

Abstract
TGF-β is abundantly produced in the skeletal system and plays a crucial role in skeletal homeostasis. E-selectin ligand-1 (ESL-1), a Golgi apparatus-localized protein, acts as a negative regulator of TGF-β bioavailability by attenuating maturation of pro–TGF-β during cartilage homeostasis. However, whether regulation of intracellular TGF-β maturation by ESL-1 is also crucial during bone homeostasis has not been well defined. Here, we show that Esl-1−/− mice exhibit a severe osteopenia with elevated bone resorption and decreased bone mineralization. In primary culture, Esl-1−/− osteoclast progenitors show no difference in osteoclastogenesis. However, Esl-1−/− osteoblasts show delayed differentiation and mineralization and stimulate osteoclastogenesis more potently in the osteoblast–osteoclast coculture, suggesting that ESL-1 primarily acts in osteoblasts to regulate bone homeostasis. In addition, Esl-1−/− calvaria exhibit an elevated mature TGF-β/pro–TGF-β ratio, with increased expression of TGF-β downstream targets (plasminogen activator inhibitor-1, parathyroid hormone-related peptide, connective tissue growth factor, and matrix metallopeptidase 13, etc.) and a key regulator of osteoclastogenesis (receptor activator of nuclear factor κB ligand). Moreover, in vivo treatment with 1D11, a pan–TGF-β antibody, significantly improved the low bone mass of Esl-1−/− mice, suggesting that elevated TGF-β signaling is the major cause of osteopenia in Esl-1−/− mice. In summary, our study identifies ESL-1 as an important regulator of bone remodeling and demonstrates that the modulation of TGF-β maturation is pivotal in the maintenance of a homeostatic bone microenvironment and for proper osteoblast–osteoclast coupling.
Keywords: 1D11 antibody, osteoblast mineralization, osteoporosis
Bone is a highly dynamic tissue that undergoes continuous remodeling. Osteoblasts (OBs) and osteoclasts (OCs), the two cell types responsible for bone formation and bone resorption, respectively, are tightly coupled by a set of paracrine factors acting within the bone microenvironment. The dysregulation of OB–OC coupling is one of the most common causes of osteopenia/osteoporosis or other metabolic bone diseases (1).
Transforming growth factor β (TGF-β) is abundantly produced in bone. It plays a crucial role in regulating the differentiation of both OBs and OCs and mediating OB–OC coupling during bone remodeling (2, 3). Therefore, manipulating TGF-β signaling in bone offers an attractive strategy for the treatment of bone diseases, such as osteoporosis and osteolytic bone metastasis of cancers (4, 5). Recent data show that TGF-β can play either a catabolic or anabolic role in bone homeostasis, suggesting that TGF-β signaling acts in a context- and dose-dependent manner (4, 6–10). This emphasizes the need for a precise regulation of TGF-β bioavailability to maintain balanced bone remodeling. TGF-β is initially synthesized as an inactive precursor (pro–TGF-β), which undergoes cleavage/maturation by the furin protein convertase, at a specific site between the latency domain (LAP) and ligand domain. This process occurs in the Golgi apparatus and/or ECM, and is a prerequisite for TGF-β activity (2, 11–13). After cleavage, the LAP and TGF-β ligand continue to associate noncovalently within the small latency complex (SLC), which may further associate with latent TGF-β binding proteins covalently to form the large latency complex (LLC) (2, 11–13). These latent forms of TGF-β are secreted and deposited in the bone matrix, and subsequently activated by proteases, integrin, and glycosidase, etc. (11). Multiple lines of in vivo evidence have shown that dysregulated TGF-β expression, secretion, or activation from latency leads to defective bone remodeling (6, 7, 14). However, to date, to what extent bone homeostasis is modulated at the level of TGF-β maturation in the cell has not been fully defined.
We previously reported that E-selectin ligand-1 (ESL-1) (also referred to as Cysteine-rich fibroblast growth factor receptor, golgi apparatus protein 1, Latent TGF–β complexed protein-1 or MG160) (15–18) is an important regulator of TGF-β maturation in the Golgi apparatus during cartilage homeostasis (19). Loss of ESL-1 in mice led to accelerated TGF-β maturation and elevated TGF-β signaling in the growth plate. The enhanced TGF-β signaling results in a distinctive chondrodysplasia partly by disrupting the balance between parathyroid hormone-related peptide (PTHrP) and Indian hedgehog (IHH), thus impairing proliferation and terminal differentiation of chondrocytes in the growth plate (19). This mechanism was evolutionarily conserved and was functionally demonstrated in Xenopus gastrulation (19). In the present study, we found that Esl-1−/− mice develop severe osteopenia with impaired mineral deposition and elevated bone resorption. Moreover, our data indicate that, during bone remodeling, ESL-1 primarily acts within the OBs to maintain proper TGF-β bioavailability in the bone microenvironment, thus enhancing OB differentiation as well as restricting overactivation of OCs. Overall, this study suggests that attenuation of TGF-β maturation by ESL-1 is an important mechanism to maintain a balanced bone homeostasis.
Results
Loss of ESL-1 Leads to Severe Osteopenia in Mice.
Our previous studies have shown that ESL-1 regulates chondrocyte differentiation by modulating TGF-β bioavailability in the cartilage (19). Interestingly, Esl-1 is highly expressed in bone tissue and is localized in the Golgi apparatus of primary OBs (Fig. S1). Because TGF-β also plays a critical role in bone homeostasis, we further explored the extent to which ESL-1 is involved in bone homeostasis.
