

Abstract
Collaborating on studies of subchronic daily intoxication in juvenile and adult rats, we examined whether the repetitive ethanol treatments at these two life stages altered levels of key neuroinflammation-associated proteins—aquaporin-4 (AQP4), certain phospholipase A2 (PLA2) enzymes, PARP-1 and caspase-3—in hippocampus (HC) and entorhinal cortex (EC). Significant changes in the proteins could implicate activation of specific neuroinflammatory signaling pathways in these rats as well as in severely binge-intoxicated adult animals that are reported to incur degeneration of vulnerable neurons in HC and EC. Male Wistar rats, ethanol-intoxicated (3 g/kg i.p.) once daily for 6 days over an 8-day interval beginning at 37 days old and repeated at age 68–75 days, were sacrificed one hr after the day 75 dose (blood ethanol, 200–230 mg/dl). Analysis of HC with an immunoblot technique showed that AQP4, Ca+2-dependent PLA2 (cPLA2 IVA), phosphorylated (activated) p-cPLA2, cleaved (89 kD) PARP (c-PARP), and caspase-3 levels were significantly elevated over controls, whereas Ca+2-independent PLA2 (iPLA2 VIA) was reduced ~70%; however, cleaved caspase-3 was undetectable. In the EC, AQP4 was unchanged, but cPLA2 and p-cPLA2 were significantly increased while iPLA2 levels were diminished (~40%) similar to HC, although just outside statistical significance (p=0.06). In addition, EC levels of PARP-1 and c-PARP were significantly increased. The ethanol-induced activation of cPLA2 in association with reduced iPLA2 mirrors PLA2 changes in reports of neurotrauma and also of dietary omega-3 fatty acid depletion. Furthermore, the robust PARP-1 elevations accompanied by negligible caspase-3 activation indicate that repetitive ethanol intoxication may be potentiating non-apoptotic neurodegenerative processes such as parthanatos. Overall, the repetitive ethanol treatments appeared to instigate previously unappreciated neuroinflammatory pathways in vivo. The data provide insights into mechanisms of binge ethanol abuse that might suggest new therapeutic approaches to counter neurodegeneration and dementia.
Keywords: alcohol, neuroinflammation, PLA2, AQP4, neurodegeneration, arachidonic acid, PARP-1, parthanatos
Introduction
Chronic alcohol (ethanol)-induced brain degeneration comprises a major reason for cognitive deficits worldwide (Carlen et al., 1994; Eckardt and Martin, 1986; Gupta and Warner, 2008), although this fact is often overlooked in reviews of acquired brain damage and cognitive impairment. In experimental models, repetitive ethanol binging sufficient to achieve relatively sustained high blood levels cause substantial brain neurodegeneration. One in vivo approach with adult rats, the Majchrowicz model (Majchrowicz, 1975; Switzer et al., 1982), utilizes daily repetitive gavage or intubation (12–15 g/kg/d) over 4 d to cause ethanol withdrawal symptoms along with degeneration of temporal cortical (esp. entorhinal cortical) pyramidal neurons and hippocampal dentate granule cells (Collins et al., 1996). Consistent with the neuronal damage, such treated rats show impaired neurobehavior and learning (Cippitelli et al., 2010; Obernier et al., 2002b). In the above binge model or its less severe adaptations, excitotoxicity is apparently not a chief mechanism of neurodegeneration (Collins and Neafsey, 2012a); rather, neuroinflammatory and possibly neuroimmune-related pathways, often involving excessive oxidative stress, are likely to be important.
Ethanol-dependent oxidative stress of a severe nature could arise via several avenues. One possible route has been suggested to be triggered by binge ethanol-induced brain edema/swelling that could initiate, directly or indirectly, excessive phospholipase A2 (PLA2) activation and arachidonic acid (AA) mobilization (Collins and Neafsey, 2012b). There is a body of experimental evidence linking PLA2 to cell swelling and mobilized AA to downstream oxidative stress. Levels and activity of brain water channels such as aquaporin-4 (AQP4), expressed mainly in astroglia, that could regulate cellular edema are thus of specific interest. Indeed, pharmacological antagonism of AQP4 in adult rats during chronic binge ethanol intoxication suppresses edema in the brain as well as neuronal damage in the hippocampus (HC) and entorhinal cortex (EC) (Sripathirathan et al., 2009); also, repetitive ethanol treatment of organotypic rat slices of HC and EC in culture, modeling binge intoxication in vivo, increases AQP4 levels, while the AQP4 antagonist, acetazolamide, prevents slice edema and neurodegenerative sequelae. Other research has shown that knockdown or antagonism of AQP4 is neuroprotective with respect to insults producing cytotoxic (cellular) edema (Manley et al., 2000). As further proof for a key neuroinflammatory role for AQP4, the water channel was essential for significant elevations in proinflammatory cytokines after endotoxin exposure in transgenic mice (Li et al., 2011).
Asking whether repeated ethanol intoxication bouts would significantly alter in vivo levels of brain AQP4 and selected PLA2 enzymes, we examined HC and EC samples from rats that had been subjected to a relatively moderate, “binge-pattern” ethanol intoxication regimen as juveniles and again as young adults (Przybycien-Szymanska et al., 2011). Our immunoblot assays of HC and EC extracts provided levels of AQP4 and several PLA2 families often associated with neuroinflammation—Ca+2-dependent PLA2 (cPLA2 IVA), its activated (phosphorylated) p-cPLA2, and Ca+2-independent PLA2 (iPLA2 VI). Lacking histological evidence of neurodamage, we questioned whether neurodegeneration pathways were stimulated by repetitive ethanol intoxication by determining levels of poly (ADP-ribose) polymerase-1 (PARP-1), cleaved PARP (c-PARP), caspase-3 and cleaved caspase-3. PARP-1 is linked via co-activation of NF-kappaB (NF-κB) transcription factor to oxidative stress and inflammation in various tissues including brain (Hassa et al., 2002). Our results provide the first in vivo indications of PLA2-associated neuroinflammatory perturbations in susceptible brain regions of repetitive ethanol-intoxicated adult rats that might involve AQP4, and of possible instigation of neurodamaging pathways via PARP-1-related mechanisms.
