

Abstract
Myriad new applications of proteases would be enabled by an ability to fine-tune substrate specificity and activity. Herein we present a general strategy for engineering protease selectivity and activity by capitalizing on sequestration of the protease to be engineered within the yeast endoplasmic reticulum (ER). A substrate fusion protein composed of yeast adhesion receptor subunit Aga2, selection and counterselection substrate sequences, multiple intervening epitope tag sequences, and a C-terminal ER retention sequence is coexpressed with a protease library. Cleavage of the substrate fusion protein by the protease eliminates the ER retention sequence, facilitating transport to the yeast surface. Yeast cells that display Aga2 fusions in which only the selection substrate is cleaved are isolated by multicolor FACS with fluorescently labeled antiepitope tag antibodies. Using this system, the Tobacco Etch Virus protease (TEV-P), which strongly prefers Gln at P1 of its canonical ENLYFQ↓S substrate, was engineered to recognize selectively Glu or His at P1. Kinetic analysis indicated an overall 5,000-fold and 1,100-fold change in selectivity, respectively, for the Glu- and His-specific TEV variants, both of which retained high catalytic turnover. Human granzyme K and the hepatitis C virus protease were also shown to be amenable to this unique approach. Further, by adjusting the signaling strategy to identify phosphorylated as opposed to cleaved sequences, this unique system was shown to be compatible with the human Abelson tyrosine kinase.
Keywords: directed evolution, method, protein engineering
More than 600 proteases have been annotated so far, constituting the largest enzyme family in the human genome (1–3). Because of their unique ability to catalyze the hydrolysis of peptide bonds and thus activate or inactivate proteins, proteases have the potential to be used in a number of applications (4–6). For example, recombinant tissue plasminogen activator, thrombin, Factor VII, and Factor IX are approved drugs for the therapeutic modulation of thrombosis and hemostasis (7–9). Additionally, proteases find numerous applications as reagents in biotechnology, ranging from analytical to preparative biochemistry (6, 10, 11).
The list of potential practical applications of proteases can be greatly expanded, especially for therapeutic applications, once their substrate specificities and catalytic activities can be engineered as required for specific uses. Engineered proteases displaying some degree of novel specificity have been developed in a few instances either via structure-guided mutagenesis or through directed evolution (12–17). In general, though, it has proved difficult to use rational design to generate highly active proteases with a desired new substrate selectivity, as mutations in one subsite typically disrupt the structure of neighboring subsites or of residues important for catalysis. Similarly, early attempts at engineering proteases by directed evolution generally led to enzyme variants displaying relaxed, rather than truly altered, specificity (18, 19). The feat of generating highly active protease variants with new and specific substrate preferences has until now been accomplished only for the Escherichia coli outer membrane protease T (OmpT) by using a directed evolution strategy involving the electrostatic retention of fluorescent substrate cleavage products on the bacterial surface, thereby enabling multicolor FACS (15, 20–22). Unfortunately, this strategy is limited to the few bacterial enzymes that can be displayed in an active form only on the outer membrane of E. coli (23, 24).
Here we report the development of a highly versatile and general eukaryotic system for the quantitative single-cell level detection of proteolytic activity that can be exploited for the high-throughput directed evolution of substrate selectivity and activity. Briefly, a multifunctional substrate fusion polypeptide is generated with a desired protease target sequence as well as counterselection sequence(s), all of which are flanked by different antibody epitope tags (Fig. 1A). The substrate also contains an N-terminal yeast adhesion receptor subunit Aga2 domain for surface display and a C-terminal endoplasmic reticulum (ER) retention sequence (FEHDEL). The protease being engineered is also directed to the ER, where it comes in close contact with the substrate fusion polypeptide. Proteolysis of the target and/or the counterselection sequences results in the removal of respective epitope tags as well as the C-terminal ER retention signal. The remaining (N-terminal portion) is displayed on the yeast surface via the Aga2 moiety and then labeled with fluorescently conjugated antiepitope tag antibodies. Cells exhibiting a characteristic fluorescence profile reflective of selective proteolysis only at the desired substrate sequence are isolated by multicolor FACS. Because the proteolytic processing occurs within the relatively confined space of the ER, we have termed this technique yeast ER sequestration screening (YESS) (Fig. 1B).
Fig. 1.

Yeast endoplasmic reticulum sequestration screening (YESS) system. (A) Salient features of the substrate and protease fusion genes used in the YESS system. (B) Schematic showing the rationale for the YESS system. Sc, counter selection substrate; Ss, selection substrate; ERS, ER retention sequence; Aga1 and Aga2, subunits of the yeast adhesion receptor.
