EMBO J. 2007;15:3641–3652. doi: 10.1038/sj.emboj.7601788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soppe WJ, et al. The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Mol Cell. 2000;6:791–802. doi: 10.1016/s1097-2765(05)00090-0. [DOI] [PubMed] [Google Scholar]
- 30.Kakutani T, et al. Developmental abnormalities and epimutations associated with DNA hypomethylation mutations. Proc Natl Acad Sci USA. 1996;93:12406–12411. doi: 10.1073/pnas.93.22.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rangwala SH, et al. Meiotically stable natural epialleles of Sadhu, a novel Arabidopsis retroposon. PLoS Genet. 2006;2:e36. doi: 10.1371/journal.pgen.0020036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riddle NC, Richards EJ. Genetic variation in epigenetic inheritance of ribosomal RNA gene methylation in Arabidopsis. Plant J. 2005;41:524–532. doi: 10.1111/j.1365-313X.2004.02317.x. [DOI] [PubMed] [Google Scholar]
- 33.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11:204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greenberg MV, et al. Identification of genes required for de novo DNA methylation in Arabidopsis. Epigenetics. 2011;6:344–354. doi: 10.4161/epi.6.3.14242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson LM, et al. SRA-domain proteins required for DRM2-mediated de novo DNA methylation. PLoS Genet. 2008;4:e1000280. doi: 10.1371/journal.pgen.1000280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woo HR, et al. Three SRA-domain methylcytosine-binding proteins cooperate to maintain global CpG methylation and epigenetic silencing in Arabidopsis. PLoS Genet. 2008;4:e1000156. doi: 10.1371/journal.pgen.1000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan SW, et al. RNAi, DRD1, and histone methylation actively target developmentally important non-CG DNA methylation in Arabidopsis. PLoS Genet. 2006;2:e83. doi: 10.1371/journal.pgen.0020083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chan SW, et al. Two-step recruitment of RNA-directed DNA methylation to tandem repeats. PLoS Biol. 2006;4:e363. doi: 10.1371/journal.pbio.0040363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan SW, et al. RNA silencing genes control de novo DNA methylation. Science. 2004;303:1336. doi: 10.1126/science.1095989. [DOI] [PubMed] [Google Scholar]
- 40.Zilberman D, et al. ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science. 2003;299:716–719. doi: 10.1126/science.1079695. [DOI] [PubMed] [Google Scholar]
- 41.Cao X, Jacobsen SE. Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr Biol. 2002;12:1138–1144. doi: 10.1016/s0960-9822(02)00925-9. [DOI] [PubMed] [Google Scholar]
- 42.Jackson JP, et al. Control of CpNpG DNA methylation by theKRYPTONITEhistoneH3methyltransferase. Nature. 2002;416:556–560. doi: 10.1038/nature731. [DOI] [PubMed] [Google Scholar]
- 43.Lindroth AM, et al. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science. 2001;292:2077–2080. doi: 10.1126/science.1059745. [DOI] [PubMed] [Google Scholar]
- 44.Ebbs ML, Bender J. Locus-specific control of DNA methylation by the Arabidopsis SUVH5 histone methyltransferase. Plant Cell. 2006;18:1166–1176. doi: 10.1105/tpc.106.041400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ebbs ML, et al. H3 lysine 9 methylation is maintained on a transcribed inverted repeat by combined action of SUVH6 and SUVH4 methyltransferases. Mol Cell Biol. 2005;25:10507–10515. doi: 10.1128/MCB.25.23.10507-10515.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mull L, et al. A histone methylation-dependent DNA methylation pathway is uniquely impaired by deficiency in Arabidopsis S-adenosylhomocysteine hydrolase. Genetics. 2006;174:1161–1171. doi: 10.1534/genetics.106.063974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bartee L, et al. Arabidopsis cmt3 chromomethylase mutations block non-CG methylation and silencing of an endogenous gene. Genes Dev. 2001;15:1753–1758. doi: 10.1101/gad.905701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ausin I, et al. IDN1 and IDN2 are required for de novo DNA methylation in Arabidopsis thaliana. Nat Struct Mol Biol. 2009;16:1325–1327. doi: 10.1038/nsmb.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richards EJ. Inherited epigenetic variation – revisiting soft inheritance. Nat Rev Genet. 2006;7:395–401. doi: 10.1038/nrg1834. [DOI] [PubMed] [Google Scholar]
- 50.Tsukahara S, et al. Bursts of retrotransposition reproduced in Arabidopsis. Nature. 2009;461:423–426. doi: 10.1038/nature08351. [DOI] [PubMed] [Google Scholar]
- 51.Michaels SD, Amasino RM. FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell. 1999;11:949–956. doi: 10.1105/tpc.11.5.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmitz RJ, et al. Transgenerational epigenetic instability is a source of novel methylation variants. Science. 2011;334:369–373. doi: 10.1126/science.1212959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Becker C, et al. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature. 2011;480:245–249. doi: 10.1038/nature10555. [DOI] [PubMed] [Google Scholar]
