

Abstract
Homologous recombination is a major pathway for repair of DNA double-strand breaks. This repair process is initiated by resection of the 5′-terminated strand at the break site. In yeast, resection is carried out by three nucleolytic complexes: Mre11-Rad50-Xrs2, which functions at the initial step and also stimulates the two processive pathways, Sgs1-Dna2 and Exonuclease 1 (Exo1). Here we investigated the relationship between the three resection pathways with a focus on Exo1. Exo1 preferentially degrades the 5′-terminal stand of duplex DNA that is single stranded at the 3′ end, in agreement with its role downstream of the Mre11-Rad50-Xrs2 complex. Replication protein A (RPA) stimulates DNA end resection by Exo1 by both preventing nonspecific binding of Exo1 to and preventing degradation of single-stranded DNA. Nucleolytic degradation of DNA by Exo1 is inhibited by the helicase-deficient Sgs1 K706A mutant protein and, reciprocally, the nuclease-deficient Exo1 D173A mutant protein inhibits DNA unwinding by Sgs1. Thus, the activities of Sgs1 and Exo1 at DNA ends are mutually exclusive, establishing biochemically that both machineries function independently in DNA end processing. We also reconstituted Sgs1-Top3-Rmi1-RPA-Dna2 and Exo1 resection reactions both individually and combined, either with or without the Mre11-Rad50-Xrs2 complex. We show that the yeast Sgs1-Dna2 and Exo1 pathways do not stimulate one another and function as independent and separate DNA end-processing machineries, even in the presence of the stimulatory Mre11-Rad50-Xrs2 complex.
Significance
The repair of double-strand DNA breaks by homologous recombination is initiated by the nucleolytic resection of the 5′-terminated strand at the DNA break. Genetic work in Saccharomyces cerevisiae identified three DNA end resection pathways: Mre11-Rad50-Xrs2, Dna2-Sgs1-Top3-Rmi1, and Exo1. Here we investigated the relationship between the three nucleolytic complexes in vitro. With a focus on Exo1, we show that it is stimulated by the single-strand DNA-binding protein, RPA, and also by the Mre11-Rad50-Xrs2 complex. Furthermore, our analysis provides biochemical evidence for the view that Exo1 and Dna2-Sgs1-Top3-Rmi1 function downstream of Mre11-Rad50-Xrs2 as independent and mutually exclusive DNA end-processing pathways.
DNA of all living organisms is constantly being damaged by radiation, chemicals, or abortive DNA metabolism. One of the most deleterious forms of DNA damage is the double-stranded DNA break (DSB); left unrepaired, a single DSB in a haploid can be lethal (1–4) and, in a diploid, result in genomic instability (5, 6). Cells have two major pathways for DSB repair, nonhomologous end joining (NHEJ) and homologous recombination (HR). In NHEJ, both broken DNA strands are reattached together by ligation, often resulting in mutations in the vicinity of the break site (7). HR, in contrast, exploits genetic information stored in an undamaged copy of the DNA, a sister or homolog. By using it as a template for DSB repair, the HR pathway is largely error-free and may proceed by one of several mechanisms reviewed extensively elsewhere (8). The first step in DSB repair by HR is DNA end processing, which is common to all recombination subpathways (9–11). The 5′-terminated strand of a DSB must be resected to reveal a 3′-terminated single-stranded DNA (ssDNA) tail, which serves both as a substrate for the DNA strand exchange protein, Rad51, and as a primer for DNA synthesis in the downstream steps of HR. DNA end resection also commits the repair to HR, because the resected ends typically become unligatable and, thus, refractory to NHEJ. As such, DNA end resection must be tightly regulated. Furthermore, because DSBs arise in various cellular contexts, it is not surprising that cells possess multiple enzymatic machineries for DNA end resection.
The processing of DSBs in yeast can be initiated by the Mre11-Rad50-Xrs2 (MRX) complex (12, 13), with the involvement of Sae2 (14). The MRX and Sae2 proteins are recruited to a DSB within seconds and can themselves initiate nucleolytic DSB processing (15). DNA end resection by MRX and Sae2 is slow and limited to only 100–300 nt (12). MRX and Sae2 are especially important for DSBs terminated with either chemically altered nucleotides or covalently attached proteins such as topoisomerases. Processing by MRX and Sae2 is sufficient for gene conversion in some cases (13, 16). In most cases, however, recombination requires one of two processive long-range resection pathways (16). MRX was also shown to promote both long-range pathways independently of its nuclease activity: by recruiting Sgs1, an essential component of the Sgs1-Top3-Rmi1-Dna2-RPA (subsequently abbreviated here as “Sgs1-Dna2”) resection machinery, or by stimulating Exonuclease 1 (Exo1), a component of the second resection pathway (12, 17–19).
In the Sgs1-Dna2 pathway, Sgs1 helicase unwinds dsDNA from a broken DNA end and translocates on the 3′-terminated strand in a 3′→5′ direction (17, 18). The resulting single strands of DNA are then coated by the ssDNA-binding protein, RPA, which directs the Dna2 nuclease to specifically degrade the 5′-terminated strand. Dna2 physically interacts with Sgs1 and, because Dna2 nuclease is also a 5′→3′ ssDNA translocase, both proteins likely mimic the RecBCD enzyme and translocate together as a bipolar helicase complex, with each protein translocating on each of the DNA strands (20). Top3-Rmi1, a heterodimer that forms a stable complex with Sgs1, promotes DNA unwinding by Sgs1. This concerted end-processing activity produces the 3′-terminated ssDNA tail that is essential for recombination (17, 18). Exo1, the component of the second resection pathway, is a 5′→3′ dsDNA-specific nuclease (21–23). Genetic work revealed additive defects in DNA end resection in sgs1Δ exo1Δ and dna2Δ exo1Δ mutants, suggesting that Exo1 is a component of a separate resection pathway (12). Similarly to Sgs1-Dna2, Exo1 can mediate long-range resection of the 5′-terminated strand at a dsDNA end (12, 13, 24). Exo1 also functions downstream of, and is stimulated by, the MRX complex (19).
