

Abstract
Pancreatic adenocarcinoma carries an ominous prognosis and has little effective treatment. Several studies have demonstrated that the potently anti-apoptotic phosphatidyl inositol 3’-kinase (PI3K) - protein kinase B/AKT pathway is active in pancreas cancer. A recent study identified an endogenous AKT antagonist, carboxyl terminal modular protein (CTMP). CTMP inhibits the phosphorylation of AKT, preventing full activation of the kinase. We screened several cell permeable peptides from the N-terminal domain of CTMP (termed TAT-CTMP1 - 4) in vitro and found one that caused significant apoptosis in pancreatic adenocarcinoma cell lines. An inactive variant of this peptide was synthesized and used as a negative control. In all cell lines tested, TAT-CTMP4 induced a dose-dependent increase in apoptosis as detected by %-TUNEL positive cells and %-active caspase-3 (% active caspase-3 ranged from 31.2 to 61.9 at the highest dose tested (10µM)). A screening of various cell and tissue types revealed that the pro-apoptotic activity was highest in pancreatic adenocarcinoma. TAT-CTMP induced similar levels of active caspase-3 as several other known inducers of apoptosis: gemcitabine, radiation therapy, wortmannin, and recombinant tumor necrosis factor (TNF)-α. No apoptosis was observed in donor human peripheral blood mononuclear cells (PBMC, P<0.01). We further showed that TAT-CTMP4 could augment either gemcitabine chemotherapy or radiation therapy, standard therapies for pancreas cancer. Pancreatic adenocarcinoma xenografts, treated with a single dose of TAT-CTMP4 demonstrated a marked increase in caspase-3 positive tumor cells when compared to untreated controls. Additionally, pancreatic adenocarcinoma allografts treated with intratumoral TAT-CTMP and systemic gemcitabine displayed a significantly smaller tumor burden while undergoing treatment than mice in control groups (P<0.001). These data indicate that inhibiting AKT with CTMP may be of therapeutic benefit in the treatment of pancreatic adenocarcinoma and, when combined with established therapies, may result in an increase in tumor cell death.
Keywords: AKT, CTMP, Apoptosis, Pancreas Cancer
Introduction
There are few cancers as aggressive and resistant to conventional therapies as pancreatic adenocarcinoma. Pancreatic adenocarcinoma is the fourth-leading cause of cancer mortality in the US and results in greater than 37,000 deaths annually (1). Little in the way of effective therapy is available for this devastating disease (2).
Apoptosis is an essential process in cell homeostasis and eukaryotic development (3,4). The failure to execute apoptosis is both central to the development of cancer and related to resistance toward adjuvant treatments such as chemo- or radiation therapies (5–7). Thus, there is considerable interest in stimulating apoptosis and inhibiting survival machinery as components of cancer therapy (5, 8–9). Many oncogenic transformations result from the inactivation or deletion of pro-apoptotic genes or the translocation of an anti-apoptotic gene downstream of highly active promoters (10–12).
In the case of pancreatic adenocarcinoma, genes involved in AKT signaling are rarely mutated. Yet, there is mounting evidence that the AKT survival pathway plays a role in suppressing apoptosis in pancreatic adenocarcinoma (13–15). Furthermore, activating K-ras mutations are present in nearly all pancreatic adenocarcinomas (16–17). These activating mutations (in codons 12, 13, and 16) are associated with the upregulation of survival signals including the activation of the AKT survival pathway. Therefore, we devised a therapeutic strategy to target this pathway in pancreatic cancer.
Targeting the anti-apoptotic phosphatidyl inositol 3’-kinase (PI3K) - protein kinase B/AKT pathway in pancreas cancer is not a novel concept. Several investigators have undertaken the task of evaluating this therapeutic strategy. For instance, some investigators have demonstrated therapeutic benefit with the PI3K inhibitor wortmannin (18). While these studies have been encouraging, the potential ramification of inactivating an enzyme with such broad scope as PI3K maintains the potential for profound adverse events. Thus, our immediate objective was to more specifically target AKT directly.
Carboxyl terminal modular protein (CTMP) was first identified by a yeast two-hybrid assay as an AKT binding protein that both inhibited AKT phosphorylation and reverted the phenotype of v-AKT transformed cells (19). Additionally, Hwang et al recently demonstrated an anti-tumor activity of CTMP-mediated gene therapy in a lung cancer model (20). Informatics analysis of CTMP suggested that this protein has two functional domains, a structured, but functionally uncharacterized N-terminal domain and a C-terminal thioesterase domain. Based on the predicted secondary structure of N-terminal region of CTMP, we designed a panel of cell-permeable peptides (TAT-CTMPs). One of these peptides exhibited significant AKT-inhibitory activity and caused both human and murine pancreatic adenocarcinoma cell lines to undergo apoptosis in a dose-dependent fashion. Here, we assessed the pro-apoptotic activity of this CTMP-derived peptide and evaluated its therapeutic potential in multiple models of pancreatic adenocarcinoma both in vitro and in vivo.
Materials and Methods
Chemical Reagents
Unless otherwise specified, all materials were obtained from Sigma-Aldrich (Saint Louis, MO).
Cell Culture
Murine pancreatic adenocarcinoma, Panc-02, was obtained from National Cancer Institute (NCI, Frederick, MD) and maintained in supplemented RPMI 1640 containing glutamine (2mmol/L), Pyruvate (1mmol/L), penicillin and streptomycin (100 IU/mL), and 10% FBS. Human pancreatic adenocarcinoma cell lines (Panc-1, AsPC-1, CFPAC-1, and BxPC-3) were obtained from ATCC (Bethesda, MD) and maintained in Dulbecco’s modified eagle’s medium (DMEM) containing glutamine (2mmol/L), pyruvate (1mmol/L), penicillin and streptomycin (100 IU/mL), and 10% FBS. HPDE (Human Pancreas Ductal Epithelial) cells were obtained from Dr. Ming Sound Tsao and cultured in Keratinocyte serum-free (KSF) medium (Gibco/Invitrogen, Carlsbad, CA) with 50 mg/ml bovine pituitary extract (BPE), 5 ng/ml epidermal growth factor (EGF), and 1× antibiotic–antimycotic cocktail (Gibco/Invitrogen). Eso-2 esophageal adenocarcinoma and LNCaP prostate adenocarcinoma cells were provided by Peter Goedegebuure and maintained in supplemented RPMI 1640 containing glutamine (2mmol/L), pyruvate (1mmol/L), penicillin and streptomycin (100 IU/mL), and 10% FBS. HEK (Human Embryonic Kidney) 293T and AGS (gastric) cells were a kind gift of William Gillanders and maintained as described by ATCC. HT-29 colon adenocarcinoma cells were a kind gift of Nick Davidson and maintained in McCoys 5A Modified medium containing penicillin, streptomycin (100 IU/mL), and 10% FBS. All cell culture processes were carried out in a humidified atmosphere of 5% CO2 at 37 °C. All cultures were free of Mycoplasma as assayed by the Washington University Division of Comparative Medicine. Cultures were maintained for no longer than 12 weeks after recovery from frozen stocks.
Generation and preparation of TAT-CTMP peptides
Peptides were synthesized by the Tufts University Core Facility (Medford, MA) using standard HOBt/Fmoc chemistry and purified by reverse phase HPLC to >95% purity. The final amino acid compositions were verified utilizing amino acid analysis and MALDI TOF mass spectrometry. The cell permeation sequence used was previously demonstrated to have 10-fold increased intracellular concentration compared to the native TAT sequence from HIV-1 (21). We designed a negative control peptide, TAT-CTMP4-Inactive, that contained two amino acid substitutions. The design was based on the predicted helical structure of the CTMP4 peptide, replacing the two phenylalanines on one face of the helix with alanines. The amino acid sequences of these peptides are:
TAT-CTMP1: Ac-VEESSFSRLEPRPESGPLRR-RKKRR-ORN-RRR-NH2
TAT-CTMP2: Ac-WSGDECKKMFQDFLLRLDKNWSPN-RKKRR-ORN-RRR-NH2
TAT-CTMP3: Ac-DLFHTKFDQIWETPTRKYSPLRKWSGD-RKKRR-ORN-RRR-NH2
TAT-CTMP4: Ac-LDPKLMKEEQMSQAQLFTRSFDDGL-RKKRR-ORN-RRR-NH2
TAT-CTMP4 Inactive: Ac-LDPKLMKEEQMSQAQLATRSADDGL-RKKRR-ORN-RRR-NH2
Evaluation of Timecourse of Apoptosis
The timecourse of the TAT-CTMP-induced apoptosis was evaluated using the Apo-ONE homogenous caspase 3/7 Assay (Promega, Madison, WI). This assay allows for the assessment of active caspase 3/7 in a high-throughput, 96-well format that allows for the assessment of multiple samples at multiple timepoints. Briefly, Panc-02 murine pancreatic adenocarcinoma were maintained to 80% confluency in a 96-well plate. Samples were subsequently treated with PBS, TAT-CTMP4-Inactive, and TAT-CTMP4. Caspase 3/7 was then assessed at 2 hour intervals up to 20 hours after treatment as described by the manufacturer.