Esl-1−/− mice at postweaning age exhibit a markedly increased X-ray translucency throughout the skeleton, suggesting a low bone mass phenotype (Fig. 1A). Micro-computed tomography (μCT) analysis revealed that loss of ESL-1 leads to significant reduced bone volume/tissue volume (BV/TV), decreased trabecular number (Tb.N), and increased trabecular separation (Tb.Sp) in the distal femurs of 1-mo- and 3-mo-old mice, compared with wild-type (WT) littermates (Fig. 1B and Table 1). Moreover, dramatic decreases in the BV/TV and Tb.Th are also identified in 3-mo-old lumbar vertebrae 4 (L4) (Fig. 1C and Table 1). In addition, cortical thickness (Cortical.Th) in Esl-1−/− mice is significantly decreased. The µCT data also revealed that 1-mo Esl-1−/− mice have significantly decreased midfemur cortical diameter (WT vs. Esl-1−/−: 1.29 vs. 1.01 mm, a 22% decrease), and lower bone mineral density (BMD) in the midfemur cortical bone (WT vs. Esl-1−/−: 890.6 vs. 820.3 mg HA/ccm, a 8.5% decrease) (Fig. 1B, Table 1, and Fig. S2 A and B). The decreased BMD, Cortical.Th, and cortical diameter suggest that Esl-1−/− mice may have defective mechanical properties. This notion is further supported by a three-point bending test on 1-mo femurs. As expected, the reduction in bone mass resulted in significantly diminished maximum load and energy to failure for the Esl-1−/− animals. However, the ultimate strength is not significantly changed in the Esl-1−/− femurs, and the elastic modulus is increased (Table S1). These data indicate that loss of ESL-1 results in an alteration in the composition of bone matrix, which affects the material properties of the bone. In summary, the early onset and sustained low bone mass in both the long bones and vertebrae of Esl-1−/− mice suggests that ESL-1 is an important regulator of bone homeostasis, acting in both axial and appendicular skeleton.
Fig. 1.

ESL-1 is expressed in OBs and loss of ESL-1 leads to a severe osteoporotic phenotype. (A) One-month-old Esl-1−/− skeleton shows increased X-ray translucency. (B) µCT images of Esl-1−/− vs. WT femurs. (Upper) Longitudinal sections of 1-mo-old male distal femurs. (Lower) The transverse sections right below the minor trochanter of the same femurs. (C) Decreased trabecular bone of 3-mo-old vertebra L4. (Scale bars: 1 mm.)
Table 1.
Quantification of µCT scanning reveals a severe osteoporosis in Esl-1−/− bones
| Bone samples | Genotypes | BV/TV | BS/BV | Tb.N | Tb.Th | Tb.Sp | Cortical.Th |
| WT | 0.1095 ± 0.0294 | 56.9721 ± 4.6514 | 3.0659 ± 0.5731 | 0.0353 ± 0.0030 | 0.3006 ± 0.0652 | 0.1201 ± 0.0009 | |
| 1-mo femurs | Esl-1−/− | 0.0508 ± 0.0103 | 68.0776 ± 0.9726 | 1.7237 ± 0.3313 | 0.0294 ± 0.0004 | 0.5706 ± 0.1387 | 0.0873 ± 0.0008 |
| P | 0.0054 | 0.0017 | 0.0030 | 0.0051 | 0.0029 | 0.0004 | |
| WT | 0.1476 ± 0.0033 | 43.2653 ± 1.5988 | 2.8626 ± 0.2660 | 0.0483 ± 0.0007 | 0.3081 ± 0.0342 | 0.2143 ± 0.0191 | |
| 3-mo femurs | Esl-1−/− | 0.0703 ± 0.0221 | 43.9756 ± 1.1180 | 1.5533 ± 0.5343 | 0.0448 ± 0.0013 | 0.6447 ± 0.2021 | 0.1657 ± 0.0085 |
| P | 0.0039 | 0.5693 | 0.0191 | 0.0134 | 0.0466 | 0.015 | |
| WT | 0.2948 ± 0.0279 | 33.2170 ± 0.9125 | 4.9064 ± 0.5747 | 0.0602 ± 0.00017 | 0.1457 ± 0.0242 | N/A | |
| 3-mo vertebrae L4 | Esl-1−/− | 0.1481 ± 0.0068 | 42.7234 ± 1.7368 | 3.1603 ± 0.1113 | 0.0469 ± 0.0019 | 0.2698 ± 0.0109 | N/A |
| P | 0.0009 | 0.0011 | 0.0067 | 0.0008 | 0.0013 | N/A |
The 1-mo-old and 3-mo-old femurs and the 3-mo-old L4 vertebrae were scanned and quantified by µCT. Quantified parameters, including BV/TV (bone volume/tissue volume), BS/TV (bone surface/tissue volume), Tb.N (trabecular bone number), Tb.Th (trabecular bone thickness), Tb.Sp (trabecular bone separation), and Cortical.Th (cortical bone thickness), indicate a distinctive osteoporotic phenotype in Esl-1−/− long bones and vertebrae.
Esl-1−/− Low Bone Mass Is Caused by Abnormal Activity of both OBs and OCs.