The mechanisms of DNA end processing are largely conserved between yeast and humans, with a few apparent differences. Most notably, the human homolog of Sgs1, the bloom (BLM) helicase, was shown to stimulate the nucleolytic activity of the human EXO1 (25), suggesting that BLM can participate in a nonessential manner in the EXO1 pathway (24). In vitro reconstitution experiments showed that humans possess two end-processing machineries: BLM-DNA2 and EXO1-BLM, with the latter using BLM only for a stimulatory role. Thus, BLM and EXO1 function in both common and separate pathways (26). Here we investigated the relationship between the Exo1 and Sgs1 pathways in Saccharomyces cerevisiae, using purified recombinant proteins. Our results strongly suggest that, in yeast, Exo1 and Sgs1-Dna2 represent independent and mutually exclusive resection pathways.
Results
RPA Blocks Degradation of ssDNA by Exo1.
To characterize the role of S. cerevisiae Exo1 in DNA end resection, we expressed a C-terminally FLAG-tagged wild-type Exo1 and the nuclease-deficient Exo1 D173A mutant (21) in Spodoptera frugiperda (Sf9) cells. The proteins were purified by affinity and ion exchange chromatography (Fig. 1A). Given the physiological abundance of the ssDNA-binding protein, RPA and its involvement in the Sgs1-Dna2 resection pathway (17, 18), we first tested the effect of RPA on the nuclease activity of Exo1. Using an equimolar concentration of 5′-radiolabeled ssDNA (50 nt) and dsDNA (50 bp), we followed loss of the 5′-end label due to the 5′→3′ exonuclease activity of Exo1. In the absence of RPA, Exo1 initiated degradation of both ssDNA and dsDNA in a concentration-dependent manner (Fig. 1 B and C). However, in the presence of a saturating amount of RPA, the dsDNA substrate was fully degraded whereas the ssDNA substrate remained intact (Fig. 1 B and C). The effect of RPA on the nuclease activity of Exo1 was further analyzed using a Y-structure substrate that mimics DNA unwound by a helicase (Fig. S1). For this substrate, RPA blocked exonucleolytic degradation of the 5′-ssDNA arm, but it had no effect on the dsDNA region of the substrate (Fig. S1 A–F).
Fig. 1.

RPA blocks degradation of ssDNA by Exo1. (A) FLAG-tagged wild-type Exo1 (210 ng) and FLAG-tagged nuclease-dead Exo1 (D173A, 140 ng) used in this study. (B) ssDNA and dsDNA (1 nM each), 32P labeled at the 5′ end, were used to analyze the nuclease activity of Exo1 (0.1, 0.2, 0.4, 0.8, 1.5, and 3 nM, respectively) in the absence or presence of RPA (22.5 nM). (C) Quantification of experiments as shown in B. Error bars show SE.
Next, we tested the ability of Exo1 to degrade plasmid-length (2.7 kb) dsDNA. Because Exo1 degrades DNA in a 5′→3′ direction, the substrate was radiolabeled at its 3′ ends to allow detection of reaction intermediates. Resection of the DNA resulted in the time-dependent formation of intermediates (Fig. 2 A and B). In the presence of RPA, in particular, a notable intermediate was produced after a few minutes that migrated with a mobility faster than that of the full-length ssDNA marker (Fig. 2 A and B, lane 5), but that comigrated with half-length (∼1,350 nt) ssDNA (Fig. S2). Because the concentration of Exo1 was saturating with respect to DNA ends, we infer that Exo1 initiated DNA degradation from each DNA end simultaneously, with exonucleolytic processing of the 5′-terminated strands proceeding toward the middle of the DNA molecule (Fig. 2C). When both Exo1 molecules meet at the approximate middle of the substrate, the product is the observed half-length ssDNA intermediate (Fig. 2C). In agreement with the data presented in Fig. 1, RPA blocks subsequent degradation of the ssDNA intermediate by Exo1 (Fig. 2B, lanes 5–10), and the half-length ssDNA persists for the remainder of the 30-min time course. However, without RPA (Fig. 2A, lanes 5–10), Exo1 continues to more slowly degrade the ssDNA intermediates, resulting in progressively smaller reaction products. These two outcomes are schematically illustrated in Fig. 2C.
Fig. 2.
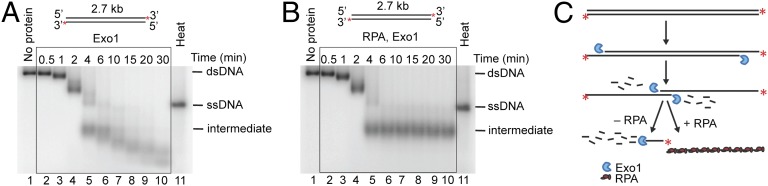
In the presence of RPA, resection of linear plasmid-length dsDNA by Exo1 produces kilobase-sized ssDNA. (A) Nuclease activity of Exo1 (2.7 nM) as a function of time (0.5, 1, 2, 4, 6, 10, 15, 20, and 30 min). pUC19 dsDNA (blunt, 1 nM), 32P labeled at the 3′ end, was the substrate. “Heat” refers to ssDNA produced by heat denaturation of the pUC19 dsDNA. (B) Reaction as in A carried out in the presence of RPA (0.4 μM). (C) Illustration summarizing results from A and B, showing the intermediates and products of dsDNA degradation by Exo1 in the presence or absence of RPA.
In contrast to its vigorous exonuclease activity, Exo1 was unable to degrade covalently closed dsDNA (3.4 kb) that contained an ssDNA heteroduplex bubble of 450 nt (Fig. S3). Thus, Exo1 has no detectable endonuclease activity on circular dsDNA even with ssDNA regions. As expected, the nuclease-deficient Exo1 D173A mutant failed to degrade DNA of any type (Figs. S3 and S4), showing that the exonuclease activity of Exo1 is intrinsic to the wild-type protein. Therefore, we conclude that Exo1 requires a DNA end for nucleolytic action and that RPA blocks the action of Exo1 on ssDNA, making Exo1 an exonuclease specific strictly for one strand of dsDNA.