Materials and Methods
Materials
Antibodies used were: AQP4 (sc-20812), cPLA2 IVA (sc-454) and ser 505 p-cPLA2 IVA (sc-34391), Santa Cruz Biotechnology, Santa Cruz CA; iPLA2 VIβ (07-169), Upstate Biotech, Lake Placid NY; PARP-1 (9542s) and caspase-3 (9662), Cell Signaling, Danvers MA. Secondary antibodies were from Jackson ImmunoResearch (West Grove PA), Glyceraldehyde phosphate dehydrogenase (GAPDH; sc-166545) was from Santa Cruz Biotechnology, and luminol reagent for immunoblot detection was from Pierce Chemical Co. (Rockford IL). Other chemicals and reagents were from Sigma Chemical Co. (St. Louis MO).
Animal and animal procedure
Following a Loyola University IACUC-approved protocol, juvenile (37 d) and adult (68 d) male Wistar rats (Charles River Laboratories, Wilmington, MA) were treated intraperitoneally (i.p.) at ~10 am daily over eight (8) successive days with ethanol (3 g/kg/d) and/or an equivalent volume of isotonic saline according to Przybycien-Szymanska et al. (Przybycien-Szymanska et al., 2011), and summarized as follows:
| Juvenile (days old) | Adult | |
|
|
||
| Group | (37→39 [40–41 saline] 42→44) | (68→70 [71–72 salinel 73→75) |
| Juvenile+Adult repetitive intoxication | Ethanol 3g/kg ip once a day | Ethanol 3g/kg ip once a day |
| Controls for above | Isotonic saline ip once a day | Isotonic saline ip once a day |
Rats were sacrificed by decapitation one hr after the last injection and trunk blood was collected for assay of blood ethanol levels (BEL) by spectrophotometry using a Pointe Scientific alcohol reagent kit (Canton MI). Intact brains were removed and frozen for later dissection. Hippocampus (HC) and entorhinal cortex (EC) were removed and homogenized for 15–30 sec in 500 ul RIPA buffer (25 mM Tris, 150 mM NaCl, 1% Triton X100, 1% sodium deoxycholate (SDS), and 0.1% sodium dodecyl sulfate (SDS)) containing protease inhibitor and phosphatase inhibitor cocktails (#p8340 and #p5726, respectively; Sigma Chemical Co., St. Louis MO). Homogenates were centrifuged 10 min @ 13,000 rpm and supernatants were collected.
Immunoblot analyses
Immunoblots of proteins in supernatants from HC and EC homogenates were performed according to standard protocols. Protein concentrations of each sample were determined with the bicinchoninic acid method. An aliquot containing 40 μg total protein/sample was applied to each lane and separations were achieved with 4–12% SDS PAGE. Proteins were transferred electrophoretically to PVDF membranes. Non-specific binding was blocked either by 1% bovine serum albumin or 5% nonfat dry milk for 1 hr at room temperature in 1X tris-buffered saline-Tween 20 (TBST), pH 8.0. Subsequently, membranes were incubated overnight at 4°C with primary antibodies of AQP4 (1:1000), cPLA2 (1:500), p-cPLA2 (1:500), iPLA2 (1:200), sPLA2 (1:200), PARP (1:1000) or Caspase-3 (1:500). Membranes were washed three times with 1X TBST for 10 min each. Either anti-goat, anti-rabbit or anti-mouse horse radish peroxidase-conjugated IgG or IgM were used as secondary antibodies, and luminol reagents were used as a tracer to detect the bands on the blots. To facilitate calculation of levels, membranes were stripped in Tris-HCl, SDS and mercaptoethanol, and reprobed with GAPDH (1:1000) as standard. The intensity of immunostaining was analyzed by scanning the images with LABwork 4.5 image acquisition and analysis software from Ultra Violet Products (Upland CA).
Statistical analyses
Immunoblot intensity results were expressed as means +/− sem, and analyzed for stastical significance (p<0.05 or p<0.01) by Tukey's t-test and one-way analysis of variance (ANOVA) with completely randomized design.
Results
In juvenile+adult ethanol-intoxicated rats one hour after the last adult injection of 3 g/kg ethanol, the blood ethanol level (BEL) range was 200–230 mg/dl (Przybycien-Szymanska et al., 2011), which is in the mid-range of clinical blood levels reported in binging alcoholics (Adachi et al., 1991).
AQP4
Figure 1 shows representative immunoblots of AQP4 and GAPDH in HC and EC; quantitation of blots showed that in repetitive ethanol (E)-intoxicated rats the AQP4 levels in HC were ~50% higher than controls (*p<0.05) an hour following the last ethanol treatment, whereas in EC the levels of AQP4 at this timepoint were not different between ethanol and control rats.
Figure 1. Repetitive daily ethanol intoxication increases levels of AQP4 in adult rat HC.
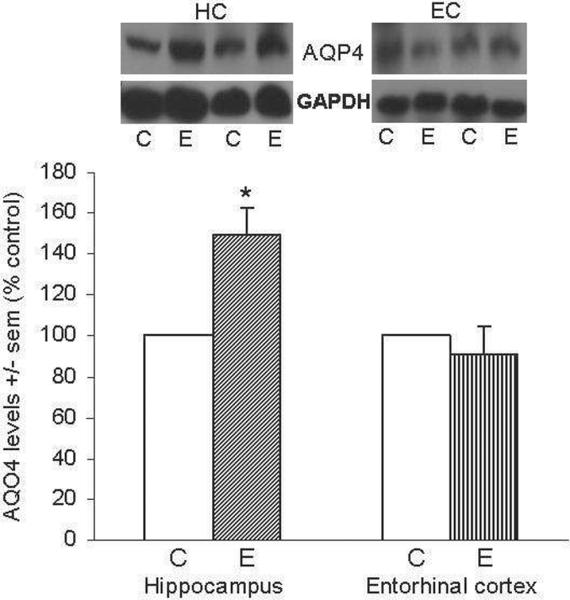
Quantitation of AQP4 in HC and EC of juvenile+adult ethanol-intoxicated (E) and control (C) rats, with representative immunoblots shown for AQP4 and GAPDH loading standard. Results from controls are set at 100%. Data are means ± sem (n = 5–6 per group); *p<0.05 vs. control (one-way ANOVA, HC, F=16.1, p=0.001 and EC, F=0.4).