The low bone mass phenotype can be a result of impaired bone formation, overactive bone resorption, or a combinatorial effect of each. To differentiate among these possibilities, trabecular parameters of 3-mo-old male Esl-1−/− vs. WT mouse vertebra (L4) were assessed by bone histomorphometry (Fig. 2). We found that the OB surface per bone surface (Ob.S/BS), and the number of OBs per bone perimeter (N.Ob/B.Pm) are not significantly changed, suggesting that the proliferation of OBs is unaffected and does not account for the osteopenia in Esl-1−/− mice (Fig. 2A). Next, to understand whether loss of ESL-1 influences osteoblastic activity in the production and mineralization of bone matrix, we examined OB function by calcein double-labeling analysis. The bone formation rate per bone surface (BFR/BS) shows a trend toward a decrease in the Esl-1−/− mice, although not statistically significant. However, the mineralization apposition rate (MAR) is significantly decreased in the KO mice, suggesting that loss of ESL-1 decreases OB activity in producing mineralized bone matrix (Fig. 2B). Moreover, we found that the OC surface/bone surface (Oc.S/BS) and OC number/bone perimeter (N.Oc/B.Pm) are increased by ∼30% in the mutant mice (Fig. 2C). These data suggest that loss of ESL-1 leads to an overall increase in OC differentiation. Overall, our results imply that the Esl-1−/− osteopenia phenotype is due to the combinatorial effects of both decreased bone mineralization and elevated bone resorption.
Fig. 2.

Osteopenia in Esl-1−/− mice is the result of elevated bone resorption and decreased bone mineralization. Bone histomorphometric analysis of 3-mo-old Esl-1−/− vs. WT L4 vertebra. (A) The OB number and surface are not changed significantly. The OBs on bone surfaces are underscored with red curves. (Scale bar: 100 µm.) OB number per bone surface (Ob.S/BS) and OB number per bone perimeter (N.Ob/B.Pm) are quantified at Right. (B) Calcein double labeling with a 4-d interval. (Scale bar: 100 µm.) Bone dynamic indices are shown at Right. Bone formation rate/bone surface (BFR/BS) are trending toward but not significantly decreased, whereas mineral apposition rate (MAR) is significantly decreased in Esl-1−/− mice. (C) The OC numbers and surfaces are significantly increased in Esl-1−/− L4 vertebrae. (Scale bar: 50 µm.) OCs were identified by TRAP staining. OC surface per bone surface (Oc.S/BS) and OC number per bone perimeter (N.Oc/B.Pm) are quantified at Right. n = 5 per group. *P < 0.05.
Loss of ESL-1 in OBs Inhibits Their Maturation and Enhances OB-Dependent Osteoclastogenesis.
To further test whether ESL-1 plays a role in OB proliferation, primary OBs from Esl-1−/− or WT P1 calvaria were cultured and assessed for cell proliferation (Fig. 3A). The proliferation rate of ESL-1−/− OB is unchanged compared with the WT, consistent with the observation in the histomorphometric analysis (Fig. 2A). However, the decreased MAR in the Esl-1−/− vertebrae suggests that ESL-1 may play an important role in OB differentiation (Fig. 2B). To assess this, bone marrow stromal cells (BMSCs) were cultured in osteogenic medium containing ascorbic acid and β-glycerol phosphate. The cultures were assayed for early OB differentiation by alkaline phosphatase (ALP) staining at day 6 and for late OB differentiation/mineralization by Alizarin Red S staining at day 21, respectively. Our results showed that ALP-positive areas are not significantly changed in Esl-1−/− OBs (Fig. 3B). However, Alizarin Red S staining for mineralized matrix is dramatically decreased compared with WT cells (Fig. 3C), suggesting that ESL-1 is involved in the late differentiation and mineralization of OBs.
Fig. 3.

Loss of ESL-1 in osteoblastic lineage cells attenuates late OB differentiation and enhances OB-dependent osteoclastogenesis. (A–C) Function of ESL-1 in OB proliferation and differentiation. Loss of ESL-1 does not affect primary calvarial OB proliferation (A) or early OB differentiation of BMSCs cultured in osteogenic medium as indicated by ALP staining (day 6) (B), but significantly attenuates late OB differentiation and mineralization as indicated by Alizarin Red S staining (day 21) (C). (D) BMMCs derived from WT vs. Esl-1−/− mice show comparable osteoclastogenesis upon treatment with M-CSF and RANKL. (E) In BMSC/splenocyte cocultures, Esl-1−/− BMSCs more strongly induce OC formation than WT BMSCs, irrespective of the genotype of splenocytes. SP, splenocytes. n = 3 per group; *P < 0.05, **P < 0.01; NS, nonsignificance.
Elevated N.Oc/B.Pm and Oc.S/BS in Esl-1−/− bone suggest that ESL-1 may play a role in osteoclastogenesis. OC formation is influenced by intrinsic factors such as colony stimulating factor 1 receptor (CSF1R) or receptor activator of nuclear factor κB (RANK) in the OC progenitors, or by extrinsic stimulators such as macrophage colony-stimulating factor (M-CSF) or RANK ligand (RANKL) derived from OBs or other cell lineages within the bone microenvironment (20). First, to evaluate the intrinsic function of ESL-1 in osteoclastogenesis, bone marrow monocytes (BMMCs) derived from bone marrows of 4-wk-old WT or Esl-1−/− mice were treated with M-CSF and RANKL to induce OC differentiation. We found that Esl-1−/− BMMCs show no distinctive difference in osteoclastogenesis compared with WT controls (Fig. 3D), suggesting that loss of ESL-1 does not potentiate the response of BMMCs to the stimulation of M-CSF and RANKL. Hence, the elevated OC formation in the Esl-1−/− bone may be due to the altered signaling context in the microenvironment of Esl-1−/− bone, likely through an OB–OC coupling mechanism. To assess this possibility, we cocultured WT vs. Esl-1−/− splenocytes (used as the OC progenitors) with Esl-1−/− vs. WT BMSCs, to detect whether loss of ESL-1 increases the potency of OB lineage cells in the induction of osteoclastogenesis, as well as whether loss of ESL-1 increases the response of OC progenitors in forming mature OCs. The results showed that, compared with WT BMSCs, the Esl-1−/− BMSCs are able to induce greater OC formation irrespective of the genotypes of the splenocytes. Meanwhile, WT and Esl-1−/− splenocytes show no difference in osteoclastogenesis when cocultured with the BMSCs of the same genotypes (Fig. 3E). These data suggest that the ESL-1 acts in the osteoblastic lineage cells, likely by modulating paracrine signals in the bone microenvironment, to indirectly regulate OC differentiation.