Tobacco Etch Virus protease is a cysteine endopeptidase encoded by the Nuclear Inclusion a (NIa) gene of the tobacco etch virus. It displays stringent substrate specificity and is widely used for biotechnology research, particularly for the processing of fusion proteins (11), and it has been engineered for the activation of modified proenzymes (6, 10, 25). Because the original wild-type protease undergoes strong autolysis, a well-characterized variant containing a S219P mutation to significantly increase its stability was used in our research, which is annotated as Tobacco Etch Virus protease (TEV-P) (26). TEV-P recognizes the peptide sequence ENLYFQ↓S/G (27), where Q and S/G correspond to the P1 and P1′ residues, respectively. Even though cleavage sequence mapping studies using peptide combinatorial libraries have shown that the highest stringency was seen for Gln at P1 and that the P2, P4, and P5 positions can be occupied by a number of amino acids, no adventitious cleavage of full-length proteins at sites other than the canonical ENLYFQ↓S motif have been reported (28–30). We used the YESS system for the directed evolution of a TEV-P mutant library against a library of substrate constructs containing ENLYFX↓S, where X is any amino acid. The canonical ENLYFQ↓S sequence preferred by the wild-type enzyme was used as the counterselection substrate. TEV-P variants that can recognize selectively 6 different amino acids at the P1 position were isolated. Two variants, specific for Glu and His at P1, respectively, were subjected to random mutagenesis and screening by YESS to select for higher catalytic activity. In this manner we isolated highly active TEV-P variants displaying 5,000-fold and 1,100-fold change in catalytic selectivity for Glu or His P1 residue, respectively, relative to Gln at P1. In addition, a TEV-P variant exhibiting approximately 4-fold higher proteolytic activity toward its ENLYFQ↓S substrate was obtained using the YESS system. The YESS system was also shown to be compatible with another viral protease [the hepatitis C virus (HCV) protease] as well as with the human protease granzyme K (GrK). The YESS system was also found to work with the human Abelson tyrosine kinase (AblTK) after adjusting the signaling strategy to identify phosphorylated as opposed to cleaved sequences on the yeast surface.
Results
Quantitative Detection of Protease Activity at the Single-Cell Level, Using the YESS System.
In the YESS system (Fig. 1), a substrate fusion construct is used that contains a selection proteolysis substrate sequence as well as one or more counterselection proteolysis substrate sequences, flanked by different antibody epitope tags, all of which are fused to the Aga2 protein to enable display on the surface of Saccharomyces cerevisae. In this format (Fig. 1A), the one or more counterselection substrate sequences are placed N-terminal to the desired selection substrate sequence. A key feature of the YESS system is the fusion of a C-terminal ER retention sequence (FEHDEL) on the substrate and protease constructs to increase their respective ER residence times (31, 32). The bidirectional galactose (GAL) induced GAL1-GAL10 hybrid promoter, in which the GAL1 promoter has a similar individual promoter strength to that of the GAL10 promoter, is used to drive relatively high-level expression of both the protease and the substrate constructs. If the protease cleaves the substrate construct within the counterselection and/or selection substrate sequences, any epitope tag(s) C-terminal to the cleavage site, as well as the ER retention sequence, are removed. Following transit through the ER, the processed substrate fusion polypeptide is anchored onto the surface of S. cerevisae via the Aga2 moiety (33). The cells are probed with phycoerythrin (PE)-labeled anti-FLAG and FITC-labeled anti-6×His antibodies. Cells exhibiting relatively high fluorescence in both the PE and the FITC channels, or little fluorescence with either fluorophore, are assumed to indicate either no cleavage or cleavage at the undesired counterselection substrate sequence, respectively, so they are discarded. Cells exhibiting relatively high PE fluorescence, but little or no FITC fluorescence, are assumed to indicate specific cleavage at only the desired new substrate sequence and are isolated. In this way, rare cells that harbor a protease capable of specifically cleaving at only the desired new sequence are enriched. Therefore, the YESS system provides a means to address and overcome a major issue in protease engineering, and in directed evolution in general, which is that overwhelmingly, mutations that increase the catalytic activity toward a desired substrate also result in relaxed specificity or higher catalytic promiscuity (34, 35). The protease itself can be thought of as an effective counterselection substrate in the sense that any protease variant with specificity relaxed to the point that it efficiently cleaves itself will not exhibit a positive signal.
The ER retention sequence plays an important role in modulating the sensitivity and dynamic range of the YESS system. In our time-course experiments, two ER retention sequences, FEHDEL and KDEL, were evaluated, in which FEHDEL dramatically retains the protein substrate in the yeast ER (Fig. S1). Expression of both the protease and the substrate constructs with the ER retention signal retards their release from the ER, thus increasing the time in which they have an opportunity to react. In the absence of the ER retention signal, the contact time as well as the protease concentration is decreased, allowing selection of enzymes that process the substrate construct with higher efficiency in later rounds of directed evolution (Fig. S2).
Importantly, the expression of both the protease and the substrate as separate fusions allows for at least three different types of experiments, using the YESS format. A single new substrate can be used as the selection substrate along with one or more counterselection substrates in the presence of a protease library to isolate a protease variant with a desired new sequence specificity. Alternatively, a single protease of interest can be used with a library of substrate sequences to profile protease cleavage positional specificity. Finally, a “library against library” approach can be used in which a library of proteases is expressed in conjunction with a library of substrates, potentially increasing the odds of identifying highly active/specific new engineered protease–substrate pairs.