Evaluation of apoptosis in vitro by flow cytometry
Tumor cells were seeded at a density of approximately 0.5 × 106 cells per well in 12-well plates in 1.0 ml culture medium. Cells were then split and incubated at 37 °C in humidified 5% CO2 for more than 24 hours (Panc-02) and 48 hours (AsPC-1, BxPC-3, CFPAC-1, Panc-1) to ensure uniform growth conditions. Compounds were dissolved in PBS (TAT-CTMP compounds) or DMSO (wortmannin) and added to the culture medium at the concentrations indicated. The final concentration of DMSO in the cell culture medium was less than 1%. The cells were then incubated for 18 hours at 37 °C in humidified 5% CO2. The extent of apoptosis was subsequently measured as previously reported (22–23). Briefly, staining was performed on trypsin-EDTA treated cultures that had been fixed with 1% paraformaldehyde and 90% methanol. Cell pellets were resuspended in either TUNEL reagent (APO-BRDU kit, San Diego, CA) or FACS buffer containing the antibody recognizing cleaved caspase-3 (1:100, Cell Signaling Technology, Inc. Boston, MA) and incubated overnight. After washing, cells were resuspended in buffer containing a fluorescein-conjugated antibody (1:50) or 7-AAD buffer and incubated for 1 hour at room temperature. Cell-associated fluorescence was determined using flow cytometry (FACScan, BD Biosciences, San Jose, CA) and analyzed with CellQuest software (BD Biosciences, San Jose, CA).
Peripheral blood mononuclear cells (PBMC) were used to assess peptide activity in non-cancerous cells. PBMCs were isolated from normal laboratory volunteers as previously described (24). Briefly, fresh whole blood was obtained by venipuncture from five healthy human volunteers. PBMC were obtained after differential migration through a ficoll gradient. PBMC were quantitated and 2–3 × 106 were plated. PBMC were subsequently treated with PBS (no treatment control), TAT-CTMP4-Inactive (10 µM), TAT-CTMP4 (10 µM), or irradiated with 10,000 rads gamma radiation from a Cobalt-60 source (J.L. Shepherd and Associates). PBMCs were incubated for 18 hours at 37 °C and subsequently harvested for flow cytometric analysis. Human studies were approved by the Washington University Institutional Review Board.
TAT-CTMP4 was compared to other inducers of apoptosis. Samples were treated as outlined above in the presence of TAT-CTMP4, TAT-CTMP4 Inactive, gemcitabine (30 nM, Eli Lilly, Indianapolis, IN), wortmannin (1 µM), or recombinant human TNF-α (10 ng/ml, Cell Signal Technologies, Inc., Boston, MA). Irradiated samples were treated with 2000 rads (pancreatic adenocarcinoma) as outlined above. Apoptosis was subsequently measured 18 hours later.
Western blotting
Cells were grown to 80% confluence in complete media and then for 24 hours in serum-free DMEM (human pancreas adenocarcinoma) or RPMI (murine adenocarcinoma). Cells were then incubated in the presence of pharmacologic agents (PBS (untreated control), DMSO (vehicle control for wortmannin), wortmannin (1 µM), TAT-CTMP4-Inactive (10 µM), and TAT-CTMP4 (10 µM)). Cells were washed with PBS and then lysed with cell lysis buffer (Cell Signal Technologies, Inc. Boston, MA) as described by the manufacturer. Total protein concentrations were determined using the Pierce BCA Protein Assay Kit (Pierce, Rockford, IL). Samples of total protein (40 µg) were separated by SDS/PAGE and transferred to nitrocellulose membranes (Invitrogen, Carlsbad, CA). Blots were blocked with nonfat dry milk (5% in TBS) and incubated overnight with anti-total AKT or anti-phospho-AKT (Ser473), total GSK-3β, or phospho-GSK-3α/β (Ser21/9) antibodies (Cell Signal Technologies, Inc. Boston, MA) at 4 °C. Blots were washed and probed with species-specific secondary antibodies, and developed as described by the manufacturer.
Quantitative Detection of Phosphorylated Proteins
Cell lysates were generated and assessed utilizing the Biorad Bio-Plex™ bead suspension array system (Hercules, CA) as recommended by the manufacturer and as previously reported (25–26). This multiplexing system allows the assessment of the phosphorylation state of multiple proteins in a single sample. Briefly, samples were treated as described above and lysates were generated 18 hours later. Next, samples were incubated in the presence of Bio-Plex™ antibodies overnight followed by reporter antibodies and fluorescence was subsequently quantitated utilizing the Bio-Plex™ array reader. Analytes assessed included phospho (p)-AKT, p-GSK 3α/β, p-JNK, p-IKB α, p-ERK1/2, p-p38 MAPK, p-p70 S6 Kinase, and p-90 RSK.
Tumor Xenograft Studies
Female athymic nude mice (NCr-nu/nu) were obtained from the animal production area of the National Cancer Institute (Frederick, MD). The mice were housed and maintained in accordance with current regulations and standards of the United States Department of Agriculture, United States Department of Health and Human Services, and NIH, and their use in these experiments was approved by the Institutional Animals Care and Use Committee. The mice were used when they were 8–10 weeks of age. CFPAC-1 cells were harvested from subconfluent cultures by treatment with 0.25% trypsin and 0.02% EDTA. Trypsinization was stopped with medium containing FBS. 1 × 106 Cells were mixed with Matrigel (Collaborative Research, Bedford, MA; 3.5 mg of Matrigel in 0.5 ml of DMEM). One posterior neck tumor was generated in each mouse. Fourteen days later, the mice were randomly assigned to four treatment groups. Treatments were delivered by a single intra-tumoral injection. The treated mice were closely monitored for any signs of progressive disease and sacrificed 24 hours later for tumor removal. For immunohistochemistry and histology staining procedures, the tumor tissue was fixed in formalin and embedded in paraffin.
Immunohistochemistry
Paraffin-embedded tissues were used for identification of active caspase-3. Samples were prepared at the Washington University Morphology Core. Briefly, 4 µm-thick sections were mounted on Superfrost slides (Fisher Scientific, Houston, TX). Sections were deparaffinized in xylene, followed by three changes of 100% ethanol and pretreatment with 3% hydrogen peroxide in methanol. Retrieval was performed using Biocare Reveal reagent (Concord, CA) and a Biocare decloaking chamber. Samples were then rinsed with dH2O, placed in phosphate-buffered saline (PBS)-Tween 20 (0.05%), rinsed in PBS (pH 7.5), and incubated with anti-cleaved caspase-3 antibody (Biocare, Concord, CA) overnight at 4°C. The next day, samples were treated with Mach-2 HRP-conjugated Polymer (Biocare) and as recommended by the manufacturer. Samples were subsequently developed with DAB solution, counterstained with Hematoxylin and subsequently mounted for viewing.
Evaluation of Immunohistochemical and Histological Tissue Staining
H&E and caspase-3 stained slides were submitted for review to a patholobiologist at the Department of Comparative Medicine at Washington University. Slides were labeled numerically and subsequently submitted, in a blinded fashion, for formal interpretation as well as acquisition of representative images. H&E images are magnified by a factor of 100 and caspase 3 images by a factor of 200.
In vivo assessment of tumor growth
Female C57BL/6 mice (8–12 weeks old) were purchased from the NCI and acclimated for at least 2 weeks before tumor implantation. All mice were injected in the right flank with a 200 µl single cell suspension containing 2.5 × 105 Panc-02 cells. Prior to treatment, mice were randomized into 4 experimental groups (No treatment, gemcitabine, gemcitabine + TAT-CTMP Inactive, and gemcitabine + TAT-CTMP). Treatment of tumors started 2 weeks after tumor implantation, at which point the mean tumor diameter was approximately 5 mm. Tumor bearing mice (n=10 per group) receiving gemcitabine were treated with intraperitoneal injections (1.5 mg/mouse, days 14 and 21). Mice receiving TAT-CTMP Inactive or TAT-CTMP were treated every other day for two weeks with intratumoral injection (0.5 mg/mouse, days 14, 16, 18, 20, 22, 24, 26). Mean tumor diameter was measured three times each week. All mice were euthanized when their tumor ulcerated or reached a mean diameter of 15 mm. All studies were performed in accordance with an animal protocol approved by the Washington University Institutional Animal Care Facility.
Data Processing
Error bars, unless stated otherwise, represent means plus or minus SEM of an experiment performed with at least three technical replicates and are representative of three biological replicates. FACS results were analyzed by a two-tailed t-test. For the in vivo analyses of mean tumor diameter, statistical analyses were preformed using a linear mixed repeated measures model. A p-value of less than 0.05 was considered significant for all analyses. Figures and statistics were generated using GraphPad Prism™ software.