Changes of Bone Homeostasis-Related Genes and TGF-β Signaling in the Esl-1−/− Bone.
To further understand the molecular basis underlying the Esl-1−/− low-bone mass phenotype, as well as what specific paracrine signal(s) act(s) downstream of Esl-1 to mediate OB–OC coupling, we first compared the gene expression pattern between P1 WT vs. Esl-1−/− calvaria by quantitative RT-PCR (qPCR). We found that the early OB differentiation markers, such as Runt-related transcription factor 2 (Runx2), osterix (Osx), collagen 1a1 (Col1a1), alkaline phosphatase (Alp), and bone sialoglycoprotein (Bsp) are largely unchanged, while osteocalcin (Ocn), an OB late differentiation marker, is significantly decreased (Fig. 4A). These data suggest that loss of ESL-1 does not significantly affect early OB differentiation but inhibits OB terminal differentiation, consistent with the bone histomorphometry and primary cell culture data. Moreover, the OC differentiation markers including tartrate-resistant acid phosphatase (Trap) and matrix metalloproteinase 9 (Mmp9) are significantly increased, consistent with the elevated Oc.S/BS identified in Esl-1−/− bone (Fig. 4B). Meanwhile, RANKL, the important stimulator of osteoclastogenesis, and osteoprotegerin (Opg), the antagonist of RANKL, are both elevated in Esl-1−/− calvaria. However, the RANKL/OPG ratio is increased, suggesting that overall the bone microenvironment in the Esl-1−/− bone promotes OC formation (Fig. 4B).
Fig. 4.

Expression profile of bone homeostatic or signaling related genes in Esl-1−/− vs. WT calvaria. (A–D) qPCR analysis of gene expression (n = 5 per group; *P < 0.05, **P < 0.01; NS, nonsignificance; y axis: the fold change): gene expression of OB differentiation markers (A); expression of OC differentiation markers and key regulators of osteoclastogenesis (B); expression of TGF-β downstream target genes (C); expression of TGF-β signaling components (D). (E) Mature TGF-β is increased in the Esl-1−/− calvaria as detected by Western blot. The normalized ratios of mature/pro–TGF-β are quantified at Right.
Next, to understand whether ESL-1 regulates bioavailability of TGF-β in bone using a mechanism similar to that of cartilage, we first compared the expression of TGF-β downstream targets in P1 Esl-1−/− vs. WT mouse calvaria. TGF-β downstream targets including p21 (cyclin-dependent kinase inhibitor 1), plasminogen activator inhibitor-1 (Pai-1), Pthrp, matrix metallopeptidase 13 (Mmp13), and connective tissue growth factor (Ctgf) are significantly increased in the Esl-1−/− calvaria (Fig. 4C). The gene expression of TGF-β ligands (TGF-β1,2,3), TGF-β receptors 1, 2, and Smad2,3 (Mothers against decapentaplegic homolog 2,3) are not significantly changed in Esl-1−/− calvaria (Fig. 4D), suggesting that enhanced TGF-β signaling in Esl-1−/− bone may be due to a posttranscriptional mechanism, such as elevation in intracellular maturation of pro–TGF-β as is observed in Esl-1−/− cartilage. This notion is confirmed by Western blots showing a markedly increased mature TGF-β1 in Esl-1−/− calvarial protein in comparison with the WT controls (Fig. 4E), similar to our previous observation in the Esl-1−/− cartilage.
TGF-β Antibody Ameliorates Defective OB Differentiation and OC Induction.
These above observations suggest that the elevated active TGF-β in the bone microenvironment may be the primary causal factor for the osteopenia in Esl-1−/− mice. To test this, we first assessed to what extent blocking TGF-β availability using 1D11, a pan–TGF-β antibody, can ameliorate the defect of OB differentiation in ex vivo cultures (Fig. 5 A and B). Our data show that, in the WT BMSCs, 1D11 treatment significantly accelerates mineral deposition in the culture at an early time point (3.07-fold increase at day 8), but the effect appears to be blunted with prolonged treatment (1.19-fold increase but not significant at day 16), possibly because the number of unmineralized cells become depleted at the later time point. In contrast, 1D11 treatment shows a stronger and significant effect in accelerating mineralization of Esl-1−/− BMSCs at both time points (4.41-fold increases at day 8, and 1.75-fold at day 16), at which times the delayed Esl-1−/− OB mineralization is largely rescued and shows no significant difference to treated WT BMSCs (Fig. 5A).
Fig. 5.

Negating bioactive TGF-β effectively corrects the defects in Esl-1−/− primary cultures and osteopenia phenotypes. (A) 1D11 rescues delayed mineralization of Esl-1−/− OBs. (B) 1D11 rescues overactive osteoclastogenesis induced by Esl-1−/− BMSCs. (C) µCT images of the L4 vertebra at 8-wk-old male WT vs. Esl-1−/− mice treated with either placebo (13C4) or 1D11 for 4 wk before collection. (Scale bar: 1 mm.) (D) Quantified bone parameters, including BV/TV, Tb.N, Tb.Th, and Tb.Sp, indicate an effective correction of the Esl-1−/− osteopenia by 1D11 treatment. n = 4 per group; *P < 0.05; NS, nonsignificance.