Tajuddin et al.
cPLA2 IVA and phospho-cPLA2 (p-cPLA2)
Figure 2A shows representative immunoblots of cPLA2 IVA and GAPDH in HC and EC; levels of cPLA2 in HC and EC one hour after the last ethanol dose were elevated ~100% above controls (*p<0.05) in HC and ~45% (**p<0.01) in EC. Figure 2b shows representative immunoblots of p-cPLA2 and GAPDH in HC and EC; consistent with cPLA2 elevations, levels of activated p-cPLA2 in the HC and EC of ethanol-intoxicated rats were found to be significantly increased 30% (*p<0.05) and 118% (*p<0.05) above controls, respectively.
Figure 2. Repetitive daily ethanol exposures raise levels of cPLA2 IVA and p-cPLA2 in adult rat HC and EC.


Fig. 2a: Quantitation of cPLA2 IVA in HC and EC of juvenile+adult ethanol-intoxicated (E) and saline control (C) rats, with representative immunoblots of cPLA2 and GAPDH loading standard. Fig. 2b: Quantitation of p-cPLA2 in HC and EC of juvenile+adult ethanol-intoxicated (E) and saline control (C) rats, with representative immunoblots of p-cPLA2 and GAPDH. Results from control rats are set to 100%. Data are means ± sem (n = 5–6 per group); *p<0.05 vs. respective control; **p<0.01 vs. respective control (one-way ANOVA, cPLA2: HC, F=806, p=0.0001 and EC, F=24.5, p=0.002; p-cPLA2: HC, F=11.1, p=0.015 and EC, F=7.9, p=0.03).
Tajuddin et al.
iPLA2 VIβ
Figure 3 shows representative immunoblots of iPLA2 and GAPDH in HC and EC; ethanol-intoxicated rats displayed marked reductions in the levels of iPLA2 in HC (28% of control, **p<0.01). Also, the EC of these animals contained iPLA2 values that were ~40% below control levels, but the mean difference fell just beyond statistical significance (#p=0.06).
Figure 3. Repetitive daily ethanol intoxication reduces levels of iPLA2 VI in adult rat HC and EC.

Quantitation of iPLA2 VI in HC and EC of juvenile+adult ethanol-intoxicated (E) and control (C) rats, with representative immunoblots of iPLA2 and GAPDH loading standard. Results from control rats are set to 100%. Data are means ± sem (n = 5–6 per group); **P < 0.01 vs. respective control; # p = 0.06 vs. control (one-way ANOVA, HC, F=103.0, p=0.0001 and EC, F=2.6).
Tajuddin et al.
PARP-1 and cleaved 89 kD PARP (c-PARP)
Figure 4a shows representative immunoblots of PARP-1 and GAPDH in HC and EC; ethanol-intoxicated rats had PARP-1 levels that were ~2-fold above controls in EC (*p<0.05) but not significantly different in HC. Figure 4b shows representative immunoblots of c-PARP and GAPDH in HC and EC. The HC contained 200% increases (*p<0.05) in c-PARP, and in the EC, the two values for c-PARP levels that were available for assays were ~2-fold and ~6-fold higher than control levels in the ethanol-treated rats (average shown).
Figure 4. Repetitive daily ethanol exposure increases levels of PARP-1 and c-PARP in adult rat HC and/or EC.


Fig. 4a: Quantitation of PARP-1 in HC and EC of juvenile+adult ethanol-intoxicated (E) and control (C) rats, with representative immunoblots of PARP-1 and GAPDH. Fig. 4b: Quantitation of c-PARP in HC and EC of juvenile+adult ethanol-intoxicated (E) and control (C) rats, with representative immunoblots of c-PARP and GAPDH loading standard. Results from control rats are set to 100%. Data are means ± sem (n = 3–4 per group, with the exception of c-PARP in EC (n = 2)); *P < 0.05 vs. respective controls (one-way ANOVA, PARP-1: HC, F=6.7, p=0.06 and EC, F=7.3, p=0.035; c-PARP: HC, F=11.2, p=0.03 and EC, F=1300, p=0.0001).
Tajuddin et al.
Caspase-3 and cleaved caspase-3
Figure 5 shows that caspase-3 levels were 2-fold greater (*p<0.05) in HC of ethanol-intoxicated rats; EC tissues were not available. Important with respect to apoptotic activation, cleaved caspase-3 was undetectable in the HC of either ethanol-intoxicated or control rats.
Figure 5. Repetitive daily ethanol administration increases caspase-3 levels in adult rat HC.

Quantitation of caspase-3 in HC of juvenile+adult ethanol-intoxicated (E) and control (C) rats, with representative immunoblots of caspase-3 and GAPDH loading standard. Cleaved caspase-3 was undetectable in either brain region of C and E rats. Data are means ± sem (n = 3–4 per group); *p<0.05 vs. control (one-way ANOVA. Caspase-3: F=49.4, p=0.001)
Tajuddin et al.
Discussion
The main objective of these circumscribed assays was to ascertain whether repetitive once-daily ethanol intoxication in adult rats that produced blood ethanol levels in the mid-range of those in active binging alcoholics would potentiate selected neuroinflammatory pathways in brain regions vulnerable to binge ethanol-induced neurodegeneration. Although this is limited a descriptive study at one sacrifice timepoint, its results provide direction for our ongoing high chronic ethanol experiments with adult-age brain organotypic slice cultures in which pharmacological inhibitory approaches can clarify cause and effect relationships.
Based on experiments with severely binged adult rats and similarly treated organotypic slice cultures, we have postulated that persistent increases in AQP4 due to ethanol intoxication bouts would promote brain edema/cell (esp. astroglial) swelling, triggering increases in PLA2 levels/activity which can cause excessive AA release (discussed below), neuroinflammatory mediators including ROS and, to varying extents, neurodegeneration (Collins and Neafsey, 2012b). The expectation was that the “binge-pattern” treatment protocols here, although less intoxicating than the Majchrowicz 4-day binge treatment in adult rats which achieves BEL of or over 400 mg/dl (Collins et al., 1996), and also than the once-daily modified schedule (~5 g/kg/d) used to elicit HC and EC neurodegeneration (Collins et al., 1998), would provide supportive in vivo evidence.