Results
TAT-CTMP4 induced apoptosis in pancreatic adenocarcinoma in vitro
We synthesized four cell-permeable peptides based on the sequence of CTMP (schematically shown in Figure 1, Panels A and B) and screened them at a fixed concentration (10 µM) for the ability to induce apoptosis in Panc-1 model pancreatic adenocarcinoma (Figure 1, Panel C). Under these conditions, one candidate peptide, TAT-CTMP4, induced a 25% increase in intracellular active caspase-3 while the remaining peptides conferred no pro-apoptotic activity. The TAT-CTMP4-induced increase in apoptotic cells was comparable to the response induced by radiation therapy (2,000 rads) alone. A variant peptide was designed based on the predicted helical structure of the CTMP4 peptide; by replacing the two phenylalanines on one face of the helix with alanines, the pro-apoptotic activity was lost (Figure 1, Panel B and C). In order to further characterize this response, we utilized a screening assay that quantitates active caspase-3 and −7. Measurements were performed at multiple timepoints and the magnitude of caspase-3 activity induced by TAT-CTMP4 appeared to peak 18 hours after treatment (data not shown).
Figure 1. Schematic structure of CTMP peptides and screening of the pro-apoptotic activity of CTMP-derived peptides in model pancreas adenocarcinoma.
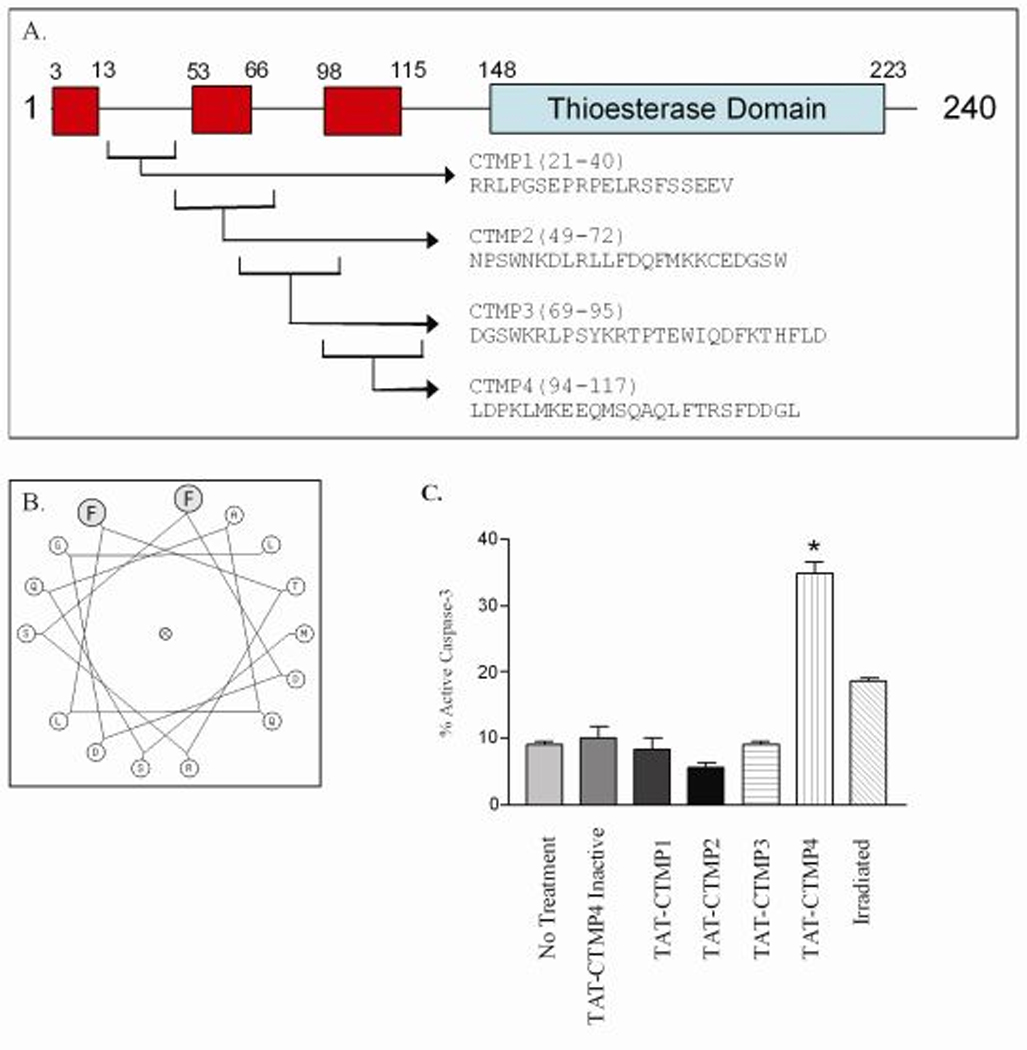
(A) Linear diagram of full-length CTMP. Areas outlined in red are predicted α-helices. Area outline in blue is a putative thioesterase domain. Cropped areas denote sequences from which CTMP peptides 1–4 were generated. (B) Helical wheel diagram of the active domain of CTMP4. The shaded residues are mutated from phenylalanine to alanine in the inactive control peptide (TAT-CTMP4 Inactive). (C) Human model pancreatic adenocarcinoma (Panc-1) were treated with TAT-CTMP peptides 1–4, and TAT-CTMP4-Inactive at a concentration of 10 µM. PBS alone and radiation therapy (2000 rads) were utilized as controls. After 18 hours of treatment, cells were harvested and % active caspase-3 was determined by flow cytometry. Each experimental group represents an n=3. Results are expressed as the mean, with bars representing standard error of the mean. *=P<0.01.
TAT-CTMP4 inhibited phosphorylation and enzymatic activity of AKT
Model human pancreatic adenocarcinoma (CFPAC-1) cell lysates were generated after treatment for 1 hour with escalating doses of TAT-CTMP4 (1 µM, 10 µM, and 100 µM). Western blots were subsequently probed for total AKT and activated, phospho-AKT (Ser473). Under these conditions, treatment with TAT-CTMP4 resulted in a dose-dependent decrease in pSer473-AKT (Figure 2, Panel A). Similar results were observed with lysates generated from the human pancreatic adenocarcinoma cell line, Panc-1 (data not shown).
Figure 2. The induction of apoptosis by TAT-CTMP4 occurs via inhibition of the AKT pathway.
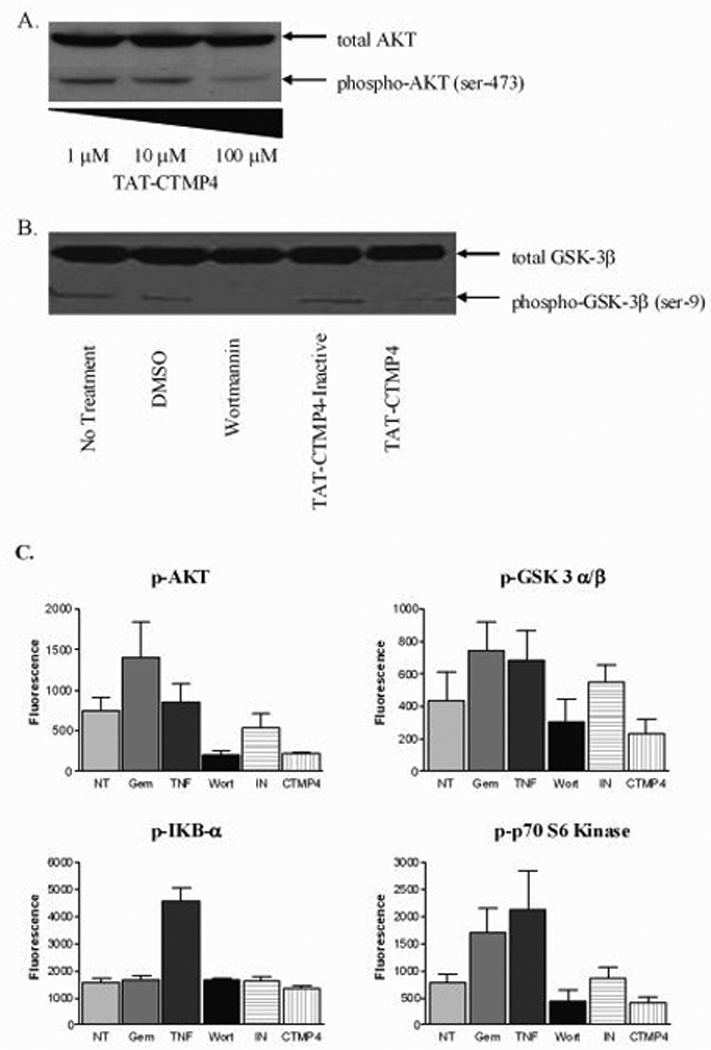
(A) Model human pancreatic adenocarcinoma (CFPAC-1) were treated with escalating doses of TAT-CTMP4. Samples were treated for 1 hour prior to preparation of cell lysates. Western blots were subsequently prepared and stained with anti-total AKT and anti-phospho-AKT (Ser473). (B) Panc-1 lysates were generated after exposure to PBS (no treatment control), DMSO (vehicle control for wortmannin), wortmannin (1 µM), TAT-CTMP4-Inactive (10 µM), and TAT-CTMP4 (10 µM) for 18 hours. Western blots were probed with anti-total GSK-3β and phospho-GSK-3α/β (Ser21/9). (C) Human model pancreatic adenocarcinoma (CFPAC-1) were treated with PBS (no treatment), gemcitabine (30 nM), recombinant human TNF-α (10 ng/ml), wortmannin (1 µM), TAT-CTMP4-Inactive (10 µM), and TAT-CTMP4 (10 µM). One hour later, cell lysates were generated and phospho-proteins were quantitated by bioplex analysis. Each experimental group represents an n=4. Results are expressed as the mean, with bars representing standard error of the mean.