Next, to evaluate to what extent increased bioactive TGF-β derived from Esl-1−/− OBs is responsible for the overactive bone resorption, we used 1D11 to treat OB–OC cocultures. The results showed that 1D11 can significantly decrease/rescue the osteoclastogenesis induced by Esl-1−/− BMSCs. Meanwhile, 1D11 treatment also shows a trend toward an attenuation of osteoclastogenesis induced by WT BMSCs. These data support that the elevated TGF-β derived from OB plays an important role in mediating the dysregulated OB–OC coupling in the osteopenia in Esl-1−/− mice (Fig. 5B).
Anti–TGF-β Antibody Effectively Improves Esl-1−/− Low Bone Mass.
To further confirm whether elevated TGF-β bioavailability in vivo is the major cause of the Esl-1−/− osteopenia, and to evaluate the therapeutic effect of 1D11 (a pan–TGF-β monoclonal antibody) on osteopenia caused by excessive TGF-β activity, we attempted to correct Esl-1−/− low bone mass with systemic treatment of 1D11. Consistent with a previous report by Edwards et al. (4), our data show that 1D11 treatment in WT mice for 4 wk resulted in a significant accrual of trabecular bone in L4 vertebra (Fig. 5C), as evidenced by a 37.8% increase in BV/TV (26.7% of the placebo group vs. 35.4% of the 1D11-treated group). In addition, an anabolic effect of 1D11 is also evident with finding of increased Tb.Th and Tb.N in conjunction with decreased Tb.Sp in the 1D11-treated WT mice, compared with placebo-treated WT mice (Fig. 5D). More importantly, 1D11 treatment exhibits an even more dramatic anabolic effect on Esl-1−/− bones: the BV/TV of L4 vertebra is significantly increased by 105% (12.5% of the placebo group vs. 25.7% of the 1D11-treated group), close to that of the placebo-treated WT group (BV/TV, 26.7%). The other bone parameters, such as BS/TV, Tb.N, Tb.Th, and Tb.Sp are also corrected to the levels of placebo-treated WT mice (Fig. 5D). Overall, 1D11 treatment vastly improves the low bone mass of the Esl-1−/− mice, suggesting that the excessive bioactive TGF-β in the bone microenvironment is a major contributor to the osteopenia in Esl-1−/− mice.
Discussion
Our data suggest that ESL-1, a Golgi-localized protein, is a crucial regulator in bone homeostasis. Loss of ESL-1 results in a severe osteopenia in mice with decreased bone formation and elevated bone resorption, due to increased TGF-β signaling in the bone. This is in accordance with our previous finding of ESL-1 as a negative regulator of TGF-β bioavailability in cartilage (19). Distinct from most other cells, in which LLC is the major form of secreted TGF-β, bone cells predominantly secrete pro–TGF-β and SLC (maturated pro–TGF-β) with high efficiency (21, 22). This suggests that SLC represents the readily available pool of TGF-β in bone, and that the SLC initially embedded in bone matrix can greatly influence the dose of bioactive TGF-β in the bone microenvironment upon bone resorption. In Esl-1−/− bone tissue, we detected an increased TGF-β maturation that may potentiate the bone matrix to release greater amounts of bioactive TGF-β during bone resorption. Moreover, significantly elevated expression of multiple TGF-β downstream targets in Esl-1−/− calvaria support this notion. TGF-β maturation is catalyzed by furin, which is a housekeeping protease, processing a broad spectrum of substrates such as VEGF and PTHrP, etc. (23, 24). We previously showed that ESL-1 binding to pro–TGF-β leads to blockade of furin on TGF-β cleavage (19). This mechanism may provide a more specific and effective control on TGF-β maturation than other mechanisms that directly act on furin production and activity.
TGF-β signaling in the bone microenvironment affects OBs and OCs, and regulates skeletal homeostasis in a context-dependent manner. The complexity of its regulation is well represented in a number of genetic models. For examples, TGF-β1−/− mice show impaired migration of mesenchymal stem cells to the newly formed bone surface during bone remodeling, thereby exhibiting a late-onset low bone mass (10). Accordingly, gain of TGF-β1 activation and secretion in Camurati–Engelmann disease causes high bone mass (9). Conversely, OB specific overexpression of mature TGF-β2 results in osteoporosis with elevated bone resorption and undermineralization of bone matrix (7). More interestingly, haploinsufficiency of Smad3, a main downstream effector of canonical TGF-β signaling, leads to high bone mass, whereas the Smad3−/− mice show low bone mass (25). This seemingly contradictory observation emphasizes that TGF-β is an important coupling factor of bone remodeling whose bioavailability/dose needs to be finely regulated at multiple levels to meet the dynamic requirement of bone remodeling at different phases. Here, our data support that ESL-1 is a crucial regulator acting at the level of TGF-β maturation before TGF-β being embedded in the bone matrix, thus preventing overactivation of TGF-β signaling during bone resorption. The importance of a regulatory mechanism for TGF-β maturation has been previously demonstrated in the control of blood pressure homeostasis as well as cartilage development (19, 26). However, it has not been appreciated in bone homeostasis and pathogenesis. Here, our finding suggests that, along with other mechanisms controlling TGF-β bioavailability during bone remodeling, including TGF-β expression, secretion, and activation from latency, the modulation of TGF-β maturation is also a pivotal and regulatable step for proper bone homeostasis.