To validate the YESS system, we elected to use the TEV-P as a model protease (Fig. 2). In this case, the substrate construct consisted of Aga2 fused at its C terminus to the HA epitope tag (for internal expression-level calibration), a flexible linker (GGGS)4, a counterselection peptide sequence [the canonical hepatitis C virus non-structural 4A/4B (NS4A/NS4B) protease (HCV-P) substrate DEMEECASHL], the FLAG epitope tag, the TEV-P preferred substrate peptide ENLYFQ↓S, the 6×His epitope tag, and finally the ER retention signal at the C terminus. Following induction of expression of the protease and substrate fusion constructs in media with galactose, the cells were incubated with the PE-labeled, anti-FLAG antibody as well as the FITC-labeled, anti-6×His antibody. When the TEV-P was not expressed, the cells were labeled with both antibodies and hence occupy the diagonal in the 2D FACS plot (Fig. 2 A and F). The presence of the TEV-P with a C-terminal ER retention sequence gave rise to a cell population exhibiting high PE but low FITC fluorescence, consistent with the expected selective cleavage at the ENLYFQ↓S sequence that results in loss of the C-terminal 6×His tag (Fig. 2B). Removal of the ER retention sequence from the C terminus of TEV-P or from both the TEV-P and the substrate construct gave rise to markedly higher FITC (6×His) fluorescence relative to that of the positive control (Fig. 2 C and D). An approximate 1:1 mixture of positive control cells with cells lacking the TEV-P gene showed a fluorescence profile identical to that of the sum of the respective single-cell populations (compare Fig. 2 A and B with E), indicating that any adventitious release of TEV-P in the culture supernatant does not lead to cleavage of the substrate construct in other cells. Further, TEV-P could not be detected in the growth medium by Western blotting presumably because, if present at all, its concentration must have been below the detection limit. After single-colony sequencing of the enriched cells, an enrichment factor of ∼600-fold was observed in a single round of YESS, using yeast cells coexpressing TEV-P and a substrate fusion polypeptide mixed with a 1,000-fold excess of cells either that lacked protease activity or in which the selection and counterselection substrate sequences were in the wrong slots (Fig. S3).
Fig. 2.

Two-color FACS analysis of cells with and without ER retention signals in the substrate fusion and protease. All cells were grown, induced, antibody labeled, and analyzed under the same conditions (details in SI Methods). (A) Cells expressing substrate fusion only (construct pESD-A in F); (B) cells coexpressing protease and substrate fusion both containing ER retention sequences (construct pESD-B in F); (C) cells as in B except that TEV-P lacks the ER retention sequence (construct pESD-C, in F); (D) cells as in B except that both TEV-P and the substrate fusion lack the ER retention sequences (construct pESD-D in F); (E) an approximate 1:1 mixture of cells from A and B above; (F) schematic of the constructs pESD-A, pESDE-B, pESD-C, and pESD-D. In the experiment constructs, the counterselection gene encodes the substrate of HCV protease (DEMEECASHL), and the selection substrate gene encodes the substrate of TEV-P (ENLYFQS). pESD, Yeast Epitope tagging vector for Suface Display; Ep, ER retention sequence at C terminal of protease; Es, ER retention sequence at C terminal of substrate.
Engineering Unique TEV-P Variants.
TEV-P displays a >500-fold preference for Gln at the P1 position of the preferred ENLYFQ↓S substrate sequence. We sought to investigate whether the YESS approach could be used to engineer the S1 subsite of TEV-P so that it could accept other P1 residues with high catalytic activity and specificity. To this end, the four residues of the TEV-P S1 pocket (T146, D148, H167, and S170; Fig. 3G) were subjected to NNS saturation mutagenesis (N is any nucleotide and S equals G/C) and screened against a library of substrate sequences in which the P1 position was also randomized. The ENLYFQS sequence was used in the counterselection slot of the substrate fusion polypeptide. As a prelude to library screening, a plasmid (pESD-L, Table S1) containing the P1 substrate construct library but lacking the TEV-P protease was constructed. Following transformation, ∼107 yeast cells were labeled with anti-6×His-FITC antibody and the top 3% of events displaying the highest FITC fluorescence were collected (Fig. S4). This step effectively removed any stop codon or frame-shift mutants from the substrate construct library that would have given false positive signals for cleavage during library screening. In addition, any substrate that is cleaved by an endogenous protease can be eliminated through this step. Plasmid DNA was extracted from the collected cells, linearized, and cotransformed into yeast with linear DNA encoding the TEV-P S1 saturation library (pESD-M, Table S1). Homologous recombination of the substrate construct and TEV-P S1 saturation libraries in S. cerevisae strain EBY100 cells resulted in 3.3 × 107 transformants. Three consecutive rounds of FACS sorting (Fig. 3 A–D) for high PE and low FITC signal intensity were then carried out.
Fig. 3.

FACS analysis of TEV-P S1 subsite library, using the YESS system. (A) Two-color FACS analysis of the library cells. (B–D) Two-color FACS of cells after the first, second, and third round of sorting. (E) The TEV-PE3 variant. (F) The TEV-PH7 variant. (G) Molecular image of the S1 pocket of the wild-type TEV protease and the amino acid substitutions introduced in the TEV-PE10 variant. The P1 residue (Gln, red) of the substrate peptide (pink) interacts with the TEV protease S1 pocket residues (emphasized in rectangular boxes) through the hydrogen bonds (blue dotted line) and the hydrophobic interactions (orange dotted line). In the TEV-PE10, mutations that are close to the new S1 pocket (P1 residue is replaced with Glu, cyan) were annotated (purple in TEV-P and green in TEV-PE10). The image was generated on the basis of the Protein Data Bank file 1LVB, amino acid substitutions were added using PyMol’s mutagenesis function, and no computer simulation was applied.