Additionally, we generated lysates with the same treatments (TAT-CTMP4 (10 µM), TAT-CTMP4-Inactive (10 µM), wortmannin (1 µM), or vehicle control) after 18 hours of exposure. We then probed these samples for a proxmal substrate and downstream effector of the AKT survival pathway, total GSK-3β and phospho-GSK3β (Ser9). Treatment with TAT-CTMP4 at this interval also demonstrated a reduction in the amount of pSer9-GSK3β (Figure 2, Panel B).
In order to quantitate these observed phosphorylation-state changes, we utilized a Bio-Plex™ suspension array system. This antibody-based system allows the assessment of the phosphorylation state of multiple intracellular signaling mediators in a single sample. For this assessment, we generated lysates of human pancreatic cancer cell lines treated with PBS (no treatment control), gemcitabine (30 nM), recombinant human TNF-α, wortmannin (1 µM), TAT-CTMP4-Inactive (10 µM) and TAT-CTMP4 (10 µM). As demonstrated in Figure 2, Panel C, samples treated with TAT-CTMP4 demonstrated a decrease in phospho-AKT and phospho- GSK3β, comparable to the effect caused by direct inhibition of the upstream kinase, PI3k, with wortmannin. Additionally, phospho-p70 S6 kinase, another downstream mediator of the AKT pathway, was found to be reduced. These data strongly indicate that the activity of TAT-CTMP4 occurs via inhibition of the AKT survival pathway. Importantly, no changes in the phosphorylation state of the pro-inflammatory mediator phospho-IKB-α was observed (Figure 2, Panel C). Additionally, the phosphorylation state of phospho-JNK, phospho-ERK1/2, phospho-RSK, and phospho-p38 MAPK was also assessed and found to be unchanged when compared to no-treatment and TAT-CTMP4-Inactive controls (data not shown).
Consistent with previous reports, treatment with gemcitabine induced phosphorylation of Akt and its downstream effectors (Fig 2C), indicating that the cytotoxic effects of gemcitabine may be augmented by concomitant treatment with Akt inhibitors. The therapeutic utility of coadministering gemcitabine with an Akt inhibitor was first suggested by the findings of Ng et al (27). In that study, wortmannin co-administered with gemcitabine, had a significant effect compared to gemcitabine alone. RNA silencing studies have also implied a role for Akt in gemcitabine sensitivity of several pancreas adenocarcinoma cell lines (28), but siRNA treatments do not result in immediate down-regulation and may have off-target effects that mask the role of the targeted gene. The paucity of reagents that inhibit Akt directly has made it impossible to test whether Akt itself is a key gene in pancreas adenocarcinoma pathogenesis and therefore, the CTMP4 peptide represents the most direct probe of Akt itself.
TAT-CTMP4 induced apoptosis in model pancreatic adenocarcinoma in a dose-dependent fashion
Next, we determined the apoptotic response of human pancreatic adenocarcinoma cells as a function of the concentration of TAT-CTMP4. Figure 3, Panel A displays a representative histogram demonstrating the increase in the percentage of active caspase-3 positive cells in model pancreatic adenocarcinoma treated with a fixed concentration of TAT-CTMP4 (compared to TAT-CTMP4-Inactive). TAT-CTMP4 demonstrated a dose dependent increase in apoptosis, as measured by both the percentage of cells containing active caspase-3 (Figure 3, Panel B) and the percentage of TUNEL positive cells (Figure 3, Panel C). We then determined the activity of these two peptides in human and murine pancreatic adenocarcinoma cell lines at a fixed concentration (10 µM, Figure 3, Panel D). Of the cell lines tested, AsPC-1 cells were least responsive to treatment using TAT-CTMP4 with only a 20% increase in the amount of apoptosis over baseline. This finding correlates with the diminished basal level of phospho-AKT (Ser473) in these cells as described by Bondar et al (13). The other cell lines tested were more sensitive to TAT-CTMP4 treatment with approximately 35–40% of the apoptosis observed being treatment-specific. Specifically, the range of replicates for 10 µM TAT-CTMP4-treated human Panc-1 adenocarcinomas, were 15–49%-active caspase-3 (Mean = 35%, Median = 40.9%, SEM = 4.5%, N=9) and 38–44%-TUNEL positivity (Mean = 41.5%, Median = 41.8, SEM = 1, N=6). Alternatively, the range of biological replicates for TAT-CTMP4-treated murine Panc-02 was 19–46% active caspase-3 (Mean = 34.2%, Median = 34.2%, SEM = 2.9%, N=9). As previously observed, the inactive variant peptide had no effect on any of the cell lines tested.
Figure 3. Inhibition of the AKT pathway with the novel therapeutic, TAT-CTMP4, induces apoptosis in human and murine pancreatic adenocarcinoma in a dose-dependent fashion.
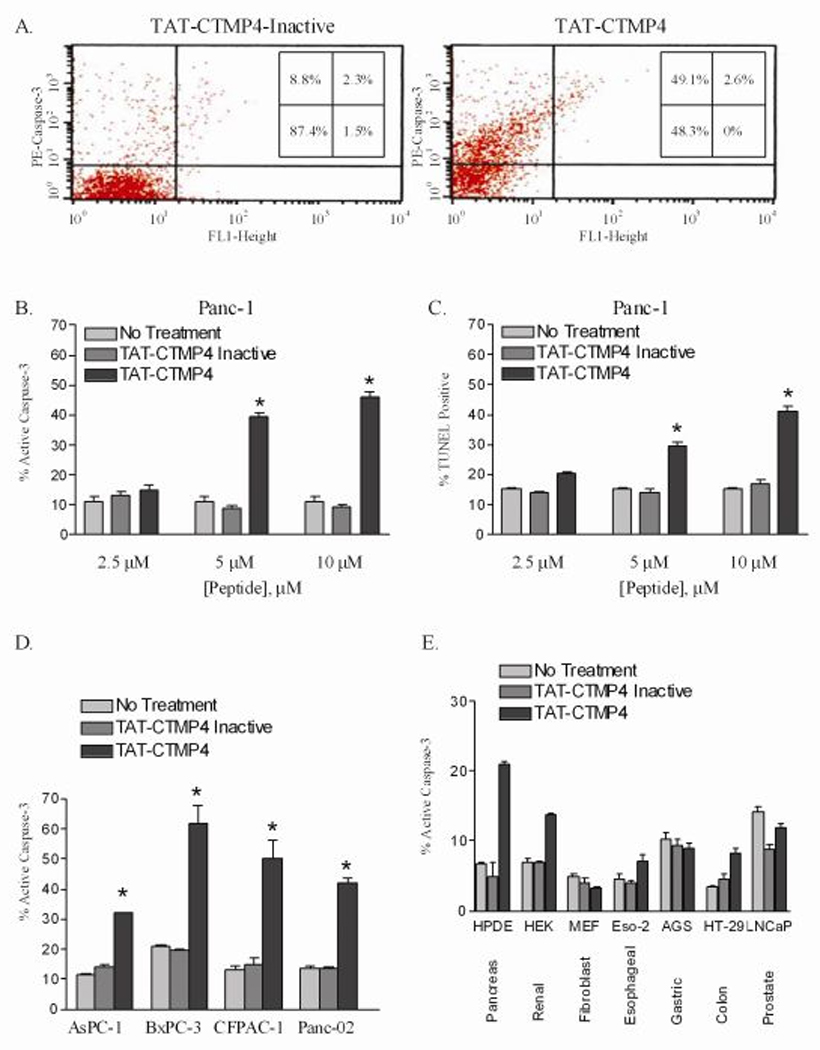
(A) Representative FACS profile of model human pancreatic adenocarcinoma (Panc-1) treated with TAT-CTMP4 Inactive (10 µM) or TAT-CTMP4 (10 µM) and subsequently stained for active caspase-3. The human pancreatic adenocarcinoma cell line, Panc-1, was treated with PBS (no treatment), TAT-CTMP4-Inactive, and TAT-CTMP4 at 2.5, 5, and 10 µM concentrations for 18 hours. Cells were subsequently harvested and (B) % active caspase-3 and (C) % TUNEL positive cells were determined by flow cytometry. Each experimental group represents an n=3. (D) Assessment of % active caspase-3 in various model human (AsPC-1, BxPC-3, CFPAC-1) and a murine (Panc-02) pancreatic adenocarcinoma cell line treated with PBS (no treatment control), TAT-CTMP4 Inactive, and TAT-CTMP4 at a concentration of 10 µM for 18 hours. In all cases: * = P< 0.03. (E) Assessment of % active caspase-3 in various cell and tissue types. Samples were treated with PBS (no treatment control), TAT-CTMP4 Inactive, and TAT-CTMP4 at a concentration of 10 µM for 18 hours. Results are expressed as the mean, with bars representing standard error of the mean.