Published data from in vivo or in vitro studies support that TGF-β decreases bone mineralization, whereas blockade of TGF-β enhances mineralization (7, 27). Previous studies also show that TGF-β signaling delays osteocalcin expression as well as OB terminal differentiation through suppressing Runx2 function (28). TGF-β also regulates expression of matrix proteins and MMPs, which play roles in matrix organization and mineralization. Esl-1−/− bone shows changes in TGF-β downstream targets, including decreased osteocalcin, and elevated CTGF and MMP13, etc. (Fig. 4 A and C), which may partly contribute to the delayed mineralization in the Esl-1−/− bone. Elevated CTGF in bone causes osteopenia by suppression of OB activity in mineralization and bone formation (29). Loss of MMP13 increases trabecular bone mass by attenuating matrix resorption during endochondral ossification (30).
That TGF-β stimulates RANKL production has been revealed in vivo. For example, excessive TGF-β bioavailability in the bone of Fibrillin 2 knockout (Fbn2−/−) mice significantly elevates OB-derived RANKL level and leads to osteoporosis (31). In our previous studies, we identified that PTHrP, a TGF-β downstream target, is significantly up-regulated in Esl-1−/− cartilage and accounts for the shortening of the growth plate (19). Interestingly, we found that PTHrP is also significantly elevated along with other TGF-β downstream targets in the bone. PTHrP plays a dual role during bone remodeling. The OB-specific PTHrP deficiency leads to osteoporosis with decreased OB survival, but the OC formation is also impaired (32). A prolonged and continuous infusion of PTHrP stimulates bone resorption and bone loss (33). Similarly, humoral hypercalcemia of malignancy (HHM) occurs in patients with tumor cells expressing a large amount of PTHrP into the circulation causing overactive osteoclastogenesis and osteoporosis (34). The augmented PTHrP production we observed in the Esl-1−/− bone may partly resemble the prolonged and continuous PTHrP administration or HHM.
Blockade of TGF-β signaling in vivo by 1D11 or SD-208 treatment (an inhibitor of the TGF-β type 1 receptor) has been shown to exert significant anabolic effects on bone accrual (4, 5). In our 1D11 treatment groups, the BV/TV in Esl-1−/− vertebrae is elevated to a greater extent than that in WT controls (105% vs. 37.8% increase), implying that TGF-β signaling is a major downstream target of ESL-1 during bone homeostasis. Meanwhile, although the 1D11-treated Esl-1−/− BV/TV is corrected to a comparable level of the untreated WT BV/TV (25.6% vs. 26.7%), it is still significantly lower than 1D11-treated WT BV/TV (25.6% vs. 35.4%). This is likely due to the substantially different BV/TV baseline between Esl-1−/− to WT bone, intrinsic limitations of the treatment protocol (dosage or duration), or the undefined TGF-β–independent ESL-1 function(s) in bone homeostasis. Nevertheless, these data suggest that 1D11 can be potentially used for treating low bone mass caused by altered TGF-β signaling either directly or indirectly, such as that in Marfan syndrome (35, 36) or osteolytic bone metastasis of cancers (27).
Because ESL-1 and TGF-β are also expressed in the OC and its progenitors, it is possible that ESL-1 may play a similar role in regulating TGF-β maturation in these cells during bone homeostasis. However, due to the relatively smaller population of the monocyte/OC lineage compared with the OB lineage in the bone microenvironment, their contribution to the overall TGF-β bioavailability may be limited. Moreover, loss of ESL-1 in OC precursors apparently does not affect osteoclastogenesis (Fig. 3 D and E), suggesting that the intrinsic function of ESL-1 in monocytic cells may not be directly involved in OC differentiation. To definitively elucidate the relative contribution of OB or OC-derived ESL-1 in regulating bone homeostasis, a cell-specific ESL-1 conditional knockout allele needs to be established and studied. Moreover, our three-point bending test on Esl-1−/− femurs show largely similar results to TGF-β bone specific transgenic mice, with the exception of an elevated elastic modulus (25). This difference may be caused by different test methods used, i.e., whole-bone fracture test vs. nanoindentation and/or wet bone vs. dry bone. However, it is also possible that ESL-1 may regulate matrix composition in a TGF-β–independent manner, which deserves further study.
In summary, ESL-1 plays a crucial role in bone remolding by modulating TGF-β maturation in cells of OB lineage, which ultimately influences TGF-β bioavailability in the bone microenvironment and affects the differentiation of OBs and OCs as well as their coupling (as illustrated in Fig. 6) . This study reveals that TGF-β maturation is a critical and regulatable step during bone homeostasis. Targeting TGF-β maturation may provide a new perspective to develop therapies for skeletal or other disorders related to dysregulated TGF-β signaling.
Fig. 6.
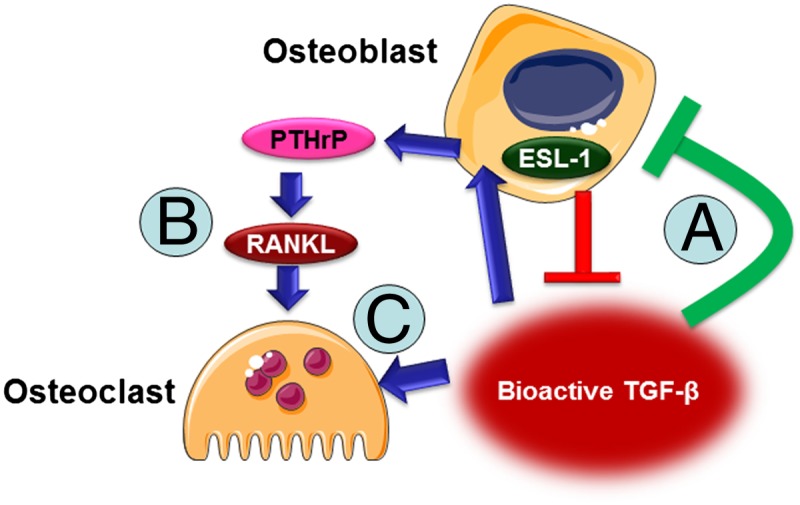
A schematic of ESL-1 function in bone remodeling. ESL-1 acts in OBs to suppress production of bioactive TGF-β in the bone microenvironment, which inhibits OB mineralization (A), increases PTHrP and RANKL production from OB to activate osteoclastogenesis (B), and facilitates OC differentiation (C).