The AQP4 elevations in the HC are consistent with the above view, although whether transcriptional induction (or suppression) is responsible for changes in this and the other proteins needs to be established. AQP4 is the major aquaporin form expressed in brain, and primarily in astroglia (Gunnarson et al., 2004); activated microglia also express AQP4 in some studies but not others (Li et al., 2011; Tomas-Camardiel et al., 2004). Increased brain AQP4 levels and activity are important mediators of cytotoxic (cellular) edema in acquired neurodegenerative conditions such as ischemia, stroke and trauma (Roberta and Rossella, 2010; Saadoun and Papadopoulos, 2010). In that regard, moderate brain edema is present in repetitive binge ethanol-treated adult rats and organotypic HC+EC slice cultures, and is reduced by an AQP4 inhibitor (Collins et al., 1998; Sripathirathan et al., 2009). A strong argument for AQP4's intrinsic neuroinflammatory potential is its required astroglial presence, concomitant with cell swelling, for augmentation of proinflammatory cytokines due to neuroimmune insults such as endotoxin (Li et al., 2011). It is possible that the lack of observable AQP4 increases in EC indicates that changes in water channels occur within a timeframe missed in this study. The binge-pattern treatments also could be insufficient to cause observable water channel augmentation in this region. However, the significant cPLA2 activation in the EC (e.g., ratio of p-cPLA2/cPLA2), lends support the former possibility.
In addition to stimulating proinflammatory cytokine elevations, an earlier documented consequence of brain cytotoxic swelling is volume- or stretch-initiated PLA2 activation, which causes excessive AA mobilization (Koivusalo et al., 2009; Lambert et al., 2006; Lehtonen and Kinnunen, 1995). The metabolism and auto-oxidation of excessively released AA, and even its activation of NADPH oxidase, are major ROS sources during neurodegenerative insults (Bobba et al., 2008; Dana et al., 1998; Friis et al., 2008). It also is tenable that AQP4 could increase cPLA2 levels/activity secondary to stimulating elevations in pro-inflammatory cytokines, since these proteins have been reported to stimulate PLA2 expression in various stress situations (Adibhatla and Hatcher, 2007; Kramer et al., 1996). Although we did not assay cytokines, studies have linked pro-inflammatory cytokines/chemokines (and reduced anti-inflammatory cytokines) with alcohol-dependent neuroinflammation and neurodamage in adult rodents (Alfonso-Loeches et al., 2010; Crews et al., 2011; Zou and Crews, 2010).
The increased brain cPLA2 and p-cPLA2 levels in the two regions of ethanol-intoxicated rats agree with considerable literature documenting inflammatory roles for hyperactivated PLA2 superfamily members (Adibhatla and Hatcher, 2008; Sun et al., 2010). In a range of neuroinflammatory insults, cPLA2 has been found to be a dominant activity releasing AA (Ong et al., 2010). With respect to ethanol, in vitro exposure to high concentrations increased AA-specific PLA2 in neuroblastoma cultures (Basavarajappa et al., 1997) as well as AA release in our organotypic brain slice cultures (Brown et al., 2009). With primary astrocyte cultures, exposure to moderate ethanol concentrations for 2 days augmented cPLA2 activity (Floreani et al., 2010), but apparently not to high ethanol concentrations for 24 hr (Luo et al., 2001). Evidence in vivo for chronic/subchronic ethanol effects on PLA2 in adult brain is surprisingly limited. The activity of an AA-specific PLA2 with characteristics of cPLA2 was increased in brains of chronic ethanol-treated mice (Basavarajappa et al., 1998), but prolonged ethanol intake via liquid diet did not alter rat cPLA2 mRNA expression in rat brain cortex and HC (Simonyi et al., 2002)—although it increased message for cyclooxygenase-2, the enzyme converting AA to pro-inflammatory eicosanoid precursors. Parenthetically, members of another large Ca+2-dependent PLA2 family, small secreted PLA2's (sPLA2), sometimes are triggers for AA release by activating a signaling kinase cascade upstream of cPLA2 (Hernandez et al., 1998; Kolko et al., 2003). sPLA2 members also have been implicated as neuroinflammatory stimuli upregulated in neurodegeneration (Goracci et al., 2010) including Alzheimer's disease (Moses et al., 2006). Although not assayed in these juvenile+adult intoxicated rats, in our ethanol-treated organotypic HC-EC slice cultures sPLA2 inhibitors are neuroprotective (Moon et al., submitted), indicating that sPLA2 is important in binge ethanol-induced neurodamage.
The increased cPLA2 levels and activation in this study of intoxicated rats were not unexpected, but the large reduction in iPLA2 in the two regions were surprising. As the dominant PLA2 activity in rat brain (Yang et al., 1999), iPLA2 has been considered an essential housekeeping and “protective” enzyme within mitochondria (Ong et al., 2010; Seleznev et al., 2006); as such, the enzyme's loss in disease states or the inhibition of its activity can be deleterious to mitochondrial and neuronal function (Ma et al., 2011). Other evidence shows that iPLA2 is important in suppressing lipid peroxidation (Kinsey et al., 2008), and its deficiency has been associated with neuroaxonal dystrophy in mice (Shinzawa et al., 2008). Of particular importance is that iPLA2 is the primary regulator of the release and turnover of docosahexaenoic acid (DHA) in brain phospholipids (Strokin et al., 2006). This suggests reduced endogenous turnover of this important brain omega-3 fatty acid, which might lead to diminished mobilization of/signaling by neuroprotective DHA and/or its anti-neuroinflammatory metabolites such as resolvins and neuroprotectins (Bazan et al., 2011). Indeed, counteraction of such events is a recent explanation for clozapine's upregulation of decreased rat brain iPLA2 expression seen in bipolar disorder and thus the mood stabilizer's anti-inflammatory and neuroprotective effects (Kim et al., 2012).