Sequencing of 50 of the selected clones led to the identification of 35 different TEV-P variant–substrate combinations that contained Pro, Thr, Asn, Leu, Glu, or His at P1 (Fig. S5 and Table S2). Notably, no TEV-P variant–substrate combinations encoding the wild-type preferred sequence ENLYFQ↓S were isolated, highlighting the utility of the counterselection substrate. The TEV-P variants PE3 (P1 = E) (Fig. 3E) and PH7 (P1 = H) (Fig. 3F) displayed the highest PE/FITC fluorescence ratio by FACS and were subjected to further optimization by directed evolution. In particular, the PE3 and PH7 TEV-P genes were subjected to random mutagenesis by error-prone PCR (1.5–3.0% error rate) using the designed promers (Table S3). To increase the stringency of the sorting, the ER retention sequence was removed from the protease, thereby limiting contact time and the protease concentration in the ER in an effort to identify the most active variants in the libraries. Following homologous recombination in yeast, 5 × 107 and 2 × 107 cells for the TEV-PE3 and TEV-PH7 libraries, respectively, were subjected to five rounds of FACS sorting (Fig. S6) and 17 different TEV-P clones were obtained (Table S2). FACS analysis of single clones identified the TEV-PE10 and TEV-PH21 clones as displaying the highest PE vs. FITC fluorescence (Fig. S5).
The TEV-PE10 and TEV-PH21 variants, each with a 6×His N-terminal peptide, were fused to maltose binding protein (MBP), to increase their solubility in E. coli. The sequences ENLYFES or ENLYFHS, which comprise the respective selection substrates, were inserted between MBP and the 6×His-TEV-P moiety so that the latter could be released by autocatalytic cleavage and purified by immobilized metal ion affinity chromatography (IMAC) (Fig. S7 A and B). Enzyme kinetics for purified protease variants were determined with peptide substrates encoding Gln, Glu, or His as the P1 substrate residue, as appropriate (Table 1 and Fig. S8). The kcat/KM value determined for TEV-P reacting with its preferred TENLYFQSGTRRW substrate with its cleavage after Q (underlined) is 1.20 ± 0.09 mM−1⋅s−1, which is 375-fold and 150-fold greater than the values determined for TEV-P using TENLYFESGTRRW (3.14 ± 0.45 × 10−3 mM−1⋅s−1) and TENLYFHSGTRRW (7.55 ± 0.68 × 10−3 mM−1⋅s−1), respectively. The TEV-PE10 variant exhibited a 13-fold higher kcat/KM value (2.06 ± 0.46 mM−1⋅s−1) for TENLYFESGTRRW vs. TENLYFQSGTRRW (0.16 ± 0.02 mM−1⋅s−1), resulting in a 5,000-fold reversal of substrate specificity compared with TEV-P. Similarly, TEV-PH21 exhibited a 7-fold higher kcat/KM value for TENLYFHSGTRRW (0.15 ± 0.02 mM−1⋅s−1) vs. TENLYFQSGTRRW (2.07 ± 0.13 × 10−2 mM−1⋅s−1), a 1,100-fold reversal of substrate specificity compared with TEV-P. To assess the activities of the TEV-P, TEV-PE10, and TEV-PH21 variants with protein (as opposed to peptide) substrates, three different MBP-GST protein fusions were created containing the sequences ENLYFQS (MBP-ENLYFQS-GST), ENLYFES (MBP-ENLYFES-GST), and ENLYFHS (MBP-ENLYFHS-GST), within a linker inserted between the MBP and the GST (Fig. S7 A and B). Under conditions where the TEV-P cleaved 95% of the MBP-ENLYFQS-GST fusion, ∼3% cleavage was seen with either the MBP-ENLYFES-GST or the MBP-ENLYFHS-GST fusion protein substrates (Fig. 4). TEV-PE10 caused more than 99% cleavage of the MBP-ENLYFES-GST construct and TEV-PH21 gave close to 50% cleavage of the MBP-ENLYFHS-GST construct under these conditions. Importantly, in the latter case, longer incubation led to complete cleavage. In addition, after 1 h incubation at 30 °C, TEV-PE10 presented close to 60% and 5% cleavage against protein substrates MBP-ENLYFQS-GST and MBP-ENLYFHS-GST, respectively, whereas TEV-PE21 presented close to 10% and 3% cleavage against protein substrates MBP-ENLYFQS-GST and MBP-ENLYFES-GST, respectively (Fig. S7C). The TEV-PE10 and TEV-PH21 variants exhibited a pH dependence that is qualitatively similar to that of the parental enzyme, exhibiting the highest activity at pH 8.0 and slightly decreased activity at pH 7.2 and pH 6.5 (Fig. S7 C–E).
Table 1.