Materials and Methods
Animal Model.
Esl-1−/− mice were generated as previously described (19), and maintained and used following the Animal Care and Use Committee (IACUC) at the Baylor College of Medicine.
Bone Tissue Preparation, μCT Analysis, and Bone Histomorphometry.
Mice were injected with calcein (1.5 mg/mouse) twice at 6 d and 2 d before killing. Femurs and lumbar vertebrae were collected and scanned by μCT (μCT-40; Scanco) for quantification of trabecular and cortical bone parameters. The scanned bones were subsequently embedded in resin for sectioning. Toluidine blue staining, TRAP staining, and calcein double labeling are used for visualization of OBs, OCs, and the dynamics of bone accrual, respectively; histomorphometry data were analyzed using a Bioquant Osteo system.
Treatment with 1D11 Antibody.
The 4-wk-old male Esl-1−/− and WT mice on C57/B6 background were injected with 1D11 antibody at 10 µg/g body weight for 4 wk, three times each week. The placebo controls were injected at the same condition with 13C4 antibody (nonspecific mouse IgG). The lumbar vertebrae were collected for µCT analysis. For primary OB culture or OB–OC coculture, 1D11 or 13C4 were added into the corresponding culture medium at 10 µg/mL.
Statistics.
Student t tests were used to determine whether the difference between two groups of data are normally distributed or not. P values less than 0.05 are considered statistically significant.
Supplementary Material
Acknowledgments
We thank Drs. Kuber Sampath and Patrick Finn (Genzyme Corporation) for providing 1D11 antibody and Brian Dawson and Mike Starbuck for assistance and consultation in µCT and bone histology. This work is supported by an Arthritis Foundation postdoctoral fellowship (to T.Y.), a pilot grant from The Rolanette and Berdon Lawrence Bone Disease Program of Texas (to T.Y.), National Institutes of Health Grants 5P01HD070394 and DE016990 (to B.L.), and the Howard Hughes Medical Institute Foundation (B.L.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1219748110/-/DCSupplemental.
References
- 1.Feng X, McDonald JM. Disorders of bone remodeling. Annu Rev Pathol. 2011;6:121–145. doi: 10.1146/annurev-pathol-011110-130203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Janssens K, ten Dijke P, Janssens S, Van Hul W. Transforming growth factor-beta1 to the bone. Endocr Rev. 2005;26(6):743–774. doi: 10.1210/er.2004-0001. [DOI] [PubMed] [Google Scholar]
- 3.Serra R, Chang C. TGF-beta signaling in human skeletal and patterning disorders. Birth Defects Res C Embryo Today. 2003;69(4):333–351. doi: 10.1002/bdrc.10023. [DOI] [PubMed] [Google Scholar]
- 4.Edwards JR, et al. Inhibition of TGF-β signaling by 1D11 antibody treatment increases bone mass and quality in vivo. J Bone Miner Res. 2010;25(11):2419–2426. doi: 10.1002/jbmr.139. [DOI] [PubMed] [Google Scholar]
- 5.Mohammad KS, et al. Pharmacologic inhibition of the TGF-beta type I receptor kinase has anabolic and anti-catabolic effects on bone. PLoS One. 2009;4(4):e5275. doi: 10.1371/journal.pone.0005275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dabovic B, et al. Bone abnormalities in latent TGF-[beta] binding protein (Ltbp)-3-null mice indicate a role for Ltbp-3 in modulating TGF-[beta] bioavailability. J Cell Biol. 2002;156(2):227–232. doi: 10.1083/jcb.200111080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erlebacher A, Derynck R. Increased expression of TGF-beta 2 in osteoblasts results in an osteoporosis-like phenotype. J Cell Biol. 1996;132(1–2):195–210. doi: 10.1083/jcb.132.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Filvaroff E, et al. Inhibition of TGF-beta receptor signaling in osteoblasts leads to decreased bone remodeling and increased trabecular bone mass. Development. 1999;126(19):4267–4279. doi: 10.1242/dev.126.19.4267. [DOI] [PubMed] [Google Scholar]
- 9.Janssens K, et al. Mutations in the gene encoding the latency-associated peptide of TGF-beta 1 cause Camurati-Engelmann disease. Nat Genet. 2000;26(3):273–275. doi: 10.1038/81563. [DOI] [PubMed] [Google Scholar]
- 10.Tang Y, et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med. 2009;15(7):757–765. doi: 10.1038/nm.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116(Pt 2):217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 12.Dubois CM, Laprise MH, Blanchette F, Gentry LE, Leduc R. Processing of transforming growth factor beta 1 precursor by human furin convertase. J Biol Chem. 1995;270(18):10618–10624. doi: 10.1074/jbc.270.18.10618. [DOI] [PubMed] [Google Scholar]
- 13.Miyazono K, Thyberg J, Heldin CH. Retention of the transforming growth factor-beta 1 precursor in the Golgi complex in a latent endoglycosidase H-sensitive form. J Biol Chem. 1992;267(8):5668–5675. [PubMed] [Google Scholar]
- 14.Le Goff C, et al. ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nat Genet. 2008;40(9):1119–1123. doi: 10.1038/ng.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burrus LW, Zuber ME, Lueddecke BA, Olwin BB. Identification of a cysteine-rich receptor for fibroblast growth factors. Mol Cell Biol. 1992;12(12):5600–5609. doi: 10.1128/mcb.12.12.5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steegmaier M, et al. The E-selectin-ligand ESL-1 is a variant of a receptor for fibroblast growth factor. Nature. 1995;373(6515):615–620. doi: 10.1038/373615a0. [DOI] [PubMed] [Google Scholar]