In order to further characterize the specificity of the TAT-CTMP response, we evaluated several other cell lines under similar conditions. The non-cancerous Human Pancreatic Ductal Epithelia cell lines, perhaps the closest possible normal control, did display a modest response to TAT-CTMP4 (Figure 3, Panel E) when compared to controls. This response was less than the least sensitive pancreatic adenocarcinoma cell line, AsPC-1. Still, this result may predict a broad-based pancreatic toxicity. Notably, the fibroblasts and gastric adenocarcinoma did not elicit a response while renal, esophageal, colon, and prostatic demonstrated modest responses with respect to increases in caspase-3 activity.
TAT-CTMP4 treatment does not induce apoptosis in human peripheral blood mononuclear cells, but does induce levels of apoptosis comparable to other known apoptosis-inducing agents
A feasible, translatable therapy for pancreas cancer ideally is non-toxic to normal, non-malignant cells. In order to address this question, we treated normal, human PBMC with PBS (no treatment control), TAT-CTMP4 Inactive, TAT-CTMP4, and with radiotherapy. As demonstrated in Figure 4, Panel A, TAT-CTMP4 does not induce apoptosis in these normal cells. Specifically, the range of replicates for TAT-CTMP4 treated PBMC was 3.7–8.9% apoptosis (Mean = 5.4%, Median = 4.6%, SEM = 0.6%, N=10).
Figure 4. TAT-CTMP4 treatment does not induce apoptosis in human peripheral blood mononuclear cells (PBMC, A.), but does induce levels of apoptosis comparable to other known apoptosis-inducing agents (B.).
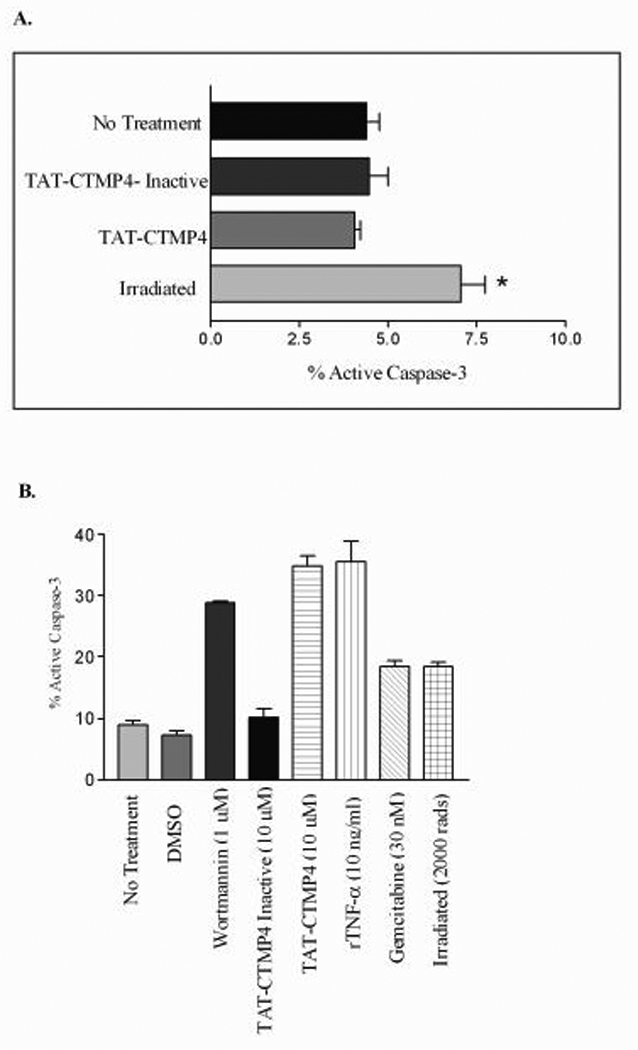
Human peripheral blood mononuclear cells were isolated from normal laboratory volunteers. 2.0 – 3.0 × 106 PBMC were incubated for 18 h with PBS (no treatment control), TAT-CTMP4-Inactive (10 µM), TAT-CTMP4 (10 µM), or after being irradiated at 10,000 rads. % active caspase-3 was subsequently measured by flow cytometry. Each experimental group represents an n=5. Results are expressed as the mean, with bars representing standard error of the mean. * = p<0.01. (B) Human model pancreatic adenocarcinoma (Panc-1) were treated with PBS (no treatment), DMSO (vehicle control for wortmannin), wortmannin (1 µM), TAT-CTMP4-Inactive (10 µM), TAT-CTMP4 (10 µM), recombinant human TNF-α (10 ng/ml), gemcitabine (30 nM), and radiation therapy (2000 rads). After 18 hours of treatment, cells were harvested and % active caspase-3 was determined by flow cytometry. Each experimental group represents an n=3. Results are expressed as the mean, with bars representing standard error of the mean.
We next sought to compare the magnitude of apoptosis induced by TAT-CTMP4 (at sub maximal doses) to established doses of other known inducers of apoptosis in model pancreatic adenocarcinoma cells (Panc-1). In this example, treatment with TAT-CTMP4 induced a similar degree of apoptosis as established doses of wortmannin, gemcitabine, recombinant TNF-α, or radiation (Figure 4, Panel B). Neither vehicle-treated nor TAT-CTMP4-Inactive treated cells displayed an increase in the percentage of apoptotic cells compared to untreated cells in culture.
Gemcitabine or radiation sensitized human pancreatic adenocarcinoma cells to TAT-CTMP4 treatment
Of the cell lines tested, the human pancreas adenocarcinoma AsPC-1 was the most resistant to TAT-CTMP4-specific apoptosis (Figure 3, Panel D). Therefore, we next sought to determine whether we could augment the pro-apoptotic response of TAT-CTMP4 by treating these relatively resistant model adenocarcinoma with submaximal doses of TAT-CTMP4 and conventional chemotherapy (gemcitabine) or radiation therapy. When given together at submaximal concentrations, gemcitabine and TAT-CTMP4 induce an additive response that is greater than either agent in isolation (Figure 5, Panel A). Additionally, the response observed after the treatment of model pancreatic adenocarcinoma with TAT-CTMP4 and radiation is also clearly greater than either agent alone (Figure 5, Panel B). It is important to note that the response observed to TAT-CTMP4-Inactive and gemcitabine (Figure 5, Panel A) or TAT-CTMP4-Inactive and radiation (Figure 5, Panel B) is equivalent to the response observed to gemcitabine or radiation therapy alone.
Figure 5. Treatment of the chemo-resistant cell line, AsPC-1, utilizing combination therapy with TAT-CTMP4 in the presence of gemcitabine (A) and radiation therapy (B) results in an enhanced induction of apoptosis in model human pancreatic adenocarcinoma.
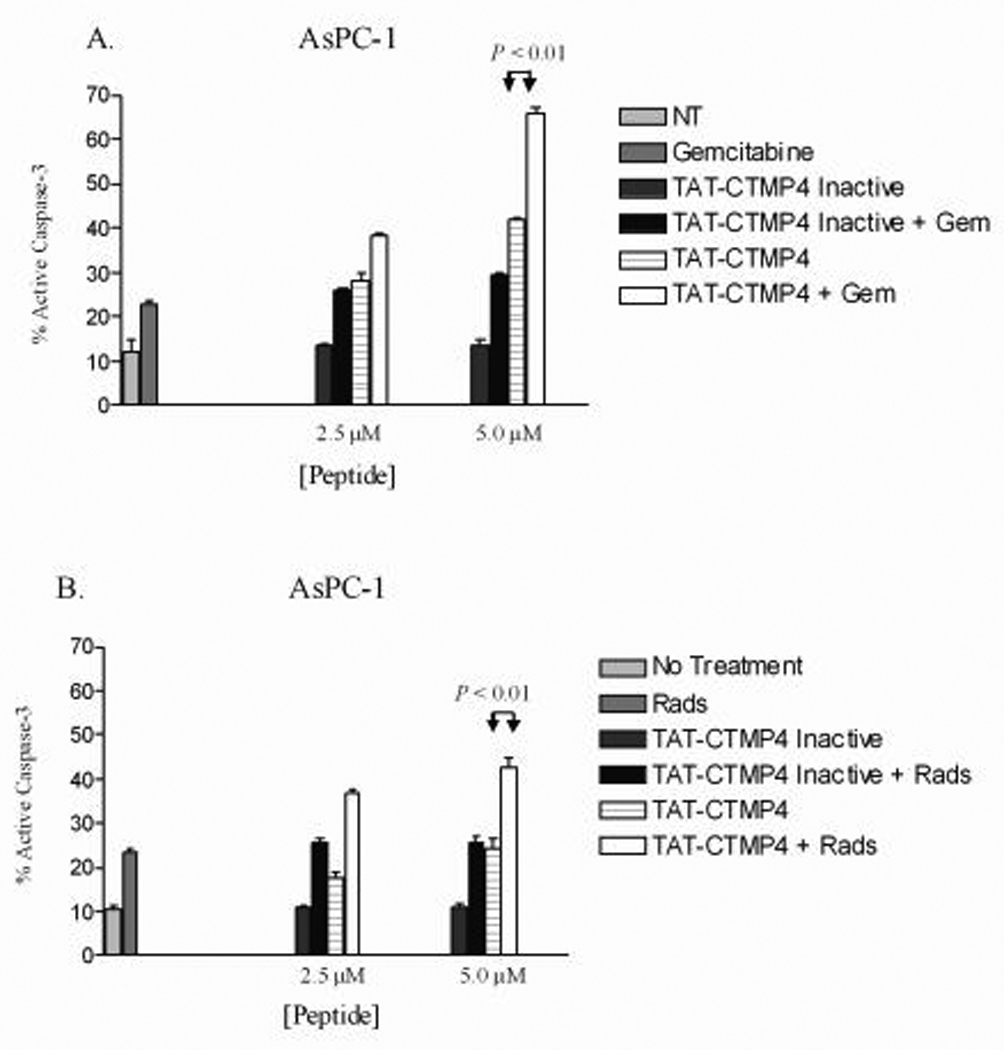
The human pancreatic adenocarcinoma cell line, AsPC-1, was treated with PBS (no treatment), and submaximal doses of TAT-CTMP4 (2.5 and 5 µM), TAT-CTMP4-Inactive (2.5 and 5 µM) in the presence of gemcitabine (A, 30 nM) or radiation therapy (B, 2000 rads). After 18 hours of treatment, cells were harvested and % active caspase-3 was determined by flow cytometry. Results are expressed as the mean, with bars representing standard error of the mean.