Furthermore, the ethanol-induced reductions of brain iPLA2 congruent with cPLA2 and p-cPLA2 elevations mirror alterations in the brains (cortex) of rats chronically fed omega-3 fatty acid-deficient diets (Rao et al., 2007). Consistent with iPLA2 reductions of the above-mentioned study, the laboratory previously demonstrated that brain DHA turnover was suppressed by dietary omega-3 deficiency. Rat brain iPLA2 depletion also occurred due to fluid percussion trauma and was countered by DHA supplementation (Wu et al., 2011). Whether the ethanol-evoked iPLA2 reductions are due to decreased brain expression or to overt brain cell loss is open to question, although the PARP-1 results imply the occurrence of neurodegenerative processes (further discussed below). At this stage it is reasonable to consider that depletion of iPLA2 due to repetitive ethanol intoxication, in concert with cPLA2 activation and potentially excessive AA mobilization, could contribute appreciably to initiation of neuroinflammation as well as incipient oxidative-stress dependent neuronal damage.
While histological assessment of degenerating neurons in HC and EC was not done, the finding of large elevations in PARP-1 and cleaved (89Kd) PARP (c-PARP) in repetitively intoxicated rats points to ongoing neurodegenerative processes. However, negligible caspase-3 activation (no detectable cleaved caspase-3) after ethanol suggests limited classical apoptosis, consistent with a reported lack of brain TUNEL staining in the more severe Majchrowicz binge intoxication rat model (Obernier et al., 2002a). Accordingly, the 89 kD c-PARP fragment could arise from activities of proteases other than activated caspase-3, such as calpains, cathepsins, and/or metalloproteinases—and knowing which could help to identify specific forms of cell death (Chaitanya et al., 2010). Thus the results indicate alternative possibilities that non-apoptotic cell death programs linked to PARP-1—for example, parthanatos (David et al., 2009)—are triggered by the ethanol treatments. Parthanatos is a programmed cell death process distinct from apoptosis and conventional necrosis (Galluzzi et al., 2012; Wang et al., 2009). That nuclear PARP-1 is activated by oxidative DNA damage (Pacher and Szabo, 2008) is consistent with evidence for (a) excessive brain oxidative stress in chronic ethanol-exposed rodents (Collins and Neafsey, 2012a), (b) brain elevations in NF-κB mediated pathways by binge ethanol (Crews et al., 2011), and (c) cerebral oxidative damage in alcoholic brain (Gotz et al., 2001). Nevertheless, whether parthanatos is a neuronal death mechanism activated by chronic (binge) ethanol in adult brain requires further molecular and pharmacological validation. If the brain PARP-1 pathway is upregulated in human alcoholics, studies with inhibitors of PARP-1 activation, currently of therapeutic interest in other neurodegenerative diseases and insults (Andrabi et al., 2011; Chaitanya et al., 2010), could be extended to chronic ethanol models.
By way of a summary, Figure 6 depicts a potential transduction scheme relating the proteins changed in adult brain by repetitive ethanol treatment in vivo to oxidative stress, neuroinflammation and neurodamage. Reiterating, analyses of the brain regions known to be vulnerable to neurodegeneration show elevations in neuroinflammation-linked proteins—AQP4, cPLA2, and activated p-cPLA2. Increased cPLA2 activity would generate excessive AA release and pro-inflammatory eicosanoid production, and oxidative stress, concurrent with reduced iPLA2 which could lessen mitochondrial integrity, mitochondrial membrane potential (MMP), and neuroprotective DHA turnover. Consequently, PARP-1, increased along with c-PARP by repetitive ethanol intoxication, may be responding to oxidative stress (ROS) to drive degenerative pathways in neurons such as parthanatos or necroptosis. As further speculation, neuroimmune routes involving toll-like receptor-4 (TLR4), NF-κB, and pro-inflammatory cytokines, also linked to neuropathology during chronic alcohol exposure (Alfonso-Loeches et al., 2010; Crews et al., 2011), are integrated into this scheme in Figure 6. Whether these neuroimmune molecular events are entirely upstream of and responsible for AQP4 and PLA2 perturbations is open to investigation, although as mentioned, AQP4 is required for full pro-inflammatory cytokine responses to neuroinflammatory stimulation (Li et al., 2011). Furthermore, the cellular environments for these processes are not known; however, astrocytes, being highly enriched with AQP4, are seen as important target cells upstream of ROS- and PARP-1-mediated damage to neurons. The in vivo findings here are not inconsistent with the view (Collins and Neafsey, 2012b) that AQP4 could be a binge ethanol-responsive sensor linked to neuroinflammatory/neuroimmune cascades, especially those involving the PLA2 families but also possibly TLR4 and NF-κB.
Figure 6. Scheme interrelating repetitive daily ethanol-induced changes in AQP4, PLA2 families, ROS, and PARP-1 to proposed associations with neuroinflammation and neurodamage.
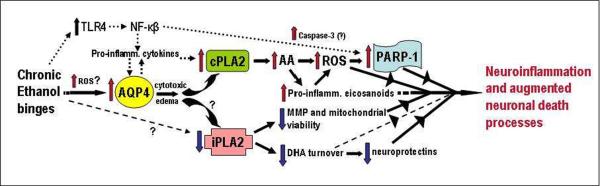
Assuming that transcriptional rather than turnover alterations are principally responsible for changes in levels, AQP4 elevations due to repeated daily ethanol intoxication, possibly stimulated by early ROS bursts, could potentiate pro-inflammatory cytokines and trigger increased cPLA2 levels/activity and AA mobilization. AA is known to generate ROS nonenzymatically and via eicosanoid biosynthesis. Pro-inflammatory eicosanoids such as leukotrienes could also have receptor-mediated neuroinflammatory effects. PARP-1 elevations could be dependent on ROS, but also on increased NF-kappaB activity. The NF-kappaB pathway is stimulated by toll-like receptor 4 (TLR4), which is known to be activated as well by chronic ethanol binges. How ethanol might decrease iPLA2 levels is not evident (question marks); nevertheless, significant loss of the enzyme may lead to decreased mitochondrial membrane permeability (MMP) and viability and reduced docosahexaenoic acid (DHA) turnover. Diminished DHA could lead to less pro-survival effects, including neuroprotectin (e.g., NPD-1) formation. See text for supporting references.
Tajuddin et al.
