

Abstract
Progression of solid tumors to the metastatic stage is accountable for the majority of cancer-related deaths. Further understanding of the molecular mechanisms governing metastasis is essential for the development of antimetastatic regimens. Here, we aimed to identify Rac activators that could promote metastasis downstream of human epithelial growth factor receptor 2 (HER2). We investigated if Dedicator of Cytokinesis 1 (DOCK1), based on its evolutionarily conserved role in receptor tyrosine kinases (RTKs)-mediated Rac activation and cell invasion, could be a regulator of metastasis. We report that high expression of DOCK1 in HER2+ and basal breast cancer subtypes inversely correlates with human patients’ survival. Mechanistically, DOCK1 interacts with HER2 and promotes HER2-induced Rac activation and cell migration. To gain further insight, we developed a HER2 breast cancer mouse model with mammary-gland–specific inactivation of DOCK1. In this in vivo model, a significant decrease in tumor growth and metastasis in lungs was found in animals where DOCK1 is inactivated. Furthermore, we found that DOCK1 is required for maximal activation of two HER2 effectors, c-JUN and STAT3. Using an unbiased gene profiling approach, we identified a mammary tumor DOCK1-associated gene signature enriched for genes implicated in response to IFN type I. This analysis revealed a unique set of genes, including Receptor Transporter Protein 4 (RTP4) and STAT1, for which the expression levels can be used to independently predict breast cancer outcome in HER2+ patients. Our work demonstrates DOCK1–Rac signaling as an HER2 effector pathway essential for HER2-mediated breast cancer progression to metastasis and offers a therapeutic opportunity to limit the spread of metastatic breast cancers.
Keywords: ErbB2, DOCK180, RhoGEF, tumorigenesis
Despite breakthroughs in the treatment of breast cancer, the most prevalent cancer in women, progression of the disease remains an important cause of death. Virtually all fatalities can be attributed to complications due to the appearance of secondary tumors at distant sites. The identification and therapeutic targeting of proteins regulating the metastatic step is therefore a priority for improving the lifespan of afflicted patients (1). Two major breast cancer subtypes, basal-like and HER2+, are linked to aggressive and recurrent primary and metastatic tumors, and ultimately, to poor survival (2). HER2 is a member of the EGF receptor family of receptor tyrosine kinases (RTKs) also comprising HER1, HER3, and HER4 (3). Amplification of the HER2 locus, or aberrant expression of its protein product, is observed in nearly 20% of human breast cancers (4). Mouse models expressing various forms of HER2 in the mammary gland recapitulate most aspects of the human disease, including metastasis, and are powerful tools to gain insight into signaling pathways controlling tumorigenesis in vivo (5). Nonetheless, regulators of HER2-mediated metastasis remain poorly characterized.
Metastasis is a complex and deadly, yet inefficient, step in breast cancer that involves cancer cells leaving the primary tumor, entering blood vessels, and exiting the circulation to colonize foreign soils (6). Aggressive migration and invasion behaviors of breast cancer cells are presumed to be essential for metastatic dissemination. Rho GTPases are established as central signaling intermediates controlling the migration of cancer cells (6). Rac promotes mesenchymal cell movement (7), but also contributes to cell proliferation and survival. Whereas HER2 can activate Rac in cell lines (8), the exact guanine exchange factors (GEFs) responsible for its activation downstream of HER2 remain poorly defined in vivo. The Dedicator of Cytokinesis 1 (DOCK1) family GEFs are an important class of cytoskeletal regulators controlling cell migration (9). In oogenesis, the Drosophila ortholog Myoblast City acts downstream of the RTK PDGF/VEGF receptor to control the invasive migration of the border cell cluster (10). Studies in cell lines, including breast cancer, also established that mammalian DOCK1 is a regulator of cell migration and invasion downstream of integrin signaling (11). The DOCK1–Rac pathway was uncovered to promote glioblastoma cell migration driven by oncogenic RTKs including the PDGFR and the EGFR variant type III (12, 13). Both receptors orchestrate tyrosine phosphorylation of DOCK1 to increase its GEF activity. These data establish the DOCK1-Rac pathway as a potential drug target to limit the spread of brain cancer.
In search of RacGEFs promoting metastasis downstream of HER2, we hypothesized that DOCK1 could be such a regulator. We report that DOCK1 enters in a complex with HER2 to promote Rac activation and cell migration. Mammary-gland–specific inactivation of DOCK1 decreased tumor growth and metastasis to lungs. We also identified a DOCK1-associated gene signature that predicts overall survival in human breast cancer patients. Collectively, this work demonstrates DOCK1–Rac signaling as an HER2 effector pathway essential for breast cancer metastasis.