Exo1 Preferentially Degrades the 5′-Terminal Strand of dsDNA with a 3′-ssDNA Overhang.
In wild-type cells, dsDNA breaks in yeast can be initially resected by the MRX complex and Sae2 (12, 13). Following a limited nucleolytic degradation of the 5′-terminated strand, either the Exo1 or the Sgs1-Dna2 pathway can further resect these processed DNA ends. To test whether Exo1 might show a preference for 3′-ssDNA tailed duplex DNA that would result from initial reaction by MRX and Sae2, we generated dsDNA substrates with blunt ends or with 5′- or 3′-terminated ssDNA (4 nt long). As shown in Fig. 3A and Fig. S5A, in the presence of RPA, Exo1 preferentially degraded DNA with the 3′ overhangs (i.e., possessing 5′-recessed ends), followed by the blunt-ended DNA, and finally the DNA with the 5′-overhangs. The concentration dependence suggested that Exo1 bound most poorly to the DNA with the 5′ overhangs.
Fig. 3.

Exo1 preferentially degrades dsDNA resected at the 5′ end to produce an ssDNA tail at the 3′ end. (A) Quantification of Exo1 nuclease activity, in the presence of RPA (3 μM), using unlabeled pUC19 dsDNA (7.6 nM) that either was blunt or contained an ssDNA overhang (4 nt) at either 3′ or 5′ ends. Error bars indicate SE. (B) A representative experiment showing degradation of pUC19 dsDNA with a 5′-ssDNA overhang of 3 nt (7 nM, 32P labeled at the 3′ end) by Exo1 (0.05, 0.15, 0.45, 1.3, 4, and 12 nM, respectively) in the presence of RPA (2.8 μM). “D173A”: The nuclease-deficient Exo1 D173A mutant was used instead of wild-type Exo1 (12 nM). “Heat”: Heat-denatured substrate. (C) Illustration summarizing results from B showing degradation by Exo1 of a dsDNA substrate with 5′-end ssDNA overhangs. (D) Quantification of Exo1 nuclease activity on 3.0 kb unlabeled dsDNA (7.6 nM) containing an ssDNA overhang of 4 nt at both 5′ ends (squares) vs. dsDNA with a 4-nt 5′ overhang at one end and a 3′ (circles) or 5′ (triangles) overhang of ∼100 nt at the other end. The reactions were carried out in the presence of RPA (3 μM). Error bars show SE.
In the case of the low-affinity 5′-tailed DNA substrate, a markedly unique reaction intermediate that comigrated with full-length ssDNA was produced (Fig. 3B, compare lanes 7 and 10). Even at the highest concentration of 12 nM Exo1 (Fig. 3B, lane 7), the half-length ssDNA intermediate that had been seen during processing of the blunt substrate (Fig. 2) was not apparent; instead, full-length ssDNA was being produced. Our interpretation was that, during initial end processing, one end of the low-affinity 5′-tailed DNA was being slowly converted into a high-affinity 5′-recessed DNA by the 5′→3′ nuclease activity of Exo1. The resulting DNA, now with just one 5′-recessed end, represents a much better substrate for the subsequent degradation by Exo1 than any of the 3′-overhanging ends, resulting in repeated and full degradation of the entire length of one DNA strand and producing the full-length ssDNA intermediate (Fig. 3C). This interpretation is consistent with the low processivity of the human EXO1 (27).
To further test this interpretation, and because the MRX and Sae2 proteins resect up to ∼100–300 nt of the 5′-terminated strand in vivo (12), we investigated the processing of substrates with longer (∼100 nt) 5′- or 3′-ssDNA overhangs. As above, Exo1 showed a clear preference for the substrate containing the longer 3′-terminated ssDNA tail (Fig. 3D), even in the absence of RPA (Fig. S5 B–E). In summary, these results indicate that Exo1 acts preferentially on a dsDNA end possessing a 3′-ssDNA overhang (i.e., 5′-recessed end), such as those produced in vivo by the MRX-dependent pathway.
Single-Stranded DNA-Binding Proteins Stimulate Degradation of a Single Strand of Duplex DNA by Exo1.
We had noted that saturating concentrations of RPA stimulated DNA end resection, especially at high ratios of DNA to Exo1 (Fig. S6). RPA also stimulated resection by Sgs1-Dna2, and the effect was specific to yeast RPA (17). In the case of Exo1, however, the resection was not species specific: When RPA was substituted with Escherichia coli ssDNA-binding (SSB) protein, we observed a similar stimulation of DNA degradation by Exo1 (Fig. S7). To understand the basis for the stimulation by ssDNA-binding proteins, we reasoned that the ssDNA produced by Exo1 might act as an inhibitor by sequestering free Exo1 and thus prevent its distributive action on the remaining dsDNA. To test this hypothesis, circular ssDNA, which is refractory to cleavage by Exo1, was added to standard reactions. Indeed, the ssDNA dramatically inhibited resection of the dsDNA that was dependent on the amount of ssDNA added (Fig. S8, lanes 3–6). Importantly, RPA completely abolished the inhibitory effect of the circular ssDNA (Fig. S8, lanes 7–10). Thus, RPA stimulates Exo1 by preventing the formation of the nonproductive ssDNA-Exo1 complex (Fig. S8, compare lanes 6 and 10). These results collectively indicate that the ssDNA-binding proteins stimulate the nucleolytic degradation of dsDNA by nonspecifically preventing the binding and sequestration of Exo1 to ssDNA.
RPA Promotes DNA End Resection by Mre11-Rad50-Xrs2 and Exo1.
Recent results established that the Mre11-Rad50-Xrs2 protein complex promoted DNA degradation by Exo1 (19). To further investigate the functional interactions between MRX, Exo1, and RPA, we expressed the Mre11-Rad50-Xrs2 complex and purified it as a heterotrimer from Sf9 cells (Fig. S9). MRX clearly stimulated nucleolytic processing of DNA by Exo1 (Fig. 4A), as reported previously (19). We next examined the effect of RPA; however, in contrast to the prior report, the stimulation by MRX was noticeably greater in the presence of RPA (Fig. 4 B and C). Thus, under physiological conditions when RPA is present, Exo1 and MRX comprise a very effective complex for the processing of DNA ends.