Treatment of mice bearing heterotopic pancreatic adenocarcinoma with TAT- CTMP4 results in increased tumor apoptosis and tumor growth inhibition
In order to quantitate an in vivo response to TAT-CTMP4 treatment, a murine tumor xenograft model was developed. Athymic nude mice bearing human pancreatic adenocarcinoma were maintained, and established xenografts were treated with a single intra-tumoral dose of TAT-CTMP4 or appropriate controls. Tumors were then harvested and processed for routine histology and immunohistochemical staining. As shown in Figure 6, tumors harvested after a single treatment with TAT-CTMP4 demonstrated a marked increase in tumor apoptosis as demonstrated by increased caspase-3 staining of paraffin-embedded specimens.
Figure 6. Treatment of mice bearing heterotopic pancreatic adenocarcinoma xenografts with TAT-CTMP4 results in increased tumor apoptosis as detected by caspase-3 staining.
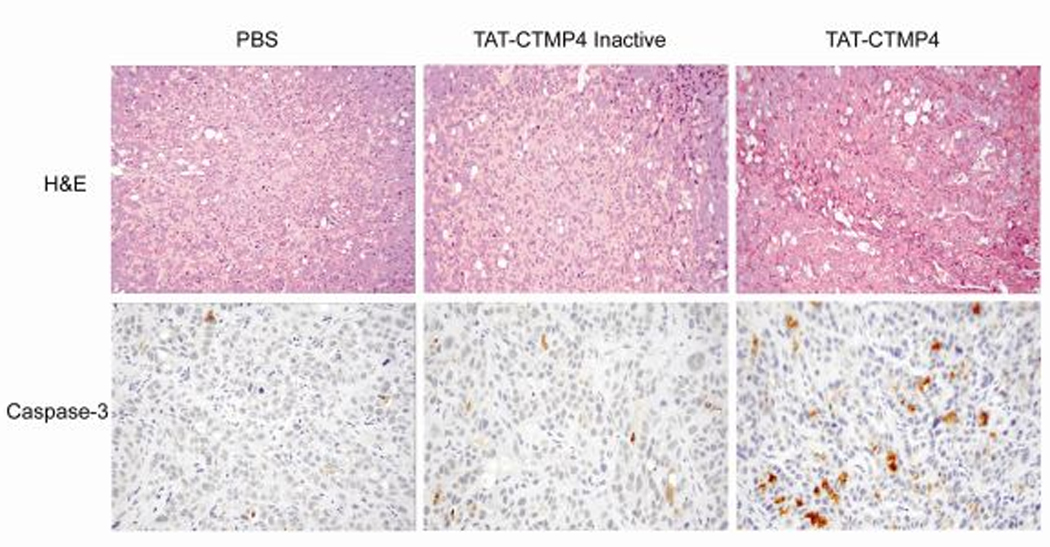
The human pancreatic adenocarcinoma cell line, CFPAC-1, was implanted into the neck of nude mice (1 × 106). After 10 days (mean tumor volume approx. 200 mm3), xenografts were treated with a single dose of intratumor PBS (200 µl), TAT-CTMP4 Inactive (400 µg), or TAT-CTMP4 (400 µg). Tumors were harvested 24 hours later and H&E and caspase-3 staining was subsequently performed on paraffin-embedded samples. Images presented were selected in a blinded fashion by an independent pathobiologist.
We further characterized the therapeutic efficacy of our pro-apoptotic agent by treating murine pancreatic adenocarcinoma allografts. Based on our in vitro data, we tested TAT-CTMP4 in combination with conventional chemotherapy in a manner intended to optimize our ability to elicit a positive response. Our treatment regimen was designed to closely mimic the clinical standard of weekly gemcitabine administration. As demonstrated in Figure 7, mice treated with weekly, systemic gemcitabine (2 doses), and every other day TAT-CTMP4 (intratumoral, 7 doses) demonstrated a statistically significant reduction in mean tumor diameter compared to no treatment, gemcitabine-only, and gemcitabine +TAT-CTMP4 Inactive controls. No gross toxicity was observed in mice treated with combination gemcitabine/TAT-CTMP4 or gemcitabine-alone. Our data strengthen the assertions of Ng et al and for the first time provide unambiguous evidence for the role of Akt, divorced from PI3-kinase, in pancreas adenocarcinoma pathogenesis.
Figure 7. Treatment of mice bearing heterotopic pancreatic allografts with weekly gemcitabine and every other day TAT-CTMP results in inhibition of tumor growth compared to no treatment and gemcitabine-only controls.
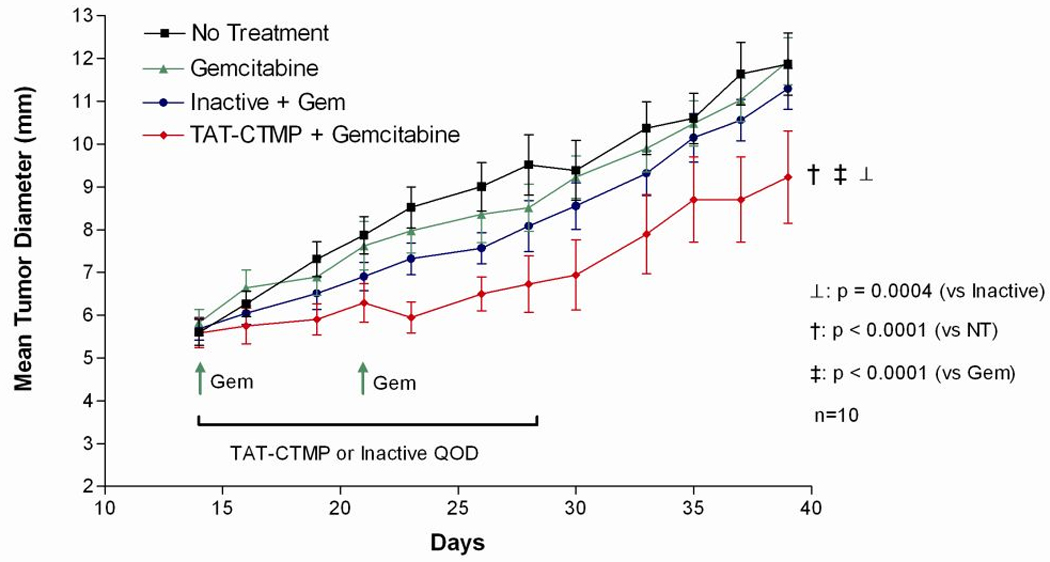
The murine pancreatic adenocarcinoma cell line, Panc-02, was implanted into the right flank of C57/BL6 mice (2.5 × 105). After 14 days (mean tumor diameter approx 5 mm), treatment was initiated. Tumor bearing mice (n=10 per group) receiving gemcitabine were treated with intraperitoneal injections (1.5 mg/mouse, days 14 and 21). Mice receiving TAT-CTMP Inactive or TAT-CTMP were treated every other day for two weeks by intratumoral injection (0.5 mg/mouse, days 14, 16, 18, 20, 22, 24, 26). Results are expressed as the mean, with bars representing standard error of the mean.
Discussion
This report demonstrates that inhibition of AKT phosphorylation with the cell-permeable peptide TAT-CTMP4 results in a dose-dependent induction of apoptosis in model pancreatic adenocarcinoma. This appears to be an effect that is limited to specific cell types, as PBMC, fibroblasts, and some adenocarcinomas are unaffected by treatment with this peptide. Additionally, these data support the concept of utilizing biologic therapy (AKT inhibition) combined with chemo- or radiotherapy to further augment an anti-tumor response in resistant malignancies.