Results
Expression of DOCK1 Is Associated with an Adverse Clinical Outcome for HER2+ and Basal Breast Cancer Patients.
Immunohistochemistry (IHC) was performed on a panel of breast tumors to assess if DOCK1 is expressed in this disease. DOCK1 protein was detectable in the tumor epithelial cells of 144/145 samples (Fig. S1A). We examined the relationship between DOCK1 mRNA expression and the probability of survival of breast cancer patients according to clinical subtypes using a large set of microarray data linked to clinical outcome (n = 6,327, including 3,466 patients for whom survival data were available) (14). DOCK1 was broadly expressed but higher in estrogen receptor (ER)+/HER2− high proliferation (luminal B) subtype (Fig. S1B). As shown in Kaplan–Meier survival curves of patients classified based on DOCK1 expression, high levels of its expression was significantly associated with poor disease-free survival in HER2+ (*P = 0.048) and ER−/HER2− (*P = 0.03) subtypes (Fig. 1A); association with survival was not observed for the luminal subtypes (ER+/HER2− low and high proliferation tumors; Fig. S1C). These analyses indicated that DOCK1 expression is linked to survival of patients afflicted with aggressive breast cancers and provided the impetus for investigating the mechanism(s) whereby this GEF contributes to breast cancer.
Fig. 1.

DOCK1, a negative prognostic factor for human breast cancer survival, activates Rac and promotes cell migration downstream of HER2. (A) High levels of DOCK1 expression are associated with a poor prognostic for HER2+ and basal-like breast cancer patients. (B) DOCK1 is found in a complex with HER2 upon treatment of T47D cells with HRG (n = 3). (C) DOCK1 is phosphorylated on Y1811 in T47D breast cancer cells upon treatment with 20 ng/mL HRG for 15 min (n = 5). (D) DOCK1 is phosphorylated on Y1811 by SRC kinase. In vitro kinase assay was performed using recombinant SRC and purified GST or GST-DOCK11228–1865 proteins (n = 3). (E and F) Pharmacological inhibition of DOCK1 impairs HRG-mediated Rac activation and migration in T47D cells. (E) Cells were treated with 20 ng/mL HRG for 15 min in the presence of 100 μM CPYPP and active Rac was measured by precipitation with the purified p21-Binding Domain of PAK protein kinase expressed as a GST fusion (GST-PAK-PBD) (n = 5). (F) Migration of T47D cells toward 20 ng/mL HRG was measured in a Boyden chamber assay in the presence or absence of 100 μM CPYPP (n = 7). (G) Migration of T47D cells transfected with 100 µM NON-targeting or ON-target smart pool DOCK1 siRNA toward 20 ng/mL HRG was measured in Boyden chamber assay (n = 7). (H) Expression profiling uncovers DOCK1 as a highly expressed RacGEF in HER2-induced mouse mammary tumors. Data were expressed as mean ± SD (n = 4). One-way ANOVA followed by a Bonferroni test calculated the P values; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and **** P ≤ 0.0001.
DOCK1, a Target of HER2, Promotes Heregulin-Mediated Rac Activation.
Because DOCK1 binds RTKs and activate Rac during cell motility (10, 12, 13), we examined whether it can enter in a protein complex with HER2. In human ductal breast epithelial tumor cell line T47D, endogenous HER2 coimmunoprecipitated with DOCK1 only when cells were treated with Heregulin β1 (HRG) (Fig. 1B). We also expressed Flag-DOCK1 and an oncogenic and activated form of rat HER2 (NeuNT) and performed coimmunoprecipitation assays. A robust coprecipitation of NeuNT was detected in Flag-DOCK1 immunoprecipitates (Fig. S2A). Treatment of T47D cells with HRG, or overexpression of NeuNT, resulted in the phosphorylation of DOCK1 on a regulatory site, Y1811, known to promote its RacGEF activity (Fig. 1C and Fig. S2B) (12). Phosphorylation on Y1811 was not directly regulated by HER2 but instead mediated by SRC protein kinases (Fig. 1D and Fig. S2 C and D). HRG-mediated activation of HER2 is reported to activate Rac and increase cell motility (8). To examine whether DOCK1 can facilitate HRG-induced Rac activation and cell migration, its GEF activity was inhibited via a small molecule, 4-[3′-(2″-chlorophenyl)-2′-propen-1′-ylidene]-1-phenyl-3,5-pyrazolidinedione (CPYPP) (15). Treatment of T47D cells with HRG induced a ninefold increase in Rac activation that was blocked in the presence of the inhibitor (Fig. 1E). Whereas treatment of T47D cells with HRG induced cell migration, this effect was abrogated either in the presence of CPYPP or when DOCK1 expression levels were reduced using RNAi (Fig. 1 F and G). These results demonstrate that DOCK1 is activated and mediates Rac activation and migration downstream of HER2.