Fig. 4.

Mre11-Rad50-Xrs2 complex stimulates resection of dsDNA by Exo1. (A) A representative experiment with blunt-ended pUC19 dsDNA (1 nM, 32P labeled at the 3′ end) showing degradation by Exo1 (0.4 nM, where indicated) and its stimulation by MRX [1, 3, 10, 30, and 100 nM (lanes 2–6) and 1, 3, 10, and 30 nM (lanes 8–11), respectively]. (B) Reaction as in A carried out in the presence of RPA (0.4 μM). (C) Quantitation of experiments as in A and B. Error bars show SE.
Exo1 and Sgs1-Dna2 Represent Separate DNA End-Processing Pathways.
We next investigated biochemically the relationship between the Exo1 and Sgs1-Dna2 pathways of DNA end resection. First we asked whether Sgs1 could stimulate Exo1 activity, to determine whether it mimicked the behavior of human BLM helicase in the stimulation of human EXO1 (25, 26). Because Sgs1 is inhibited by the 5 mM Mg2+ that is used for the Exo1 nuclease assays (28), we used the “low-salt” reaction conditions that are optimal for Sgs1 helicase activity (Methods). As presented in Fig. 5 A and B, Sgs1 did not stimulate the nucleolytic activity of Exo1, nor did Exo1 stimulate DNA unwinding by Sgs1. Sgs1 forms a stable complex with Top3-Rmi1 (29), and the heterodimer is nearly essential for DNA unwinding by Sgs1 at elevated salt concentrations (17). Consequently, we next investigated the effect of Sgs1-Top3-Rmi1 on Exo1 activity at the standard elevated Mg2+ concentration (Fig. 5 C and D). As above, we did not observe any stimulatory effect of Sgs1-Top3-Rmi1 on Exo1 or vice versa.
Fig. 5.

Sgs1 does not stimulate resection of dsDNA by Exo1. (A) Nuclease assays with Exo1 (0.35, 0.53, 0.8, 1.2, and 1.8 nM), RPA (0.4 μM), and either without (lanes 2–6) or with Sgs1 (0.1 nM, lanes 8–13) in low-salt buffer. Blunt-ended pUC19 dsDNA (1 nM), 32P labeled at the 3′ end, was used. (B) Quantification of experiments as shown in A. Error bars show SE. (C) Nuclease assays with Exo1 (0.53, 0.8, 1.2, 1.8, and 2.7 nM), RPA (0.4 μM), and either without (lanes 2–6) or with Sgs1 (0.5 nM) and Top3-Rmi1 (5 nM, lanes 9–14, respectively), in standard buffer. Substrate is as in A. (D) Quantification of experiments as shown in C. Error bars show SE. (E) Nuclease assay carried out with Exo1 (0.5, 1, 2, 3, and 4 nM), RPA (0.4 μM), and either without (lanes 2–6) or with helicase-dead Sgs1 K706A (20 nM, lanes 8–12). Substrate is as in A. (F) Increasing amounts of nuclease-dead Exo1 D173A (0.53, 0.8, 1.2, 1.8, 2.7, 4, and 8 nM) were added to reactions containing Sgs1 (0.5 nM) and/or Top3-Rmi1 (5 nM), as indicated, in the presence of RPA (0.4 μM). Substrate is as in A.
One possible limitation of the above experiments is that, intentionally, neither Exo1 nor Sgs1 was used at concentrations that would have fully saturated the DNA ends. These substoichiometric reaction conditions might have limited a potential stimulatory effect. However, due to the high specific activity of the proteins, the use of saturating enzyme concentrations would result in complete processing of the DNA in a timeframe too short for analysis. To circumvent this problem, we used catalytically inactive mutants of either Sgs1 or Exo1 to determine whether the presence of one had an effect on the activity of the other wild-type protein. These mutants retain DNA-binding capacities, but are devoid of helicase and nuclease activities, respectively, and can thus be used at saturating concentrations (21, 28). Strikingly, the helicase-deficient Sgs1 (K706A) inhibited the exonucleolytic activity of Exo1 (Fig. 5E) in a concentration-dependent manner (Fig. S10). Similarly, the nuclease-deficient Exo1 (D173A) inhibited DNA unwinding by Sgs1-Top3-Rmi1 in a concentration-dependent manner (Fig. 5F).
Having established that Exo1 and Sgs1 seem to operate rather independently, we wanted to investigate the relationship between Exo1 and the Sgs1-Top3-Rmi1-Dna2 resection machinery. We first used blunt-ended dsDNA (167 bp) and monitored release of the 32P label from the 5′ end in the presence of RPA (Fig. S11A). Within 1 min, the Sgs1-Top3-Rmi1-Dna2 machinery processed ∼45% of the DNA ends; under these conditions, Exo1 alone resected ∼58% of the DNA ends. When we used both Exo1 and Sgs1-Top3-Rmi1-Dna2, ∼70% of DNA ends were processed (Fig. S11A). Identical analysis after 3 min (Fig. S11B) revealed that the reaction saturates with ∼90% of DNA ends processed, showing that the reaction was not saturated at the 1-min time point. The slightly less than additive effect suggests that Exo1 and Sgs1-Top3-Rmi1-Dna2 resection machineries do not synergize with one another. Next, we monitored resection of plasmid dsDNA, using identical protein complexes (Fig. 6A). As is apparent from the quantification, the combined activities of the Exo1 and Sgs1-Dna2 reactions were less than additive; in fact, the combined reaction was less efficient than processing by Exo1 alone (Fig. 6B). This finding confirms our previous interpretations that the activities of Sgs1 and Exo1 at DNA ends are mutually exclusive (Fig. 5 E and F) and shows that Exo1 and Sgs1-Dna2 operate in alternative pathways. Similar results were obtained when reactions were supplemented with Mre11-Rad50-Xrs2 (Fig. 6 C and D). Collectively, our data are in accord with the genetic interactions (12) and strongly indicate that, in yeast, Exo1 and Sgs1-Top3-Rmi1-Dna2 represent independent and separate pathways for the processing of DNA ends.