- 54.Eichten SR, et al. Heritable epigenetic variation among maize inbreds. PLoS Genet. 2011;7:e1002372. doi: 10.1371/journal.pgen.1002372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shaw RG, et al. Spontaneous mutational effects on reproductive traits of Arabidopsis thaliana. Genetics. 2000;155:369–378. doi: 10.1093/genetics/155.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ossowski S, et al. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science. 2010;327:92–94. doi: 10.1126/science.1180677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lister R, et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Penterman J, et al. Genetic interactions between DNA demethylation and methylation in Arabidopsis thaliana. Plant Physiol. 2007;145:1549–1557. doi: 10.1104/pp.107.107730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saze H, et al. Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat Genet. 2003;34:65–69. doi: 10.1038/ng1138. [DOI] [PubMed] [Google Scholar]
- 60.Mosher RA, et al. Uniparental expression of PolIV-dependent siRNAs in developing endosperm of Arabidopsis. Nature. 2009;460:283–286. doi: 10.1038/nature08084. [DOI] [PubMed] [Google Scholar]
- 61.Slotkin RK, et al. Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell. 2009;136:461–472. doi: 10.1016/j.cell.2008.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Teixeira FK, et al. A role for RNAi in the selective correction of DNA methylation defects. Science. 2009;323:1600–1604. doi: 10.1126/science.1165313. [DOI] [PubMed] [Google Scholar]
- 63.Zhang X, et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell. 2006;126:1189–1201. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 64.Zilberman D, et al. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2006;39:61–69. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]
- 65.Chodavarapu RK, et al. Relationship between nucleosome positioning and DNA methylation. Nature. 2010;466:388–392. doi: 10.1038/nature09147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pagnussat GC, et al. Genetic and molecular identification of genes required for female gametophyte development and function in Arabidopsis. Development. 2005;132:603–614. doi: 10.1242/dev.01595. [DOI] [PubMed] [Google Scholar]
- 67.Vongs A, et al. Arabidopsis thaliana DNA methylation mutants. Science. 1993;260:1926–1928. doi: 10.1126/science.8316832. [DOI] [PubMed] [Google Scholar]
- 68.Jeddeloh JA, et al. Maintenance of genomic methylation requires a SWI2/SNF2-like protein. Nat Genet. 1999;22:94–97. doi: 10.1038/8803. [DOI] [PubMed] [Google Scholar]
- 69.Cokus SJ, et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ronemus MJ, et al. Demethylation-induced developmental pleiotropy in Arabidopsis. Science. 1996;273:654–657. doi: 10.1126/science.273.5275.654. [DOI] [PubMed] [Google Scholar]
- 71.Finnegan EJ, et al. Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc Natl Acad Sci USA. 1996;93:8449–8454. doi: 10.1073/pnas.93.16.8449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jacobsen SE, et al. Ectopic hypermethylation of flower-specific genes in Arabidopsis. Curr Biol. 2000;10:179–186. doi: 10.1016/s0960-9822(00)00324-9. [DOI] [PubMed] [Google Scholar]
- 73.Miura A, et al. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature. 2001;411:212–214. doi: 10.1038/35075612. [DOI] [PubMed] [Google Scholar]
- 74.Kakutani T, et al. Meiotically and mitotically stable inheritance of DNA hypomethylation induced by ddm1 mutation of Arabidopsis thaliana. Genetics. 1999;151:831–838. doi: 10.1093/genetics/151.2.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yi H, Richards EJ. Gene duplication and hypermutation of the pathogen Resistance gene SNC1 in the Arabidopsis bal variant. Genetics. 2009;183:1227–1234. doi: 10.1534/genetics.109.105569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mirouze M, et al. Selective epigenetic control of retrotransposition in Arabidopsis. Nature. 2009;461:427–430. doi: 10.1038/nature08328. [DOI] [PubMed] [Google Scholar]
- 77.Singer T, et al. Robertson’s Mutator transposons in A. thaliana are regulated by the chromatin-remodeling gene Decrease in DNA Methylation (DDM1) Genes Dev. 2001;15:591–602. doi: 10.1101/gad.193701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McClintock B. The origin and behavior of mutable loci in maize. Proc Natl Acad Sci USA. 1950;36:344–355. doi: 10.1073/pnas.36.6.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ahmed I, et al. Genome-wide evidence for local DNA methylation spreading from small RNA-targeted sequences in Arabidopsis. Nucleic Acids Res. 2011;39:6919–6931. doi: 10.1093/nar/gkr324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lippman Z, et al. Role of transposable elements in heterochromatin and epigenetic control. Nature. 2004;430:471–476. doi: 10.1038/nature02651. [DOI] [PubMed] [Google Scholar]