Acknowledgements
The research was supported by NIH U01AA018279, R01AG033605, R21AA018398, and Loyola Alcohol Research Program (T32AA013527). Robert M. Mitchell, Ph.D., is acknowledged for helpful discussions and initial technical assistance. Grace Sun, Ph.D. is thanked for recommendations concerning PLA2 antibodies.
Abbreviations
- AA
arachidonic acid
- AQP4
aquaporin-4
- DHA
docosahexaenoic acid
- EC
entorhinal cortex
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HC
hippocampus
- PARP-1
poly (ADP-ribose) polymerase-1
- c-PARP
cleaved PARP
- PLA2
phospholipase A2
- cPLA2
Ca+2-dependent PLA2
- p-cPLA2
phospho-cPLA2
- iPLA2
Ca+2-independent iPLA2
- sPLA2
secretory PLA2
- ROS
reactive oxygen species
Footnotes
Authors' contributions NFT and MMP: experimental development and/or data acquisition; EJN, TP and MAC: experimental design, data interpretation and analysis and manuscript preparation. All authors read and approved the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
In memory of Dr. Robert D. Myers, founding editor, ALCOHOL journal
Competing interests All of the authors declare that they have no competing interests.
References
- Adachi J, Mizoi Y, Fukunaga T, Ogawa Y, Ueno Y, Imamichi H. Degrees of alcohol intoxication in 117 hospitalized cases. J. Stud. Alcohol. 1991;52:448–453. doi: 10.15288/jsa.1991.52.448. [DOI] [PubMed] [Google Scholar]
- Adibhatla RM, Hatcher JF. Secretory phospholipase A2 IIA is up-regulated by TNF-alpha and IL-1alpha/beta after transient focal cerebral ischemia in rat. Brain Res. 2007;1134:199–205. doi: 10.1016/j.brainres.2006.11.080. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Adibhatla RM, Hatcher JF. Phospholipase A(2), reactive oxygen species, and lipid peroxidation in CNS pathologies. BMB Rep. 2008;41:560–567. doi: 10.5483/bmbrep.2008.41.8.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, Sanchez-Vera I, Guerri C. Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J. Neurosci. 2010;30:8285–8295. doi: 10.1523/JNEUROSCI.0976-10.2010. (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Kang HC, Haince JF, Lee YI, Zhang J, Chi Z, West AB, Koehler RC, Poirier GG, Dawson TM, et al. Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nature Med. 2011;17:692–699. doi: 10.1038/nm.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavarajappa BS, Cooper TB, Hungund BL. Effect of chronic ethanol exposure on mouse brain arachidonic acid specific phospholipase A2. Biochem. Pharmacol. 1998;55:515–521. doi: 10.1016/s0006-2952(97)00501-7. [DOI] [PubMed] [Google Scholar]
- Basavarajappa BS, Saito M, Cooper TB, Hungund BL. Activation of arachidonic acid-specific phospholipase A2 in human neuroblastoma cells after chronic alcohol exposure: prevention by GM1 ganglioside. Alcohol. Clin. Exp. Res. 1997;21:1199–1203. [PubMed] [Google Scholar]
- Bazan NG, Molina MF, Gordon WC. Docosahexaenoic Acid signalolipidomics in nutrition: significance in aging, neuroinflammation, macular degeneration, Alzheimer's, and other neurodegenerative diseases. Annu. Rev. Nutrition. 2011;31:321–351. doi: 10.1146/annurev.nutr.012809.104635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobba A, Atlante A, Petragallo VA, Marra E. Different sources of reactive oxygen species contribute to low potassium-induced apoptosis in cerebellar granule cells. Int. J. Mol. Med. 2008;21:737–745. [PubMed] [Google Scholar]
- Brown J, 3rd, Achille N, Neafsey EJ, Collins MA. Binge ethanol-induced neurodegeneration in rat organotypic brain slice cultures: effects of PLA2 inhibitor mepacrine and docosahexaenoic acid (DHA) Neurochem. Res. 2009;34:260–267. doi: 10.1007/s11064-008-9765-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlen PL, McAndrews MP, Weiss RT, Dongier M, Hill JM, Menzano E, Farcnik K, Abarbanel J, Eastwood MR. Alcohol-related dementia in the institutionalized elderly. Alcohol. Clin. Exp. Res. 1994;18:1330–1334. doi: 10.1111/j.1530-0277.1994.tb01432.x. [DOI] [PubMed] [Google Scholar]
- Chaitanya GV, Steven AJ, Babu PP. PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Comm. Signal. 2010;8:31. doi: 10.1186/1478-811X-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cippitelli A, Zook M, Bell L, Damadzic R, Eskay RL, Schwandt M, Heilig M. Reversibility of object recognition but not spatial memory impairment following binge-like alcohol exposure in rats. Neurobiol. Learn. Mem. 2010;94:538–546. doi: 10.1016/j.nlm.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MA, Corso TD, Neafsey EJ. Neuronal degeneration in rat cerebrocortical and olfactory regions during subchronic “binge” intoxication with ethanol: possible explanation for olfactory deficits in alcoholics. Alcohol. Clin. Exp. Res. 1996;20:284–292. doi: 10.1111/j.1530-0277.1996.tb01641.x. [DOI] [PubMed] [Google Scholar]
- Collins MA, Neafsey EJ. Ethanol and adult CNS neurodamage: oxidative stress, but possibly not excitotoxicity. Front. Biosci. 2012a;4:1358–1367. doi: 10.2741/465. [DOI] [PubMed] [Google Scholar]
- Collins MA, Neafsey EJ. Neuroinflammatory pathways in binge alcohol-induced neuronal degeneration: oxidative stress cascade involving aquaporin, brain edema, and phospholipase A2 activation. Neurotoxicity Res. 2012b;21:70–78. doi: 10.1007/s12640-011-9276-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MA, Zou JY, Neafsey EJ. Brain damage due to episodic alcohol exposure in vivo and in vitro: furosemide neuroprotection implicates edema-based mechanism. FASEB J. 1998;12:221–230. doi: 10.1096/fasebj.12.2.221. [DOI] [PubMed] [Google Scholar]