Michaelis–Menten kinetics of the TEV-P and selected variants with peptide substrates
| Enzyme | Mutations | Substrate | KM, mM | kcat, s−1 | kcat/KM, mM−1⋅s−1 |
| TEV-P | S219P | TENLYFQSGTRRW | 0.11 ± 0.02 | 0.13 ± 0.01 | 1.20 ± 0.09 |
| TEV-P | S219P | TENLYFESGTRRW | 1.93 ± 0.25 | 6.09 ± 0.3 × 10−3 | 3.14 ± 0.45 × 10−3 |
| TEV-P | S219P | TENLYFHSGTRRW | 0.64 ± 0.10 | 4.93 ± 0.20 × 10−3 | 7.55 ± 0.68 × 10−3 |
| TEV-PE10 | S120R, D148R, T173A, N177K, M218I, S219P | TENLYFQSGTRRW | 0.12 ± 0.03 | 1.94 ± 0.1 × 10−2 | 0.16 ± 0.02 |
| TEV-PE10 | TENLYFESGTRRW | 1.28 ± 0.18 × 10−2 | 2.55 ± 0.08 × 10−2 | 2.06 ± 0.46 | |
| TEV-PH21 | T17A, T146A, D148P, S153C, S168T, S170A, T173A, S219P | TENLYFQSGTRRW | 0.82 ± 0.10 | 1.71 ± 0.06 × 10−2 | 2.07 ± 0.13 × 10−2 |
| TEV-PH21 | TENLYFHSGTRRW | 0.25 ± 0.02 | 3.75 ± 0.08 × 10−2 | 0.15 ± 0.02 | |
| TEV-Fast | G79E, T173A, S219V | TENLYFQSGTRRW | 6.50 ± 0.80 × 10−2 | 0.30 ± 0.02 | 4.61 ± 0.11 |
Fig. 4.

Digestion of protein fusion substrates by engineered TEV-P variants. Reactions were incubated at 30 °C, pH 8.0, for 1 h with 5 µg protein substrate mixed with or without 0.1 µg protease in 20 µL reaction buffer. Lane 1, molecular mass ladders; lane 2, the MBP-ENLYFQS-GST substrate only; lane 3, the MBP-ENLYFES-GST substrate only; lane 4, the MBP-ENLYFHS-GST substrate only; lane 5, the MBP-ENLYFQS-GST substrate incubated with the TEV-P; lane 6, the MBP-ENLYFES-GST substrate incubated with the TEV-P; lane 7, the MBP-ENLYFHS-GST substrate incubated with the TEV-P; lane 8, the MBP-ENLYFES-GST substrate incubated with the TEV-PE10; lane 9, the MBP-ENLYFHS-GST substrate incubated with the TEV-PH21; lane 10, the MBP-ENLYFHS-GST substrate incubated with the TEV-PH21 for 3 h.
In complementary studies, we demonstrated that the YESS system could be used for the engineering of TEV-P having higher catalytic activity toward its native preferred peptide substrate, ENLYFQ↓S. To increase the dynamic range of the YESS assay, the ER-retention sequences were omitted from both the protease and the substrate fusion construct. By decreasing the amount of time the protease and substrate have to interact in the ER, this more stringent version of YESS should favor the isolation of more catalytically efficient protease variants. Following random mutagenesis starting with a previously reported TEV-P variant that contains the S219V mutation (26), and five rounds of FACS enrichment (Fig. S9B), a TEV-P variant (TEV-Fast) containing three amino acid substitutions (G79E/T173A/S219V) and displaying high PE fluorescence but little FITC fluorescence was isolated and characterized (Fig. S9 C and D). TEV-Fast cleaved the ENLYFQ↓S peptide with a kcat/KM = 4.61 ± 0.11 mM−1⋅s−1, a value approximately fourfold greater relative to that of the TEV-P (Table 1 and Fig. S9E), and also displayed higher proteolytic efficiency in the cleavage of fusion proteins (Fig. S9F). The Val mutation at position 219 had been previously identified as increasing stability of TEV-P, which cleaves the TENLYFQSGTRRW substrate with a kcat/KM of 2.96 ± 0.23 mM−1.s−1.
Using Other Enzymes with the YESS System.
To assess the generality of the YESS system for other proteases, analogous constructs were created in which the HCV-P and the human GrK protease were used in conjunction with their preferred substrate sequences. As seen in Fig. 5 A and B, yeast cells expressing the HCV-P and GrK protease with their preferred substrates displayed relatively similar PE but low FITC signals by FACS compared with the controls lacking proteases. Thus, it appears that the YESS system will be generally applicable to a variety of different proteases.
Fig. 5.
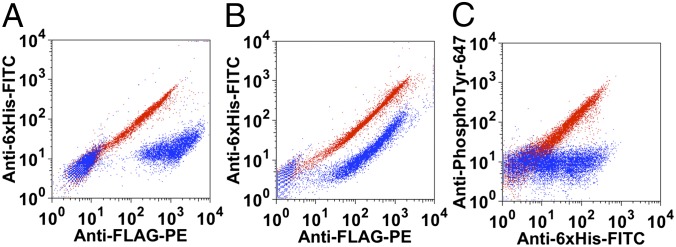
Detection of human GrK HCV protease, and human AblTK by YESS. (A) HCV protease was assayed in the YESS system. Red data: negative control consisting of the HCV protease preferred substrate construct with no HCV protease (construct pESD-N, Table S1). Blue data: HCV protease expressed along with its preferred substrate construct (construct pESD-O, Table S1). (B) Human GrK was assayed in the YESS system. Red data: negative control consisting of the human GrK preferred substrate construct with no human GrK (construct pESD-P, Table S1). Blue data: human GrK expressed along with its preferred substrate construct (construct pESD-Q, Table S1). (C) Human AblTK was assayed in the YESS system. Red data: human AblTK expressed along with its preferred substrate construct (construct pESD-R, Table S1). Blue data: negative control consisting of the human AblTK preferred substrate construct with no human AblTK (construct pESD-S, Table S1).