- 81.Hollister JD, Gaut BS. Epigenetic silencing of transposable elements: a trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 2009;19:1419–1428. doi: 10.1101/gr.091678.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hollister JD, et al. Transposable elements and small RNAs contribute togene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc Natl Acad Sci USA. 2011;108:2322–2327. doi: 10.1073/pnas.1018222108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ito H, et al. An siRNA pathway prevents transgenerational retrotransposition in plants subjected to stress. Nature. 2011;472:115–119. doi: 10.1038/nature09861. [DOI] [PubMed] [Google Scholar]
- 84.Tanurdzic M, et al. Epigenomic consequences of immortalized plant cell suspension culture. PLoS Biol. 2008;6:2880–2895. doi: 10.1371/journal.pbio.0060302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Johannes F, et al. Epigenome dynamics: a quantitative genetics perspective. Nat Rev Genet. 2008;9:883–890. doi: 10.1038/nrg2467. [DOI] [PubMed] [Google Scholar]
- 86.Roux F, et al. Genome-wide epigenetic perturbation jumpstarts patterns of heritable variation found in nature. Genetics. 2011;188:1015–1017. doi: 10.1534/genetics.111.128744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Riddle NC, Richards EJ. The control of natural variation in cytosine methylation in Arabidopsis. Genetics. 2002;162:355–363. doi: 10.1093/genetics/162.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Woo HR, et al. VIM1, amethylcytosine-binding proteinrequired for centromeric heterochromatinization. Genes Dev. 2007;21:267–277. doi: 10.1101/gad.1512007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Melquist S, et al. Arabidopsis PAI gene arrangements, cytosine methylation and expression. Genetics. 1999;153:401–413. doi: 10.1093/genetics/153.1.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Melquist S, Bender J. Transcription from an upstream promoter controls methylation signaling from an inverted repeat of endogenous genes in Arabidopsis. Genes Dev. 2003;17:2036–2047. doi: 10.1101/gad.1081603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schmitz RJ, Zhang X. High-throughput approaches for plant epigenomic studies. Curr Opin Plant Biol. 2011;14:130–136. doi: 10.1016/j.pbi.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang X, et al. Genome-wide analysis of mono-, di- and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol. 2009;10:R62. doi: 10.1186/gb-2009-10-6-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang X, et al. Whole-genome analysis of histone H3 lysine 27 trimethylation in Arabidopsis. PLoS Biol. 2007;5:e129. doi: 10.1371/journal.pbio.0050129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bernatavichute YV, et al. Genome-wide association of histone H3 lysine nine methylation with CHG DNA methylation in Arabidopsis thaliana. PLoS ONE. 2008;3:e3156. doi: 10.1371/journal.pone.0003156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Clark SJ, et al. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;22:2990–2997. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Frommer M, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hayatsu H, et al. Reaction of sodium bisulfite with uracil, cytosine, and their derivatives. Biochemistry. 1970;9:2858–2865. doi: 10.1021/bi00816a016. [DOI] [PubMed] [Google Scholar]
- 98.Shapiro R, et al. Deamination of cytosine derivatives by bisulfite. Mechanism of the reaction. J Am Chem Soc. 1974;96:906–912. doi: 10.1021/ja00810a043. [DOI] [PubMed] [Google Scholar]
- 99.Shapiro R, et al. Nucleic acid reactivity and conformation. II. Reaction of cytosine and uracil with sodium bisulfite. J Biol Chem. 1973;248:4060–4064. [PubMed] [Google Scholar]
- 100.Hayatsu H. Bisulfite modification of nucleic acids and their constituents. Prog Nucleic Acid Res Mol Biol. 1976;16:75–124. doi: 10.1016/s0079-6603(08)60756-4. [DOI] [PubMed] [Google Scholar]
- 101.Wang RY, et al. Comparison of bisulfite modification of 5-methyldeoxycytidine and deoxycytidine residues. Nucleic Acids Res. 1980;8:4777–4790. doi: 10.1093/nar/8.20.4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Deal RB, Henikoff S. A simple method for gene expression and chromatin profiling of individual cell types within a tissue. Dev Cell. 2010;18:1030–1040. doi: 10.1016/j.devcel.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Deal RB, Henikoff S. The INTACT method for cell type-specific gene expression and chromatin profiling in Arabidopsis thaliana. Nat Protoc. 2011;6:56–68. doi: 10.1038/nprot.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Birnbaum K, et al. A gene expression map of the Arabidopsis root. Science. 2003;302:1956–1960. doi: 10.1126/science.1090022. [DOI] [PubMed] [Google Scholar]