- Crews FT, Zou J, Qin L. Induction of innate immune genes in brain create the neurobiology of addiction. Brain Behav. Immun. 2011;25(Suppl 1):S4–S12. doi: 10.1016/j.bbi.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana R, Leto TL, Malech HL, Levy R. Essential requirement of cytosolic phospholipase A2 for activation of the phagocyte NADPH oxidase. J. Biol. Chem. 1998;273:441–445. doi: 10.1074/jbc.273.1.441. [DOI] [PubMed] [Google Scholar]
- David KK, Andrabi SA, Dawson TM, Dawson VL. Parthanatos, a messenger of death. Front. Biosci. 2009;14:1116–1128. doi: 10.2741/3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckardt MJ, Martin PR. Clinical assessment of cognition in alcoholism. Alcohol. Clin. Exp. Res. 1986;10:123–127. doi: 10.1111/j.1530-0277.1986.tb05058.x. [DOI] [PubMed] [Google Scholar]
- Floreani NA, Rump TJ, Muneer PM, Alikunju S, Morsey BM, Brodie MR, Persidsky Y, Haorah J. Alcohol-induced interactive phosphorylation of Src and toll-like receptor regulates the secretion of inflammatory mediators by human astrocytes. J. Neuroimmune Pharmacol. 2010;5:533–545. doi: 10.1007/s11481-010-9213-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friis MB, Vorum KG, Lambert IH. Volume-sensitive NADPH oxidase activity and taurine efflux in NIH3T3 mouse fibroblasts. Am. J. Physiol.Cell Physiol. 2008;294:C1552–1565. doi: 10.1152/ajpcell.00571.2007. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Diff. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goracci G, Ferrini M, Nardicchi V. Low molecular weight phospholipases A2 in mammalian brain and neural cells: roles in functions and dysfunctions. Mol. Neurobiol. 2010;41:274–289. doi: 10.1007/s12035-010-8108-6. [DOI] [PubMed] [Google Scholar]
- Gotz ME, Janetzky B, Pohli S, Gottschalk A, Gsell W, Tatschner T, Ransmayr G, Leblhuber F, Gerlach M, Reichmann H, et al. Chronic alcohol consumption and cerebral indices of oxidative stress: is there a link? Alcohol. Clin. Exp. Res. 2001;25:717–725. [PubMed] [Google Scholar]
- Gunnarson E, Zelenina M, Aperia A. Regulation of brain aquaporins. Neuroscience. 2004;129:947–955. doi: 10.1016/j.neuroscience.2004.08.022. [DOI] [PubMed] [Google Scholar]
- Gupta S, Warner J. Alcohol-related dementia: a 21st-century silent epidemic? Br. J. Psychiatry. 2008;193:351–353. doi: 10.1192/bjp.bp.108.051425. [DOI] [PubMed] [Google Scholar]
- Hassa PO, Hottiger MO, Hassa PO, Hottiger MO. The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of NF-kappaB in inflammatory disorders. Cell. Mol. Life Sci. 2002;59:1534–1553. doi: 10.1007/s00018-002-8527-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez M, Burillo SL, Crespo MS, Nieto ML. Secretory phospholipase A2 activates the cascade of mitogen-activated protein kinases and cytosolic phospholipase A2 in the human astrocytoma cell line 1321N1. J. Biol. Chem. 1998;273:606–612. doi: 10.1074/jbc.273.1.606. [DOI] [PubMed] [Google Scholar]
- Kim H-W, Cheon Y, Modi HR, Rapoport SI, Rao JS. Effects of chronic clozapine administration on markers of arachidonic acid cascade and synaptic integrity in rat brain. Psychopharmacology. 2012;222:663–674. doi: 10.1007/s00213-012-2671-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey GR, Blum JL, Covington MD, Cummings BS, McHowat J, Schnellmann RG. Decreased iPLA2gamma expression induces lipid peroxidation and cell death and sensitizes cells to oxidant-induced apoptosis. J. Lipid Res. 2008;49:1477–1487. doi: 10.1194/jlr.M800030-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivusalo M, Kapus A, Grinstein S. Sensors, transducers, and effectors that regulate cell size and shape. J. Biol. Chem. 2009;284:6595–6599. doi: 10.1074/jbc.R800049200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolko M, Rodriguez de Turco EB, Diemer NH, Bazan NG. Neuronal damage by secretory phospholipase A2: modulation by cytosolic phospholipase A2, platelet-activating factor, and cyclooxygenase-2 in neuronal cells in culture. Neurosci. Lett. 2003;338:164–168. doi: 10.1016/s0304-3940(02)01385-x. [DOI] [PubMed] [Google Scholar]
- Kramer RM, Stephenson DT, Roberts EF, Clemens JA. Cytosolic phospholipase A2 (cPLA2) and lipid mediator release in the brain. J. Lipid Mediators Cell Signal. 1996;14:3–7. doi: 10.1016/0929-7855(96)01501-5. [DOI] [PubMed] [Google Scholar]
- Lambert IH, Pedersen SF, Poulsen KA. Activation of PLA2 isoforms by cell swelling and ischaemia/hypoxia. Acta Physiol. (Oxf) 2006;187:75–85. doi: 10.1111/j.1748-1716.2006.01557.x. [DOI] [PubMed] [Google Scholar]
- Lehtonen JY, Kinnunen PK. Phospholipase A2 as a mechanosensor. Biophys. J. 1995;68:1888–1894. doi: 10.1016/S0006-3495(95)80366-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zhang H, Varrin-Doyer M, Zamvil SS, Verkman AS. Proinflammatory role of aquaporin-4 in autoimmune neuroinflammation. FASEB J. 2011;25:1556–1566. doi: 10.1096/fj.10-177279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Lindstrom CL, Donahue A, Miller MW. Differential effects of ethanol on the expression of cyclo-oxygenase in cultured cortical astrocytes and neurons. J. Neurochem. 2001;76:1354–1363. doi: 10.1046/j.1471-4159.2001.00129.x. [DOI] [PubMed] [Google Scholar]
- Ma MT, Yeo JF, Farooqui AA, Ong WY. Role of calcium independent phospholipase A2 in maintaining mitochondrial membrane potential and preventing excessive exocytosis in PC12 cells. Neurochem. Res. 2011;36:347–354. doi: 10.1007/s11064-010-0340-y. [DOI] [PubMed] [Google Scholar]