In this report, we reasoned that the highly structured, yet functionally uncharacterized region of CTMP was responsible for the observed AKT-inhibitory activity of the full length protein. Based on criteria presented by Hemmings, and the predicted secondary structure elements of the N-terminal domain of CTMP, we designed four TAT-conjugated peptides and screened them for their ability to induce pancreas adenocarcinoma cell apoptosis. The C-terminal most peptide (TAT-CTMP4, encompassing amino acids 94–117 of the full length protein) was found to cause significant pancreatic adenocarcinoma cell apoptosis. This peptide was predicted to have a helical structure and substitution of two bulky, aromatic residues on one face of the helix with alanines resulted in an inactive variant peptide. To our knowledge, this is the first report that a short peptide from CTMP can recapitulate the AKT-inhibitory activity of the full length protein.
AKT itself is rarely mutated in pancreatic adenocarcinoma. Ras mutations, however, are among the earliest and most common genetic mutations in pancreatic cancer (16–17, 29). Specifically, activating K-ras mutations are present in nearly all pancreatic adenocarcinomas. These activating K-ras mutations result in the phosphorylation of membrane phosphatidylinositol which subsequently recruits PH domain-containing proteins, such as AKT, to the plasma membrane. This leads to the phosphorylation of AKT and the subsequent downstream activation of pro-survival signals (30). Taken together, these observations suggest that targeting AKT may provide an important checkpoint in a multimodal approach to therapy. For these reasons, we reasoned it is rational to further develop this therapeutic strategy (inhibiting AKT phosphorylation with CTMP) in pancreatic cancer.
AKT is involved in a myriad of cell signaling pathways that grossly relate to cell survival, metabolism, and growth. Inhibition of this kinase has several, potentially beneficial effects. For instance, inhibiting the AKT-mediated phosphorylation of Bad would limit its sequestration and promote Bcl-2 activation leading to apoptosis (31–32). Similarly, the AKT-mediated inhibition of GSK-3β may have therapeutic implications; GSK-3β is a core component of two pathways involved in cell fate determination and morphology: Wnt and Hedgehog. Both or these pathways are well characterized mediators of several cancers (33).
It is important to distinguish this approach from a PI3K-targeted therapy. Although much enthusiasm exists for PI3K-targeted therapeutics, there also exists a potential for broad-based effects due to the wide-ranging intracellular actions of PI3K. In contrast, AKT, a downstream effector, has a smaller scope of action and may be better suited as a therapeutic target .
Though more thorough testing is needed to fully evaluate the potential toxicity of a CTMP-based therapeutic, the intracellular signaling profile, as measured by phospho-protein measurements, is encouraging. Namely, the absence of any change in the phosphorylation state of IKB suggests that the pro-inflammatory effectors of the NF-kB pathway are not activated as a result of this Akt-targeted therapy. Indeed, the high degree of specificity of CTMP for AKT (shown by us, Hwang et al and Maira et al) suggests that broad-based toxicity is unlikely. Recently, Ono et al have shown that CTMP may have cell context dependent activity as an Akt agonist (34). Our data suggest that this is not the case in pancreas or pancreas adenocarcinoma, however, it will be important to examine this in detail. Because our approach uses a single linear epitope from the endogenous protein, it is not likely subject to post-translational modifications that may alter its activity.
TAT-CTMP4 was effective in the treatment of both human and murine pancreatic adenocarcinoma. This phenomenon was observed in the setting of only a 68% sequence homology between the murine and human sequences. This suggests that TAT-CTMP4 treatment may be broadly applied to pancreatic cancers and may imply applicability to other forms of malignancies in which the AKT survival pathway is highly active, including cancers of the ovary, breast, lung, thyroid, and gastrointestinal tract (35, 20, 36–37) as well as our demonstration of activity in cancers of the kidney, colon and esophagus. Importantly, the residues mutated to make the inactive peptide are also conserved between murine and human systems.
The data presented here further suggest that radiation therapy may be useful to sensitize pancreatic malignancies to biologic therapy with TAT-CTMP4, which could be directly injected following irradiation of metastatic lesions, for example. This may facilitate a new role for radiation therapy in the treatment of pancreatic neoplasms.
It should be stressed that while our overall objective is to eradicate pancreas cancer with this biologic agent or a combination of conventional and biologic agents, such a therapy may also serve to downsize patients’ tumors such that they become candidates for curative resection. Alternatively, a targeted therapy based on our CTMP compound may be highly useful in the treatment of focal recurrences, especially when combined with gemcitabine or radiotherapy.
We recognize that this treatment modality, directly translated, may be of most interest in other directly-injectable malignancies (such as extremity sarcoma, malignant skin lesions, etc.) that can be easily accessed and do not require systemic targeting. With that said, it may be possible to inject TAT-CTMP4 into pancreatic malignancies via CT-guidance (metastatic disease) or with Endoscopic Ultrasound (EUS). Furthermore, since activity appears high in pancreatic tissue, there may exist a role in infiltrating the surgical bed (at the time of resection) with such a therapeutic given the high rates of local recurrence in surgical patients (38).
Alternatively, one may envision a therapeutic strategy that combines the pro-apoptotic therapy of CTMP with other inducers of apoptosis. Combination strategies could include TAT-BIM which targets intrinsic apoptotic pathway via antagonizing anti-apoptotic Bcl-2 family members and similarly induces apoptosis in these model systems (22). An additional pathway may also include PD98059, an inhibitor of the MAP / ERK kinase pathway. Targeting multiple pathways may result in both greater levels of apoptosis as well as the favorable prospect of utilizing lower doses of chemotherapeutic drugs. This is especially true with the highly toxic chemotherapeutic gemcitabine.
Our current goal is to engineer a compound that specifically targets pancreas cancer with the pro-apoptotic peptide CTMP4. Possible targeting strategies that may facilitate delivery of CTMP4 to pancreas cancer may include coupled ligands to any number of tumor-associated cell surface targets (39–40). We are currently synthesizing such agents and screening their ability to recapitulate the TAT-CTMP4-mediated induction of apoptosis in these model systems.
In summary, we have generated a novel therapy for the treatment of pancreatic adenocarcinoma that directly targets AKT (TAT-CTMP4). This biologic agent induced apoptosis in all pancreas adenocarcinoma cell lines and several other adenocarcinoma cells in vitro. This effect is limited in scope as important non-transformed cell types including PBMCs and fibroblasts were refractory to treatment. Certain adenocarcinomas were also resistant to CTMP-induced apoptosis. This treatment significantly augmented the effect of conventional therapies (chemo- and radiotherapy) in vitro and in vivo. These data suggest that directly inhibiting AKT with CTMP is a promising strategy for the treatment of pancreatic adenocarcinoma.
Supplementary Material
Acknowledgements
This study was supported by grants from the National Institutes of Health (T32 CA09621, P.O. Simon), GM44118, GM55194 (R.S. Hotchkiss), the American Cancer Society (MRSG-08-019-01CDD, W.G. Hawkins), the Barnes-Jewish Hospital Foundation (W.G. Hawkins), and the Alan A. and Edith L. Wolff Foundation (R.S. Hotchkiss). Histopathology specimens were prepared at the Washington University Digestive Diseases Research Core Center (DDRCC) which is supported by the National Institutes of Health (P30 DK052574). This work was presented in part at the Annual Meeting of the American Association for Cancer Research, Late Breaking Abstract Session, Los Angeles, 2007. The authors would like to thank Kim Trinkaus, PhD, for her assistance with statistical analyses, Ernesto Bernal-Mizrachi, MD and Latif Rachdi, PhD for critical discussions regarding this manuscript, and Suellen Greco, DVM DACLAM, for review and selection of pathology images. We would also like to thank Stacey Plambeck-Seuss for her technical assistance.