Quantitative Expression Profile of Rho GTPases and Their Regulators in Tumors.
Based on the clinical data and mechanistic insights above suggesting that DOCK1 is operating downstream of HER2, we aimed to investigate its function in an in vivo model of HER2 breast cancer. We first examined the major Rho GTPases, GEFs, GTPase Activating Proteins (GAPs), and guanine nucleotide dissociation inhibitors (GDIs) expressed in HER2 mouse breast tumors. mRNA deep sequencing was used to generate a quantitative transcriptome of mouse mammary tumor virus (MMTV)-Neu (variant NDL2-5) transgenic tumors (n = 4). The most expressed GEFs included LARG, DOCK9, DOCK8, TIM, α-PIX, p190RHOGEF, FARP1, and DOCK1 (Fig. S3A). α−PIX, DOCK1, β−PIX, TIAM1, and DOCK7 are the highest expressed RacGEFs (Fig. 1H). The partner of DOCK1, ELMO1, was highly expressed in comparison with ELMO2/3 in these tumors (Fig. S3B). Moreover, Rac1 was found to be the most expressed Rho GTPase in tumors (Fig. S3C). In term of negative regulators, RhoGDIs are minimally expressed (RhoGDIβ/γ; RhoGDIα was not detected) and the RhoGAP ARHGAP31/CDGAP stood out as the majorly expressed member (Fig. S3 D and E). These data provide a snapshot of the Rho regulation system and confirmed that DOCK1 is highly express in HER2 tumors.
DOCK1 Contributes to Tumor Growth.
To examine the role of DOCK1 in HER2-induced tumorigenesis in vivo, a conditional floxed (flx) mutant mouse line of DOCK1 was generated (Fig. S4 A–E). Before carrying out tumorigenesis studies, we investigated whether deletion of DOCK1 in mammary glands would have adverse effects on the development of the tissue. Whole-mount outgrowth analyses at 9, 12, and 15 wk established that DOCK1 is dispensable for mammary development (Fig. S4F). Additionally, MMTV-Cre+DOCK1flx/flx mice were capable of nursing their offsprings to maturity, suggesting that loss of DOCK1 does not impair mammary gland function.
To assess the role of DOCK1 in a HER2 model of breast cancer, Mouse Mammary Tumor Virus (MMTV)-NeuNDL2-5-Internal Ribosome Entry Site (IRES)-Cre (NIC) transgenics were intercrossed with DOCK1flx mice and tumor progression was examined in females. In this model, expression of NeuNDL2-5 and Cre recombinase are coupled and females develop ductal carcinoma in situ that progress to invasive carcinoma and lung metastasis (16). Three cohorts of mice were generated (NIC+DOCK1wt/wt, NIC+DOCK1wt/flx, and NIC+DOCK1flx/flx), and animals were monitored weekly for apparition of tumors that were next allowed to grow for 5 wk. Mice developed mammary tumors irrespective of the genotype and a small delay of tumor onset in NIC+DOCK1wt/flx and NIC+DOCK1flx/flx animals was observed (Fig. 2A). Histological analyses demonstrated that tumors from all genotypes displayed a solid adenocarcinoma appearance (Fig. 2B). We further confirmed that Cre-mediated recombination of the DOCK1 floxed allele was efficient in tumors (Fig. S4E). Furthermore, genetic ablation of DOCK1 in mammary tumors reduced the levels of DOCK1 protein as verified by Western blotting and IHC, whereas it had no impact on the expression of Neu (Fig. 2 C and D). Whereas mammary tumor initiation is normal in NIC+DOCK1flx/flx animals, they presented fewer tumor nodules as well as a significant decrease in their cumulative tumor burden after 5 wk (Fig. S5A and Fig. 2E). We observed fewer large tumors in DOCK1-null mammary glands, whereas the small and medium formed equally in all genotypes (Fig. S5B). A similar coverage of CD31+ blood vessels in DOCK1-deficient and wild-type NIC tumors was observed, ruling out a defect in angiogenesis to explain the growth defect (Fig. S5C). We next investigated an early time point of tumor onset by quantifying mammary intraepithelial neoplastic lesions (MINs) on inguinal mammary glands that did not develop tumors and noted a significant reduction in the DOCK1-deficient mammary glands (Fig. 2 F and G).