Fig. 6.

Exo1 does not stimulate DNA end resection by Dna2, Sgs1, and Top3-Rmi1. (A) Nuclease assays with Exo1 (7 nM), Dna2 (7 nM), Sgs1 (7 nM), Top3-Rmi1 (14 nM), and unlabeled pUC19 dsDNA containing 4-nt ssDNA overhangs at the 5′ ends (3.8 nM) in the presence of RPA (3 μM) for 4, 8, or 12 min. The gel is an inverted image of ethidium bromide-stained DNA. (B) Quantification of experiments as shown in A. Error bars show SE. (C) Nuclease assays with Exo1 (1 nM), Sgs1 (1 nM), Top3-Rmi1 (3 nM), Dna2 (1 nM), Mre11-Rad50-Xrs2 (25 nM), as indicated, for 1, 2, 4, 8, and 16 min in the presence of RPA (0.4 μM). Blunt-ended pUC19 dsDNA (1 nM), 32P labeled at the 3′ end, was used. (D) Quantification of experiments as shown in C. Error bars show SE.
Discussion
The main proteins involved in DNA end resection were identified genetically only a few years ago. In the years that followed, we have learned a great deal about processing mechanisms by reconstituting resection reactions in vitro. RPA was found to be an essential component of the Sgs1-Dna2 pathway. First, RPA stimulates DNA unwinding by Sgs1 in a species-specific manner (28). Second, it directs Dna2 nuclease to resect specifically the 5′-terminated ssDNA. We and others could show that RPA blocks degradation of the 3′-terminated DNA strand by Dna2 while, at the same time, it stimulates resection of 5′-terminated strand (17, 18). RPA is thus responsible for the strand bias in DNA end resection by Sgs1-Dna2, which is essential for formation of 3′-ssDNA tails. Furthermore, MRX stimulates the Sgs1-Dna2 pathway by recruiting the resection proteins to the DNA end (17, 18). Here we investigated the roles of these proteins on the alternative Exo1-dependent resection pathway. S. cerevisiae Exo1 can degrade both dsDNA and ssDNA in a 5′→3′ direction, and here we established that RPA limits the nucleolytic activity of Exo1 on ssDNA by blocking the binding of Exo1 to ssDNA. The protection of 5′-terminated ssDNA from degradation by Exo1 may not be physiologically relevant for the processing of most direct dsDNA breaks, because only 3′-terminated overhangs are expected to form during their processing in vivo. However, it may be important with regard to the intermediates formed as a consequence of blocked replication forks as, for example, during lesion bypass by template switching. Upon encountering a lesion in the leading-strand template, fork reversal models propose regression of the stalled fork to produce dsDNA that is terminated with a 5′-ssDNA tail that originates from the nascent lagging strand (30). This 5′-tailed ssDNA serves as the template for extension of the nascent leading strand and would require protection from cellular nucleases. RPA may thus protect this regressed fork structure from the degradation by Exo1 that would otherwise clearly occur in RPA’s absence. Another clear benefit of blocking the binding of Exo1 to ssDNA is that RPA enables Exo1 to resect kilobase-sized tracts of dsDNA (Figs. S6 and S8). In vitro, we established that extensive resection by Exo1 produces high concentrations of ssDNA, which sequester the Exo1 and thus reduce the effective concentration of the enzyme available for the distributive exonucleolytic degradation of dsDNA; presumably, RPA plays a similar role in vivo. By coating ssDNA, RPA prevents the nonspecific binding of Exo1 to ssDNA. In contrast to the Sgs1-Dna2 machinery, RPA stimulates Exo1 in a species-independent manner; we showed that SSB protein from E. coli elicits an identical effect (Fig. S7 A and B), indicating that the sole function of RPA in this reaction is to sequester ssDNA and that direct protein–protein interactions are not involved.
Double-stranded DNA breaks may form directly as a result of ionizing radiation or indirectly such as when DNA replication encounters an ssDNA break. This will result in various structures at the site of the break, including 5′- and 3′-terminated ssDNA overhangs. Furthermore, the Mre11-Rad50-Xrs2 and Sae2 proteins can function before Exo1 (12, 13, 19) and can carry out a limited resection of the 5′-ended strand, resulting in DNA intermediates with 3′-ended ssDNA tails. By using substrates with blunt dsDNA ends, as well as 5′- or 3′-terminated ssDNA overhangs, we established that Exo1 prefers dsDNA with ssDNA tails at the 3′ end (Fig. 3 A and D). Thus, Exo1 may more efficiently resect DNA that had been previously processed by Mre11-Rad50-Xrs2 and Sae2. Indeed, Mre11-Rad50-Xrs2 stimulates DNA end resection by Exo1 (19), and we extended this observation by showing that this stimulatory effect is further enhanced by RPA (Fig. 4C).