Importantly, application of the YESS system is not limited to protease engineering. Initial experiments using the human AblTK indicated that the YESS system can be applied to kinases (Fig. 5C). In these experiments, yeast cells expressing the human AblTK with its preferred peptide sequence in the substrate construct were probed with an Alexa Fluor647-labeled antibody specific for phosphotyrosine. Cells expressing human AblTK displayed similar FITC (which controls for overall expression of substrate as before) but higher Alexa Fluor647 signals by FACS compared with the control cells lacking kinase, indicating substantial tyrosine phosphorylation of substrate by human AblTK in the YESS system.
Discussion
The YESS system was developed as a facile and general strategy for the engineering of protease substrate selectivity and catalytic activity that promises to open up a wide range of potential unique applications for proteases. YESS was designed to exploit the unique capabilities of yeast cells including the presence of the protein synthesis machinery required to express more complex mammalian proteases, a facile system for attaching reaction products to the outer surface, and the presence of cellular compartments, in particular the ER, which provides for the isolation of enzyme–substrate interactions away from cytosolic proteases or other interfering components present in the cellular milieu. The net result is a robust, quantitative readout of protease selectivity and activity at the single-cell level, enabling the discrimination of cells expressing enzymes with a desired catalytic activity (Fig. 1). A key feature of the YESS system is the ability to modulate the assay dynamic range, using the appropriate ER retention signals. The YESS constructs in which both the protease and the substrate fusion polypeptide contain ER retention sequences are particularly useful for detecting low catalytic activity events. Removal of one or both ER sequences progressively reduces the time during which the protease and enzyme–substrate fusion are colocalized, thus decreasing the contact time as well as the protease/substrate concentration to enable the selection of progressively faster enzymes (Fig. 2). To the best of our knowledge, such comprehensive and precise control over the dynamic range of a high-throughput enzyme library-screening assay is unique.
The YESS approach incorporates two additional powerful features for protease engineering. Studies from our laboratory and others have highlighted the importance of incorporating simultaneous counterselection substrates during the directed evolution of proteases to avoid isolating variants with relaxed specificity (14, 20, 36, 37). Any number of counterselection substrate sequences can be added to the YESS substrate fusion construct to facilitate the engineering of narrow selectivity enzymes. Additionally, because in the YESS system both the protease and the substrate fusion polypeptide are genetically encoded, either or both can be diversified at the same time to engineer proteases with new selectivities for one or multiple substrates. In this work, we used a “protease library on substrate fusion library” approach to comprehensively alter the P1 substrate specificity of the TEV-P. In this way, we show that combinatorial saturation of the residues that form the S1 subsite of TEV-P enables the isolation of enzyme variants capable of accepting 6 different amino acids other than Gln in P1. Notably, all these TEV-P mutants display selectivity for a new amino acid over the Gln that is overwhelmingly preferred by the wild-type enzyme at that position.
Following the initial FACS isolation of 35 different TEV-P variant–substrate pairs, the tunability of the YESS assay was used to advantage while screening the second-generation error-prone PCR libraries constructed from the two most promising isolated clones, one specific for Glu (TEV-PE3) and the other specific for His (TEV-PH7) at P1. In particular, by removing the ER retention sequence from the protease and/or the substrate, ER residence time will be reduced. In this way, available concentrations in the ER will be lowered, providing more stringent screening conditions. Thus, by removing the ER retention sequence from the C terminus of the protease, only variants with relatively high levels of activity produced a significant FACS signal. For example, the kcat/KM values of TEV-PE3 against substrates TENLYFQSGTRRW and TENLYFESGTRRW are 0.22 ± 0.02 mM−1⋅s−1 and 0.24 ± 0.02 mM−1⋅s−1, respectively. Once again, wild-type–preferred Gln at P1 was used as counterselection in the second library sorting. The result was two clones, TEV-PE10 and TEV-PH21, displaying substrate specificity reversals of 5,000-fold and 1,100-fold, respectively, along with relatively high overall catalytic activity. In fact, the TEV-PE10 variant displayed a kcat/KM that was roughly 2-fold higher than that of even TEV-P reacting with its preferred substrate, verifying that new specificity did not come at the expense of overall catalytic activity. As further evidence of successful protease engineering, TEV-PE10 and TEV-PH21 were shown to be efficient in the processing of GST-MBP fusions containing their preferred recognition sequences, namely MBP-ENLYFES-GST and MBP-ENLYFHS-GST, respectively (Fig. 4 and Fig. S7). Additionally, the YESS system was also applied to engineer enzymes possessing higher catalytic activity. The TEV-P was further engineered using the YESS system. The obtained TEV-Fast variant presented close to 4-fold higher overall proteolytic activity compared with the TEV-P (Fig. S9). Finally, the generality of the YESS approach was demonstrated by showing that human GrK, HCV protease, and human AblTK are also amenable to expression and quantitative assay, using the YESS system (Fig. 5).