- Majchrowicz E. Induction of physical dependence upon ethanol and the associated behavioral changes in rats. Psychopharmacologia. 1975;43:245–254. doi: 10.1007/BF00429258. [DOI] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nature Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- Moses GS, Jensen MD, Lue LF, Walker DG, Sun AY, Simonyi A, Sun GY. Secretory PLA2-IIA: a new inflammatory factor for Alzheimer's disease. J. Neuroinflamm. 2006;3 doi: 10.1186/1742-2094-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obernier JA, Bouldin TW, Crews FT. Binge ethanol exposure in adult rats causes necrotic cell death. Alcohol. Clin. Exp. Res. 2002a;26:547–557. [PubMed] [Google Scholar]
- Obernier JA, White AM, Swartzwelder HS, Crews FT. Cognitive deficits and CNS damage after a 4-day binge ethanol exposure in rats. Pharmacol. Biochem. Behav. 2002b;72:521–532. doi: 10.1016/s0091-3057(02)00715-3. [DOI] [PubMed] [Google Scholar]
- Ong WY, Farooqui T, Farooqui AA. Involvement of cytosolic phospholipase A(2), calcium independent phospholipase A(2) and plasmalogen selective phospholipase A(2) in neurodegenerative and neuropsychiatric conditions. Curr. Medicinal Chem. 2010;17:2746–2763. doi: 10.2174/092986710791859289. [DOI] [PubMed] [Google Scholar]
- Pacher P, Szabo C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am. J. Pathol. 2008;173:2–13. doi: 10.2353/ajpath.2008.080019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybycien-Szymanska MM, Mott NN, Paul CR, Gillespie RA, Pak TR. Binge-pattern alcohol exposure during puberty induces long-term changes in HPA axis reactivity. PLoS ONE. 2011;6:e18350. doi: 10.1371/journal.pone.0018350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao JS, Ertley RN, DeMar JC, Jr., Rapoport SI, Bazinet RP, Lee HJ. Dietary n-3 PUFA deprivation alters expression of enzymes of the arachidonic and docosahexaenoic acid cascades in rat frontal cortex. Mol. Psychiatry. 2007;12:151–157. doi: 10.1038/sj.mp.4001887. [DOI] [PubMed] [Google Scholar]
- Roberta A, Rossella B. Aquaporins and Glia. Curr. Neuropharmacol. 2010;8:84–91. doi: 10.2174/157015910791233178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadoun S, Papadopoulos MC. Aquaporin-4 in brain and spinal cord oedema. Neuroscience. 2010;168:1036–1046. doi: 10.1016/j.neuroscience.2009.08.019. [DOI] [PubMed] [Google Scholar]
- Seleznev K, Zhao C, Zhang XH, Song K, Ma ZA. Calcium-independent phospholipase A2 localizes in and protects mitochondria during apoptotic induction by staurosporine. J. Biol. Chem. 2006;281:22275–22288. doi: 10.1074/jbc.M604330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinzawa K, Sumi H, Ikawa M, Matsuoka Y, Okabe M, Sakoda S, Tsujimoto Y. Neuroaxonal dystrophy caused by group VIA phospholipase A2 deficiency in mice: a model of human neurodegenerative disease. J. Neurosci. 2008;28:2212–2220. doi: 10.1523/JNEUROSCI.4354-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonyi A, Woods D, Sun AY, Sun GY. Grape polyphenols inhibit chronic ethanol-induced COX-2 mRNA expression in rat brain. Alcohol. Clin. Exp. Res. 2002;26:352–357. [PubMed] [Google Scholar]
- Sripathirathan K, Brown J, Neafsey EJ, Collins MA. Linking binge alcoholinduced neurodamage to brain edema and potential aquaporin-4 upregulation: evidence in rat organotypic brain slice cultures and in vivo. J. Neurotrauma. 2009;26:261–273. doi: 10.1089/neu.2008.0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strokin M, Chechneva O, Reymann KG, Reiser G, Strokin M, Chechneva O, Reymann KG, Reiser G. Neuroprotection of rat hippocampal slices exposed to oxygen-glucose deprivation by enrichment with docosahexaenoic acid and by inhibition of hydrolysis of docosahexaenoic acid-containing phospholipids by calcium independent phospholipase A2. Neuroscience. 2006;140:547–553. doi: 10.1016/j.neuroscience.2006.02.026. [DOI] [PubMed] [Google Scholar]
- Sun GY, Shelat PB, Jensen MB, He Y, Sun AY, Simonyi A. Phospholipases A2 and inflammatory responses in the central nervous system. NeuroMolecular Med. 2010;12:133–148. doi: 10.1007/s12017-009-8092-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Switzer RC, Majchrowicz E, Weight F. Ethanol-induced argyrophilia in entorhinal cortex of rat. Anat. Rec. 1982;202:186A. [Google Scholar]
- Tomas-Camardiel M, Venero JL, de Pablos RM, Rite I, Machado A, Cano J. In vivo expression of aquaporin-4 by reactive microglia. J. Neurochem. 2004;91:891–899. doi: 10.1111/j.1471-4159.2004.02759.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Dawson VL, Dawson TM. Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp. Neurol. 2009;218:193–202. doi: 10.1016/j.expneurol.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A, Ying Z, Gomez-Pinilla F. The salutary effects of DHA dietary supplementation on cognition, neuroplasticity, and membrane homeostasis after brain trauma. J. Neurotrauma. 2011;28:2113–2122. doi: 10.1089/neu.2011.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HC, Mosior M, Ni B, Dennis EA. Regional distribution, ontogeny, purification, and characterization of the Ca2+-independent phospholipase A2 from rat brain. J. Neurochem. 1999;73:1278–1287. doi: 10.1046/j.1471-4159.1999.0731278.x. [DOI] [PubMed] [Google Scholar]
- Zou J, Crews F. Induction of innate immune gene expression cascades in brain slice cultures by ethanol: key role of NF-kappaB and proinflammatory cytokines. Alcohol. Clin. Exp. Res. 2010;34:777–789. doi: 10.1111/j.1530-0277.2010.01150.x. [DOI] [PubMed] [Google Scholar]