Yeast cells lacking either EXO1 or SGS1 do not show pronounced resection defects as measured by physical or recombination assays. However, the inactivation of both genes in exo1 sgs1 double mutants completely eliminates DNA end resection beyond ∼300 nt. Furthermore, dna2 mutants were indistinguishable from sgs1 mutants, and sgs1 dna2 mutants had the same phenotype as the individual single mutants. The synergistic genetic interactions led to the model where Exo1 and Sgs1-Dna2 were proposed to belong to independent and overlapping resection pathways (12, 13, 24). The genetic interactions seem to be conserved throughout evolution: Simultaneous knockdown of BLM (the human homolog of Sgs1) and EXO1 (human homolog of yeast Exo1) resulted in synergistic increase in DNA damage sensitivity and faulty checkpoint signaling (24). At the same time, the purified BLM helicase was found to stimulate human EXO1 in vitro, independently of its helicase activity (25), suggesting an overlap between the two pathways. Indeed, the human resection machinery was later found to consist of two essential core machineries: BLM-DNA2-RPA and EXO1-RPA (26), where BLM plays an essential role in the BLM-DNA2 pathway and a stimulatory, but nonessential structural, role in the EXO1 pathway. More recently it has been reported that hypomorphic dna2 alleles in S. cerevisiae promoted DNA end resection only in the presence of Exo1, suggesting a crosstalk between the Sgs1-Dna2 and Exo1 pathways in yeast as well (31). To clarify these issues, here we investigated the relationship between the Exo1 and Sgs1-Dna2 machineries, using purified yeast proteins. We showed that when the resection complexes were used at concentrations comparable to those of the DNA ends, the contribution of both pathways to resection was either equal to or less than additive (Fig. 6B and Fig. S11A). This implies that the Exo1 and Sgs1-Dna2 pathways are separate and do not stimulate one another. Although MRX stimulates each of the resection complexes, we also found that the relative contributions of each complex to resection were unaltered (Fig. 6D). The less than additive effect suggests that the binding of one resection complex to the DNA end excluded the other from the DNA end. By exploiting mutant enzymes, we were able to use protein concentrations sufficient to saturate DNA ends and found that both Exo1 and Sgs1 do indeed compete with one another for DNA ends. Furthermore, whereas RPA-coated ssDNA stimulates DNA degradation by Dna2 (17), it inhibits Exo1 (Fig. 1), showing that Exo1 cannot act downstream of Sgs1-catalyzed DNA unwinding. These results together suggest that Sgs1-Dna2 and Exo1 pathways are not only independent, but also mutually exclusive, and their activities in vivo must be tightly regulated. We cannot exclude the possibility that posttranslational modifications (31) or additional protein components might affect the crosstalk between the two pathways in vivo.
Methods
Expression and Purification of Recombinant Proteins.
The expression vectors for Exo1 and Exo1 D173A, both with a C-terminal FLAG epitope tag, were a gift from Michael Liskay (Oregon Health & Science University, Portland, OR) (21). Exo1 and Exo1 D173A were expressed in Sf9 cells, using the Bac-to-Bac baculovirus expression system (Invitrogen) according to the manufacturer’s instructions. The protocol describes purification from 400 mL of Sf9 cells infected with baculovirus for 52 h. All of the following steps were performed at 4 °C or on ice. Sf9 cell lysis was performed as previously described (28). The cleared cell extract was applied by gravity to a preequilibrated 0.5-mL M2–anti-FLAG affinity resin (Sigma) and then washed and eluted as described previously (21). Peak fractions (estimated by the Bradford method [32]) were pooled volume measure, and diluted by adding two volumes of dilution buffer [50 mM Tris⋅HCl, pH 7, 5 mM β-mercaptoethanol, 0.5 mM phenylmethanesulfonyl fluoride (PMSF), 6.7 μg/mL leupeptin, and 10% (vol/vol) glycerol] and three volumes of Buffer A (same as dilution buffer plus 50 mM NaCl). The diluted fractions were loaded at 0.8 mL/min on a HiTrap SP HP column (1 mL; GE Healthcare) preequilibrated with Buffer A. The column was washed with Buffer A and the protein was eluted with 5 mL gradient (0–100%) of Buffer B (same as Buffer A but with 1M NaCl). Fractions containing proteins were pooled, frozen in liquid nitrogen, and stored at −80 °C. The nuclease-dead Exo1 D173A mutant was purified exactly the same as the wild-type protein. Exo1 and Exo1 D173A concentrations were determined by densitometry by comparison with a serially diluted broad range protein marker (BioRad) on a 10% (wt/vol) polyacrylamide gel stained with Coomassie Blue. Protein concentrations obtained were ∼131–393 nM for Exo1 and 89 nM for Exo1 D173A. We found Exo1 to be quite unstable. To maintain high specific activity, protein preparation must be carried out promptly upon cell lysis without any delay.
The Mre11-Rad50-Xrs2 complex, containing a His tag and a FLAG tag at the C terminus of Mre11 and Xrs2, respectively, was expressed by coinfecting Sf9 cells with recombinant baculoviruses, using the Bac-to-Bac expression system (Invitrogen) and vectors pTP391 and pTP694 (Mre11 and Xrs2 expression vectors, a gift from Tanya Paull, University of Texas at Austin, Austin, TX) and pFastBac-Rad50 (pFB-Rad50). The pFB-Rad50 vector was constructed by amplifying the RAD50 ORF sequence using PCR and then by cloning the sequence into a pFastBac1 expression vector (Invitrogen), using BamHI and XhoI restriction endonucleases (both from New England Biolabs). The vector pGPGK-Rad50 (a gift from Patrick Sung, Yale University, New Haven, CT) and the primers, Rad50-Forward (CCGTTCGGGCCCGGATCCATGAGCGCTATCTATAAATTA) and Rad50-Reverse (GAACCTCTCGAGTCAATAAGTGACTCTGTTAA) were used for PCR. The infected Sf9 cell pellets from 1.6 L of media (72 h postinfection) were resuspended in lysis buffer [50 mM Tris⋅HCl (pH 7.5), 1 mM EDTA, 2 mM β-mercaptoethanol, 20 mM imidazole, 1:500 dilution of protease inhibitor mixture (Sigma; P8340), 30 μg/mL leupeptin, and 1 mM PMSF] up to a volume of 64 mL. The previous and all subsequent steps were performed at 4 °C or on ice. The sample was mixed slowly with a stir bar for 20 min, and then 32 mL of 50% (vol/vol) glycerol and 6.2 mL of 5M NaCl were added slowly and sequentially while mixing. The suspension was further incubated for 30 min and mixed occasionally. The cell suspension was centrifuged at 55,000 × g for 30 min to obtain soluble extract. The cleared extract was batch incubated with preequilibrated Ni2+-nitrilotriacetic acid (NTA) resin (Qiagen) for 1 h. The resin was washed extensively with wash buffer [30 mM Tris⋅HCl, pH 7.5, 300 mM NaCl, 10% (vol/vol) glycerol, 1 mM β-mercaptoethanol, 20 mM imidazole, 1:1,000 dilution of protease inhibitor mixture, 15 μg/mL leupeptin, and 0.5 mM PMSF] batch-wise and then on column. MRX was eluted with elution buffer (same as wash buffer, but with 400 mM imidazole). The eluate was then diluted fivefold with dilution buffer [30 mM Tris⋅HCl, pH 7.5, 300 mM NaCl, 1 mM β-mercaptoethanol, 10% (vol/vol) glycerol, 15 μg/mL leupeptin, and 0.5 mM PMSF] and batch incubated with M2-anti-FLAG affinity resin (Sigma; 0.8 mL) for 40 min. The resin was then washed with 20 mL of dilution buffer in a column and eluted with elution buffer [the same as dilution buffer, but with 200 μg/mL FLAG-peptide (Sigma)]. Fractions containing recombinant MRX complex were pooled, aliquoted, and stored at −80 °C. The protein concentration was estimated by the Bradford method (32).