It is tempting to speculate as to the origin of substrate specificity in the isolated enzyme variants (Fig. 3G). Molecular modeling of TEV-PE10 in comparison with wild-type TEV protease indicates that the negatively charged D148 in the TEV-P S1 subsite was mutated to the positively charged residue Arg, likely favoring interaction with the negatively charged Glu residue at P1. We note that another mutant with Glu specificity in P1 contained a Lys at the 148 position (Table S2). In addition to the D148R replacement, two other mutations, T173A and N177K, might be also involved in the recognition of Glu at P1 by TEV-PE10 (Fig. 3G). Likewise, in all mutants capable of accepting a His at P1 of the substrate, including TEV-PH21, residues T146 and D148 were replaced by a small amino acid (Ala, Cys, and Ser) or Pro, with the net effect of likely opening up the S1 site for the somewhat larger His residue. Although we did not have direct evidence of the specificity of these two evolved variants at residues flanking P1 in the substrate, we speculate that the TEV-PE10 and TEV-PE21 possess similar specificity to the TEV-P because of the lack of amino acid substitutions near the P1′, P3, and P6 binding subsites (27) in the variants. The ability to engineer human proteases is particularly attractive as it opens the possibility of cleaving and therefore deactivating disease target proteins in catalytic fashion.
Methods
Protocols for vector construction, library construction, yeast cell sorting, and protease characterization are described in SI Methods. All the constructs used in this study are listed in Table S1. The primers used for library construction in this study are listed in Table S3.
Supplementary Material
Acknowledgments
We thank Dr. Dane K. Wittrup (Massachusetts Institute of Technology) and Dr. Edward W. Marcotte (University of Texas at Austin) for the generous contribution of plasmids. This work was supported by the Clayton Foundation (B.L.I.) as well as by US National Institutes of Health Grants R01 GM065551 and R01 GM073089 (to B.L.I. and G.G.).
Footnotes
Conflict of interest statement: The authors have a filed patent application for the YESS system related to this paper.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1215994110/-/DCSupplemental.
References
- 1.Marnett AB, Craik CS. Papa’s got a brand new tag: Advances in identification of proteases and their substrates. Trends Biotechnol. 2005;23(2):59–64. doi: 10.1016/j.tibtech.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 2.Overall CM, Blobel CP. In search of partners: Linking extracellular proteases to substrates. Nat Rev Mol Cell Biol. 2007;8(3):245–257. doi: 10.1038/nrm2120. [DOI] [PubMed] [Google Scholar]
- 3.Schilling O, Overall CM. Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat Biotechnol. 2008;26(6):685–694. doi: 10.1038/nbt1408. [DOI] [PubMed] [Google Scholar]
- 4.Chanalia P, Gandhi D, Jodha D, Singh J. Applications of microbial proteases in pharmaceutical industry: An overview. Rev Med Microbiol. 2011;22(4):96–101. [Google Scholar]
- 5.Gupta R, Beg QK, Lorenz P. Bacterial alkaline proteases: Molecular approaches and industrial applications. Appl Microbiol Biotechnol. 2002;59(1):15–32. doi: 10.1007/s00253-002-0975-y. [DOI] [PubMed] [Google Scholar]
- 6.Wehr MC, et al. Monitoring regulated protein-protein interactions using split TEV. Nat Methods. 2006;3(12):985–993. doi: 10.1038/nmeth967. [DOI] [PubMed] [Google Scholar]
- 7.Collen D, Lijnen HR. Basic and clinical aspects of fibrinolysis and thrombolysis. Blood. 1991;78(12):3114–3124. [PubMed] [Google Scholar]
- 8.Craik CS, Page MJ, Madison EL. Proteases as therapeutics. Biochem J. 2011;435(1):1–16. doi: 10.1042/BJ20100965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drag M, Salvesen GS. Emerging principles in protease-based drug discovery. Nat Rev Drug Discov. 2010;9(9):690–701. doi: 10.1038/nrd3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gray DC, Mahrus S, Wells JA. Activation of specific apoptotic caspases with an engineered small-molecule-activated protease. Cell. 2010;142(4):637–646. doi: 10.1016/j.cell.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waugh DS. An overview of enzymatic reagents for the removal of affinity tags. Protein Expr Purif. 2011;80(2):283–293. doi: 10.1016/j.pep.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hedstrom L, Szilagyi L, Rutter WJ. Converting trypsin to chymotrypsin: The role of surface loops. Science. 1992;255(5049):1249–1253. doi: 10.1126/science.1546324. [DOI] [PubMed] [Google Scholar]
- 13.Lim EJ, et al. Swapping the substrate specificities of the neuropeptidases neurolysin and thimet oligopeptidase. J Biol Chem. 2007;282(13):9722–9732. doi: 10.1074/jbc.M609897200. [DOI] [PubMed] [Google Scholar]
- 14.Sellamuthu S, et al. Engineering of protease variants exhibiting altered substrate specificity. Biochem Biophys Res Commun. 2008;371(1):122–126. doi: 10.1016/j.bbrc.2008.04.026. [DOI] [PubMed] [Google Scholar]