- 17.Olofsson A, et al. Latent transforming growth factor-beta complex in Chinese hamster ovary cells contains the multifunctional cysteine-rich fibroblast growth factor receptor, also termed E-selectin-ligand or MG-160. Biochem J. 1997;324(Pt 2):427–434. doi: 10.1042/bj3240427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stieber A, Mourelatos Z, Chen YJ, Le Douarin N, Gonatas NK. MG160, a membrane protein of the Golgi apparatus which is homologous to a fibroblast growth factor receptor and to a ligand for E-selectin, is found only in the Golgi apparatus and appears early in chicken embryo development. Exp Cell Res. 1995;219(2):562–570. doi: 10.1006/excr.1995.1265. [DOI] [PubMed] [Google Scholar]
- 19.Yang T, et al. E-selectin ligand-1 regulates growth plate homeostasis in mice by inhibiting the intracellular processing and secretion of mature TGF-beta. J Clin Invest. 2010;120(7):2474–2485. doi: 10.1172/JCI42150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423(6937):337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 21.Bonewald LF, et al. Latent forms of transforming growth factor-beta (TGF beta) derived from bone cultures: Identification of a naturally occurring 100-kDa complex with similarity to recombinant latent TGF beta. Mol Endocrinol. 1991;5(6):741–751. doi: 10.1210/mend-5-6-741. [DOI] [PubMed] [Google Scholar]
- 22.Dallas SL, et al. Characterization and autoregulation of latent transforming growth factor beta (TGF beta) complexes in osteoblast-like cell lines. Production of a latent complex lacking the latent TGF beta-binding protein. J Biol Chem. 1994;269(9):6815–6821. [PubMed] [Google Scholar]
- 23.Liu B, Goltzman D, Rabbani SA. Processing of pro-PTHRP by the prohormone convertase, furin: Effect on biological activity. Am J Physiol. 1995;268(5 Pt 1):E832–E838. doi: 10.1152/ajpendo.1995.268.5.E832. [DOI] [PubMed] [Google Scholar]
- 24.Siegfried G, et al. The secretory proprotein convertases furin, PC5, and PC7 activate VEGF-C to induce tumorigenesis. J Clin Invest. 2003;111(11):1723–1732. doi: 10.1172/JCI17220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balooch G, et al. TGF-beta regulates the mechanical properties and composition of bone matrix. Proc Natl Acad Sci USA. 2005;102(52):18813–18818. doi: 10.1073/pnas.0507417102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zacchigna L, et al. Emilin1 links TGF-beta maturation to blood pressure homeostasis. Cell. 2006;124(5):929–942. doi: 10.1016/j.cell.2005.12.035. [DOI] [PubMed] [Google Scholar]
- 27.Biswas S, et al. Anti-transforming growth factor ß antibody treatment rescues bone loss and prevents breast cancer metastasis to bone. PLoS One. 2011;6(11):e27090. doi: 10.1371/journal.pone.0027090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang JS, Alliston T, Delston R, Derynck R. Repression of Runx2 function by TGF-beta through recruitment of class II histone deacetylases by Smad3. EMBO J. 2005;24(14):2543–2555. doi: 10.1038/sj.emboj.7600729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smerdel-Ramoya A, Zanotti S, Stadmeyer L, Durant D, Canalis E. Skeletal overexpression of connective tissue growth factor impairs bone formation and causes osteopenia. Endocrinology. 2008;149(9):4374–4381. doi: 10.1210/en.2008-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stickens D, et al. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development. 2004;131(23):5883–5895. doi: 10.1242/dev.01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nistala H, Lee-Arteaga S, Smaldone S, Siciliano G, Ramirez F. Extracellular microfibrils control osteoblast-supported osteoclastogenesis by restricting TGFbeta stimulation of RANKL production. J Biol Chem. 2010;285(44):34126–34133. doi: 10.1074/jbc.M110.125328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miao D, et al. Osteoblast-derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1-34. J Clin Invest. 2005;115(9):2402–2411. doi: 10.1172/JCI24918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horwitz MJ, et al. A 7-day continuous infusion of PTH or PTHrP suppresses bone formation and uncouples bone turnover. J Bone Miner Res. 2011;26(9):2287–2297. doi: 10.1002/jbmr.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikeda K, et al. Identification of transcripts encoding a parathyroid hormone-like peptide in messenger RNAs from a variety of human and animal tumors associated with humoral hypercalcemia of malignancy. J Clin Invest. 1988;81(6):2010–2014. doi: 10.1172/JCI113551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grover M, et al. Assessment of bone mineral status in children with Marfan syndrome. Am J Med Genet A. 2012;158A(9):2221–2224. doi: 10.1002/ajmg.a.35540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neptune ER, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33(3):407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.