E. coli SSB, S. cerevisiae RPA, Sgs1, Sgs1 (K706A), Top3-Rmi1, and Dna2 were purified as described previously (17, 28, 33–35).
Nucleic Acid Substrates.
The oligonucleotide-based DNA substrates were described previously (28, 36). The “bubbled” substrate was prepared as described previously (37), but using the plasmids pDHJSAN+ and pDHJSAN− as building blocks (38). The 3.4-kb closed circular ssDNA was prepared as described previously (38).
The 2.7-kb dsDNA substrate with 4-nt overhangs at the 5′ end was pUC19 dsDNA linearized with HindIII (New England Biolabs). Radiolabeling of this substrate with 32P at the 3′ end with Klenow (exo−) enzyme (New England Biolabs) produced dsDNA with 3-nt overhangs. Subsequent end-filling with Klenow (exo−) enzyme (New England Biolabs) resulted in the 2.7-kb blunt-ended dsDNA substrate used in most resection experiments. Radiolabeling and end-filling was carried out according to standard protocols recommended by the enzyme manufacturer. Unlabeled 2.7-kb substrate, either blunt or containing 4 nt ssDNA overhangs at the 3′ end, was pUC19 dsDNA linearized with HincII or PstI (both New England Biolabs), respectively. The unlabeled 3.0-kb dsDNA substrate was derived from the vector pSE1 (a gift from Jody Plank, S.C.K. laboratory), which contains a tandem repeat of Nt/Nb.BbvCI restriction (or “nicking”) sites 110 nt in length. pSE1 was first linearized with HindIII (New England BioLabs) and then nicked with either Nb.BbvCI to generate the 3′-ended ssDNA overhang or Nt.BbvCI (both New England Biolabs) to generate the 5′-ended ssDNA overhang. The DNA was then diluted fivefold in water, incubated at 85 °C for 15 min, and immediately purified with a PCR purification kit (Qiagen). The 1.35-kb dsDNA was pUC19 digested with HindIII and BsrFI (New England Biolabs) and radiolabeled with 32P at the 3′ end.
Nuclease Assays.
Unless otherwise indicated, all nuclease assays (15 μL vol) were carried out using identical ionic conditions in standard buffer [25 mM Tris acetate, pH 7.5, 1 mM DTT, 5 mM magnesium acetate, 100 mM sodium acetate, 0.25 mg/mL BSA (New England Biolabs), 1 mM phosphoenolpyruvate (Sigma), 80 units/mL pyruvate kinase (Sigma), and 1 mM ATP]. Reactions conducted at low-salt conditions contained standard buffer without sodium acetate and only 2 mM magnesium acetate. Reactions were assembled on ice, initiated by adding ATP, and, unless otherwise indicated, performed at 30 °C for 30 min. Reactions were terminated by adding 5 μL of stop buffer [150 mM EDTA, 2% (wt/vol) SDS, 30% (vol/vol) glycerol, and 0.1% bromophenol blue] and 1 μL of Proteinase K (14–22 mg/mL; Roche) and were then incubated for 20 min at room temperature. When unlabeled DNA substrates were used, the reaction products were separated by a 1% agarose gel in the presence of 0.05 μg/mL ethidium bromide. The products of reactions with radiolabeled DNA substrates were analyzed as described previously (28). Closed circular ssDNA (as in Fig. S8) was incubated in reaction buffer with RPA for 5 min at room temperature before adding Exo1. MRX was incubated in reaction buffer containing RPA for 2 min at room temperature before the addition of the remaining proteins. All gels are presented as inverted images. Error bars represent SE from two to four independent experiments.
Supplementary Material
Acknowledgments
We thank Tanya Paull, Michael Liskay, Patrick Sung, and Jody Plank for constructs and reagents and members of the S.C.K. laboratory and Massimo Lopes (University of Zurich) for discussions and helpful comments on the manuscript. This work was funded by National Institutes of Health Grant GM62653 (to S.C.K.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1305166110/-/DCSupplemental.