- 15.Varadarajan N, Rodriguez S, Hwang BY, Georgiou G, Iverson BL. Highly active and selective endopeptidases with programmed substrate specificities. Nat Chem Biol. 2008;4(5):290–294. doi: 10.1038/nchembio.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verhoeven KD, Altstadt OC, Savinov SN. Intracellular detection and evolution of site-specific proteases using a genetic selection system. Appl Biochem Biotechnol. 2012;166(5):1340–1354. doi: 10.1007/s12010-011-9522-6. [DOI] [PubMed] [Google Scholar]
- 17.Villa JP, Bertenshaw GP, Bond JS. Critical amino acids in the active site of meprin metalloproteinases for substrate and peptide bond specificity. J Biol Chem. 2003;278(43):42545–42550. doi: 10.1074/jbc.M303718200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aharoni A, Amitai G, Bernath K, Magdassi S, Tawfik DS. High-throughput screening of enzyme libraries: Thiolactonases evolved by fluorescence-activated sorting of single cells in emulsion compartments. Chem Biol. 2005;12(12):1281–1289. doi: 10.1016/j.chembiol.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 19.Gould SM, Tawfik DS. Directed evolution of the promiscuous esterase activity of carbonic anhydrase II. Biochemistry. 2005;44(14):5444–5452. doi: 10.1021/bi0475471. [DOI] [PubMed] [Google Scholar]
- 20.Varadarajan N, Georgiou G, Iverson BL. An engineered protease that cleaves specifically after sulfated tyrosine. Angew Chem Int Ed Engl. 2008;47(41):7861–7863. doi: 10.1002/anie.200800736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varadarajan N, Pogson M, Georgiou G, Iverson BL. Proteases that can distinguish among different post-translational forms of tyrosine engineered using multicolor flow cytometry. J Am Chem Soc. 2009;131(50):18186–18190. doi: 10.1021/ja907803k. [DOI] [PubMed] [Google Scholar]
- 22.Yoo TH, Pogson M, Iverson BL, Georgiou G. Directed evolution of highly selective proteases by using a novel FACS-based screen that capitalizes on the p53 regulator MDM2. ChemBioChem. 2012;13(5):649–653. doi: 10.1002/cbic.201100718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pogson M, Georgiou G, Iverson BL. Engineering next generation proteases. Curr Opin Biotechnol. 2009;20(4):390–397. doi: 10.1016/j.copbio.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daugherty PS. Protein engineering with bacterial display. Curr Opin Struct Biol. 2007;17(4):474–480. doi: 10.1016/j.sbi.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 25.Stevens RC. Design of high-throughput methods of protein production for structural biology. Structure. 2000;8(9):R177–R185. doi: 10.1016/s0969-2126(00)00193-3. [DOI] [PubMed] [Google Scholar]
- 26.Kapust RB, et al. Tobacco etch virus protease: Mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14(12):993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 27.Phan J, et al. Structural basis for the substrate specificity of tobacco etch virus protease. J Biol Chem. 2002;277(52):50564–50572. doi: 10.1074/jbc.M207224200. [DOI] [PubMed] [Google Scholar]
- 28.Boulware KT, Jabaiah A, Daugherty PS. Evolutionary optimization of peptide substrates for proteases that exhibit rapid hydrolysis kinetics. Biotechnol Bioeng. 2010;106(3):339–346. doi: 10.1002/bit.22693. [DOI] [PubMed] [Google Scholar]
- 29.Dougherty WG, Carrington JC, Cary SM, Parks TD. Biochemical and mutational analysis of a plant virus polyprotein cleavage site. EMBO J. 1988;7(5):1281–1287. doi: 10.1002/j.1460-2075.1988.tb02942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kostallas G, Löfdahl PA, Samuelson P. Substrate profiling of tobacco etch virus protease using a novel fluorescence-assisted whole-cell assay. PLoS ONE. 2011;6(1):e16136. doi: 10.1371/journal.pone.0016136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pelham HR, Hardwick KG, Lewis MJ. Sorting of soluble ER proteins in yeast. EMBO J. 1988;7(6):1757–1762. doi: 10.1002/j.1460-2075.1988.tb03005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Semenza JC, Hardwick KG, Dean N, Pelham HR. ERD2, a yeast gene required for the receptor-mediated retrieval of luminal ER proteins from the secretory pathway. Cell. 1990;61(7):1349–1357. doi: 10.1016/0092-8674(90)90698-e. [DOI] [PubMed] [Google Scholar]
- 33.Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15(6):553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 34.Bloom JD, Arnold FH. In the light of directed evolution: Pathways of adaptive protein evolution. Proc Natl Acad Sci USA. 2009;106(Suppl 1):9995–10000. doi: 10.1073/pnas.0901522106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khersonsky O, Tawfik DS. Enzyme promiscuity: A mechanistic and evolutionary perspective. Annu Rev Biochem. 2010;79:471–505. doi: 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- 36.O’Loughlin TL, Greene DN, Matsumura I. Diversification and specialization of HIV protease function during in vitro evolution. Mol Biol Evol. 2006;23(4):764–772. doi: 10.1093/molbev/msj098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Varadarajan N, Gam J, Olsen MJ, Georgiou G, Iverson BL. Engineering of protease variants exhibiting high catalytic activity and exquisite substrate selectivity. Proc Natl Acad Sci USA. 2005;102(19):6855–6860. doi: 10.1073/pnas.0500063102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.