Financial Support: Simon, PO (NIH T32 CA09621), Hotchkiss, RS (NIH GM44118 and GM55194), Hawkins, WG (American Cancer Society MRSG-08-019-01CDD and the Barnes-Jewish Hospital Foundation)
Abbreviations
- CTMP
carboxyl terminal modulator protein
- IN
Inactive
- Orn
Ornithine
- Rads
Radiation
- Ser
Serine
- Wort
Wortmannin
Footnotes
Conflicts of Interest: None
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58(2):71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Sener SF, Fremgen A, Menck HR, Winchester DP. Pancreatic cancer: report of treatment and survival trends for 100,313 patients diagnosed from 1985–1995, using the National Cancer Database. J Am Coll Surg. 1999;189:1–7. doi: 10.1016/s1072-7515(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 3.Prochazkova J, Lichnovsky V, Kylarova D, Erdosova B, Vranka P. Involvement of p53 and Bcl-2 family proteins in regulating programmed cell death and proliferation in human embryogenesis. Gen Physiol Biophys. 2004;23(2):209–229. [PubMed] [Google Scholar]
- 4.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 5.Debatin KM, Krammer PH. Death receptors in chemotherapy and cancer. Oncogene. 2004;23(16):2950–2966. doi: 10.1038/sj.onc.1207558. [DOI] [PubMed] [Google Scholar]
- 6.Bergman PJ, Harris D. Radioresistance, chemoresistance, and apoptosis resistance. The past, present, and future. Vet Clin North Am Small Anim Pract. 1997;27:47–57. doi: 10.1016/s0195-5616(97)50005-2. [DOI] [PubMed] [Google Scholar]
- 7.Tan TT, Degenhardt K, Nelson DA, Beaudoin B, Nieves-Neira W, Bouillet P, Villunger A, Adams JM, White E. Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell. 2005;7:227–238. doi: 10.1016/j.ccr.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Decaudin D, Marzo I, Brenner C, Kroemer G, et al. Mitochondria in chemotherapy-induced apoptosis: a prospective novel target of cancer therapy (review) Int J Oncol. 1998;12:141–152. [PubMed] [Google Scholar]
- 9.Piro LD. Apoptosis, Bcl-2 antisense, and cancer therapy. Oncology. 2004;18:5–10. [PubMed] [Google Scholar]
- 10.Tsujimoto Y, Cossman J, Jaffe E, et al. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228:1440–1443. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- 11.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 12.Zantl N, Weirich G, Zall H, Seiffert BM, Fischer SF, Kirschnek S, Hartmann C, Fritsch RM, Gillissen B, Daniel PT, Häcker G. Frequent loss of expression of the pro-apoptotic protein Bim in renal cell carcinoma: evidence for contribution to apoptosis resistance. Oncogene. 2007;26(49):7038–7048. doi: 10.1038/sj.onc.1210510. [DOI] [PubMed] [Google Scholar]
- 13.Bondar VM, Sweeney-Gotsch B, Andreeff M, Mills GB, McConkey DJ. Inhibition of the phosphatidylinositol 3’-kinsae-AKT pathway induces apoptosis in pancreatic carcinoma cells in vitro and in vivo . Mol Cancer Ther. 2002;1(12):989–997. [PubMed] [Google Scholar]
- 14.Xiong HQ. Molecular targeting therapy for pancreas cancer. Cancer Chemother Pharmacol. 2004;54:S69–S77. doi: 10.1007/s00280-004-0890-2. [DOI] [PubMed] [Google Scholar]
- 15.Fujioka S, Sclabas GM, Schmidt C, Frederick WA, Dong QG, Abbruzzese JL, Evans DB, Baker C, Chiao PJ. Function of nuclear factor kappaB in pancreatic cancer metastasis. Clin Cancer Res. 2003;9:346–354. [PubMed] [Google Scholar]
- 16.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–1057. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 17.Leach SD. Mouse models of pancreatic cancer: the fur is finally flying! Cancer Cell. 2004;5:7–10. doi: 10.1016/s1535-6108(03)00337-4. [DOI] [PubMed] [Google Scholar]
- 18.Schultz RM, Merriman RL, Andis SL, Bonjouklian R, Grindey GB, Rutherford PG, Gallegos A, Massey K, Powis G. In vitro and in vivo antitumor activity of the phosphatidylinositol-3-kinase inhibitor, wortmannin. Anticancer Res. 1995;15(4):1135–1139. [PubMed] [Google Scholar]
- 19.Maira SM, Galetic I, Brazil DP, Kaech S, Ingley E, Thelen M, Hemmings BA. Carboxyl-Terminal Modulator Protein (CTMP), a negative regulator of PKB/AKT and v-AKT at the plasma membrane. Science. 2001;294(5541):374–380. doi: 10.1126/science.1062030. [DOI] [PubMed] [Google Scholar]
- 20.Hwang SK, Kwon JT, Park SJ, Chang SH, Lee ES, Chung YS, Beck GR, Jr, Lee KH, Piao L, Park J, Cho MH. Lentivirus-mediated carboxyl-terminal modulator protein gene transfection via aerosol in lungs of K-ras null mice. Gene Ther. 2007;14:1721–1730. doi: 10.1038/sj.gt.3303042. [DOI] [PubMed] [Google Scholar]
- 21.Gammon ST, Villalobos VM, Prior JL, Sharma V, Piwnica-Worms D. Quanitative analysis of permeation peptide complexes labeled with Technetium-99m: chiral and sequence-specific effects on net cell uptake. Bioconjug Chem. 2003;14:368–376. doi: 10.1021/bc0256291. [DOI] [PubMed] [Google Scholar]
- 22.Kashiwagi H, McDunn JE, Goedegebuure PS, Gaffney MC, Chang K, Trinkaus K, Piwnica-Worms D, Hotchkiss RS, Hawkins WG. TAT-Bim induces extensive apoptosis in cancer cells. Ann Surg Oncol. 2007;6:48. doi: 10.1245/s10434-006-9298-z. [DOI] [PubMed] [Google Scholar]
- 23.Hotchkiss RS, McConnell KW, Bullok K, Davis CG, Chang KC, Schwulst SJ, Dunne JC, Dietz GP, Bähr M, McDunn JE, Karl IE, Wagner TH, Cobb JP, Coopersmith CM, Piwnica-Worms D. TAT-BH4 and TAT-Bcl-xL peptides protect against sepsis-induced lymphocytes apoptosis in vivo. J Immunol. 2006;176:5471–5477. doi: 10.4049/jimmunol.176.9.5471. [DOI] [PubMed] [Google Scholar]
- 24.McConnell KW, Muenzer JT, Chang KC, Davis CG, McDunn JE, Coopersmith CM, Hilliard CA, Hotchkiss RS, Grigsby PW, Hunt CR. Anti-apoptotic peptides protect against radiation-induced cell death. Biochem Biophys Res Commun. 2007;355:501–507. doi: 10.1016/j.bbrc.2007.01.180. [DOI] [PubMed] [Google Scholar]
- 25.Turner TT, Lysiak JJ, Shannon JD, Nguyen QA, Bazemore-Walker CR. Testicular torsion alters the presence of specific proteins in the mouse testis as well as the phosphorylation status of specific proteins. J Androl. 2006;27:285–293. doi: 10.2164/jandrol.05134. [DOI] [PubMed] [Google Scholar]
- 26.Comer JE, Chopra AK, Peterson JW, König R. Direct inhibition of T-Lymphocyte activation by anthrax toxins in vivo. Infect Immun. 2005;73(12):8275–8281. doi: 10.1128/IAI.73.12.8275-8281.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ng SS, Tsao MS, Nicklee T, Hedley DW. Wortmannin inhibits pkb/akt phosphorylation and promotes gemcitabine antitumor activity in orthotopic human pancreatic cancer xenografts in immunodeficient mice. Clin Cancer Res. 2001;7(10):3269–3275. [PubMed] [Google Scholar]
- 28.Pham NA, Tsao MS, Cao P, Hedley DW. Dissociation of gemcitabine sensitivity and protein kinase B signaling in pancreatic ductal adenocarcinoma models. Pancreas. 2007;35(3):e16–e26. doi: 10.1097/mpa.0b013e318095a747. [DOI] [PubMed] [Google Scholar]
- 29.Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res. 2000;6:2969–2972. [PubMed] [Google Scholar]
- 30.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play of three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 31.Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4:257–262. doi: 10.1016/s1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- 32.Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of Bad. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- 33.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multitasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ono H, Sakoda H, Fujishiro M, Anai M, Kushiyama A, Fukushima Y, Katagiri H, Ogihara T, Oka Y, Kamata H, Horike N, Uchijima Y, Kurihara H, Asano T. Carboxy-terminal modulator protein induces Akt phosphorylation and activation, thereby enhancing antiapoptotic, glycogen synthetic, and glucose uptake pathways. Am J Physiol Cell Physiol. 2007;293(5):C1576–C1585. doi: 10.1152/ajpcell.00570.2006. [DOI] [PubMed] [Google Scholar]
- 35.Sun M, Wang G, Paciga JE, Feldman RI, Yuan ZQ, Ma XL, Shelley SA, Jove R, Tsichlis PN, Nicosia SV, Cheng JQ. AKT1/PKBalpha kinase is frequently elevated in human cancers and its constitutive activation is required for oncogenic transformation in NIH3T3 cells. Am J Path. 2001;159(2):431–437. doi: 10.1016/s0002-9440(10)61714-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ringel MD, Hayre N, Saito J, Saunier B, Schuppert F, Burch H, Bernet V, Burman KD, Kohn LD, Saji M. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer Res. 2001;61(16):6105–6111. [PubMed] [Google Scholar]
- 37.Michl P, Downward J. Mechanisms of disease: PI3K/AKT signaling in gastrointestinal cancers. Z Gastoenterol. 2005;43(10):1133–1139. doi: 10.1055/s-2005-858638. [DOI] [PubMed] [Google Scholar]
- 38.Kleeff J, Reiser C, Hinz U, Bachmann J, Debus J, Jaeger D, Friess H, Büchler MW. Surgery for recurrent pancreatic ductal adenocarcinoma. Ann Surg. 2007;245:566–572. doi: 10.1097/01.sla.0000245845.06772.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Argani P, Iacobuzio-Donahue C, Ryu B, Rosty C, Goggins M, Wilentz RE, Murugesan SR, Leach SD, Jaffee E, Yeo CJ, Cameron JL, Kern SE, Hruban RH. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE) Clin Cancer Res. 2001;7:3862–3868. [PubMed] [Google Scholar]
- 40.Argani P, Rosty C, Reiter RE, Wilentz RE, Murugesan SR, Leach SD, Ryu B, Skinner HG, Goggins M, Jaffee EM, Yeo CJ, Cameron JL, Kern SE, Hruban RH. Discovery of new markers of cancer through serial analysis of gene expression: prostate stem cell antigen is overexpressed in pancreatic adenocarcinoma. Cancer Res. 2001;61:4320–4324. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.