References
- 1.Aboussekhra A, Chanet R, Adjiri A, Fabre F. Semidominant suppressors of Srs2 helicase mutations of Saccharomyces cerevisiae map in the RAD51 gene, whose sequence predicts a protein with similarities to procaryotic RecA proteins. Mol Cell Biol. 1992;12(7):3224–3234. doi: 10.1128/mcb.12.7.3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malone RE, Esposito RE. The RAD52 gene is required for homothallic interconversion of mating types and spontaneous mitotic recombination in yeast. Proc Natl Acad Sci USA. 1980;77(1):503–507. doi: 10.1073/pnas.77.1.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiffenbach B, Haber JE. Homothallic mating type switching generates lethal chromosome breaks in rad52 strains of Saccharomyces cerevisiae. Mol Cell Biol. 1981;1(6):522–534. doi: 10.1128/mcb.1.6.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lettier G, et al. The role of DNA double-strand breaks in spontaneous homologous recombination in S. cerevisiae. PLoS Genet. 2006;2(11):e194. doi: 10.1371/journal.pgen.0020194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kramer KM, Haber JE. New telomeres in yeast are initiated with a highly selected subset of TG1-3 repeats. Genes Dev. 1993;7(12A):2345–2356. doi: 10.1101/gad.7.12a.2345. [DOI] [PubMed] [Google Scholar]
- 6.Sandell LL, Zakian VA. Loss of a yeast telomere: Arrest, recovery, and chromosome loss. Cell. 1993;75(4):729–739. doi: 10.1016/0092-8674(93)90493-a. [DOI] [PubMed] [Google Scholar]
- 7.Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009;417(3):639–650. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- 9.Huertas P. DNA resection in eukaryotes: Deciding how to fix the break. Nat Struct Mol Biol. 2010;17(1):11–16. doi: 10.1038/nsmb.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mimitou EP, Symington LS. Nucleases and helicases take center stage in homologous recombination. Trends Biochem Sci. 2009;34(5):264–272. doi: 10.1016/j.tibs.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 11.Mimitou EP, Symington LS. DNA end resection: Many nucleases make light work. DNA Repair. 2009;8(9):983–995. doi: 10.1016/j.dnarep.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134(6):981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455(7214):770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lengsfeld BM, Rattray AJ, Bhaskara V, Ghirlando R, Paull TT. Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol Cell. 2007;28(4):638–651. doi: 10.1016/j.molcel.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paull TT. Making the best of the loose ends: Mre11/Rad50 complexes and Sae2 promote DNA double-strand break resection. DNA Repair. 2010;9(12):1283–1291. doi: 10.1016/j.dnarep.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung WH, Zhu Z, Papusha A, Malkova A, Ira G. Defective resection at DNA double-strand breaks leads to de novo telomere formation and enhances gene targeting. PLoS Genet. 2010;6(5):e1000948. doi: 10.1371/journal.pgen.1000948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cejka P, et al. DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature. 2010;467(7311):112–116. doi: 10.1038/nature09355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niu H, et al. Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature. 2010;467(7311):108–111. doi: 10.1038/nature09318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nicolette ML, et al. Mre11-Rad50-Xrs2 and Sae2 promote 5′ strand resection of DNA double-strand breaks. Nat Struct Mol Biol. 2010;17(12):1478–1485. doi: 10.1038/nsmb.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dillingham MS, Spies M, Kowalczykowski SC. RecBCD enzyme is a bipolar DNA helicase. Nature. 2003;423(6942):893–897. doi: 10.1038/nature01673. [DOI] [PubMed] [Google Scholar]
- 21.Tran PT, Erdeniz N, Dudley S, Liskay RM. Characterization of nuclease-dependent functions of Exo1p in Saccharomyces cerevisiae. DNA Repair. 2002;1(11):895–912. doi: 10.1016/s1568-7864(02)00114-3. [DOI] [PubMed] [Google Scholar]
- 22.Fiorentini P, Huang KN, Tishkoff DX, Kolodner RD, Symington LS. Exonuclease I of Saccharomyces cerevisiae functions in mitotic recombination in vivo and in vitro. Mol Cell Biol. 1997;17(5):2764–2773. doi: 10.1128/mcb.17.5.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tishkoff DX, et al. Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proc Natl Acad Sci USA. 1997;94(14):7487–7492. doi: 10.1073/pnas.94.14.7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gravel S, Chapman JR, Magill C, Jackson SP. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22(20):2767–2772. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P, Kowalczykowski SC. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci USA. 2008;105(44):16906–16911. doi: 10.1073/pnas.0809380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nimonkar AV, et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011;25(4):350–362. doi: 10.1101/gad.2003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Modrich P. Mechanisms in eukaryotic mismatch repair. J Biol Chem. 2006;281(41):30305–30309. doi: 10.1074/jbc.R600022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cejka P, Kowalczykowski SC. The full-length Saccharomyces cerevisiae Sgs1 protein is a vigorous DNA helicase that preferentially unwinds holliday junctions. J Biol Chem. 2010;285(11):8290–8301. doi: 10.1074/jbc.M109.083196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mullen JR, Nallaseth FS, Lan YQ, Slagle CE, Brill SJ. Yeast Rmi1/Nce4 controls genome stability as a subunit of the Sgs1-Top3 complex. Mol Cell Biol. 2005;25(11):4476–4487. doi: 10.1128/MCB.25.11.4476-4487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Atkinson J, McGlynn P. Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 2009;37(11):3475–3492. doi: 10.1093/nar/gkp244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen X, et al. Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent Dna2 phosphorylation. Nat Struct Mol Biol. 2011;18(9):1015–1019. doi: 10.1038/nsmb.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 33.Harmon FG, DiGate RJ, Kowalczykowski SC. RecQ helicase and topoisomerase III comprise a novel DNA strand passage function: A conserved mechanism for control of DNA recombination. Mol Cell. 1999;3(5):611–620. doi: 10.1016/s1097-2765(00)80354-8. [DOI] [PubMed] [Google Scholar]
- 34.Kantake N, Sugiyama T, Kolodner RD, Kowalczykowski SC. The recombination-deficient mutant RPA (rfa1-t11) is displaced slowly from single-stranded DNA by Rad51 protein. J Biol Chem. 2003;278(26):23410–23417. doi: 10.1074/jbc.M302995200. [DOI] [PubMed] [Google Scholar]
- 35.Cejka P, Plank JL, Bachrati CZ, Hickson ID, Kowalczykowski SC. Rmi1 stimulates decatenation of double Holliday junctions during dissolution by Sgs1-Top3. Nat Struct Mol Biol. 2010;17(11):1377–1382. doi: 10.1038/nsmb.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jensen RB, Carreira A, Kowalczykowski SC. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. 2010;467(7316):678–683. doi: 10.1038/nature09399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsieh TS, Plank JL. Reverse gyrase functions as a DNA renaturase: Annealing of complementary single-stranded circles and positive supercoiling of a bubble substrate. J Biol Chem. 2006;281(9):5640–5647. doi: 10.1074/jbc.M513252200. [DOI] [PubMed] [Google Scholar]
- 38.Plank JL, Hsieh TS. A novel, topologically constrained DNA molecule containing a double Holliday junction: Design, synthesis, and initial biochemical characterization. J Biol Chem. 2006;281(25):17510–17516. doi: 10.1074/jbc.M602933200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.