Fig. 2.
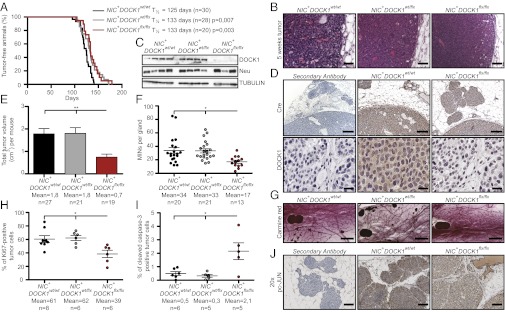
DOCK1 contributes to HER2 tumorigenesis in a mouse model of breast cancer. (A) Kaplan–Meier analysis of tumor onset. NIC+DOCK1wt/flx and NIC+DOCK1flx/flx mice display a minor delay in tumor onset. (B) H&E staining showing that all animals develop tumors with an adenocarcinoma histology. (Scale bar: 100 µm, 20×.) (C) Western blot analyses demonstrating the absence of DOCK1 and equal Neu expression in tumor lysates. (D) IHC analyses showing Cre (Upper) and DOCK1 (Lower) expression in NIC+DOCK1wt/wt and NIC+DOCK1flx/flx tumors. (Scale bar: Upper, 250 µm, 10×; Lower, 50 µm, 40×.) (E) DOCK1 contributes to HER2-induced tumor growth. Average cumulative tumor burden per animal (in cubic centimeters). (F) NIC+DOCK1flx/flx mice develop fewer mammary intraepithelial neoplasic lesions (MINs). Quantification of mammary MINs per tumor-free mammary gland 5 wk after tumor onset is presented. (G) Representative carmine red staining of mammary glands showing MINs in all genotypes. (Scale bar: 1 mm, 2.5×.) (H) DOCK1 promotes cell proliferation in HER2 tumors. Average percentage of Ki67-positive cells in tumors. (I) DOCK1 exerts an antiapoptotic role in HER2 tumors. Average percentage of activated caspase-3–positive cells in tumors. (J) Impaired c-JUN activation in DOCK1-null HER2 tumors. IHC analyses showing pc-JUN levels in MINs. (Scale bar: 100 µm, 20×.) One-way ANOVA followed by a Bonferroni test calculated the P values; *P ≤ 0.05 and **P ≤ 0.01.
The proliferative and apoptotic status of DOCK1-null mammary tumor cells was assessed to gain mechanistic insights into the tumor growth phenotype. A statistically significant decrease of Ki67-positive cells in the DOCK1flx/flx tumors was noted compared with heterozygote or wild-type tumors (Fig. 2H and Fig. S5D). Conversely, a significant increase in caspase-3 positive cells was detected in the DOCK1-mutant tumors, suggesting that DOCK1 provides a survival signal during Neu oncogenic stress (Fig. 2I and Fig. S5D). One proliferative signal downstream of HER2/Integrin β4 (Itgβ4) crosstalk is c-JUN (17). Interestingly, its activator JNK is a target of DOCK1–Rac signaling (18). We examined the levels of phosphorylated c-JUN (pc-JUN) in MINs and found a decrease in DOCK1-mutant lesions in comparison with wild-type counterparts (Fig. 2J and Fig. S6). Notably, this difference was only noticeable in MINs as pc-JUN levels were similar in fully developed wild-type and mutant tumors (Figs. S6 and S7). These results indicate that DOCK1 contributes to HER2-mediated tumor growth.
DOCK1 Is Essential for HER2-Induced Metastasis.
NIC mice develop lung metastasis and we investigated whether inactivation of DOCK1 can protect from this cancer progression step. The incidence of metastasis was decreased in lungs from NIC+DOCK1flx/flx (39%) in comparison with lungs from NIC+DOCK1wt/wt (52%) and NIC+DOCK1wt/flx (55%) mice (Fig. 3A). For animals afflicted with lung lesions, a fivefold reduction of the number of metastases was noted in NIC+DOCK1flx/flx compared with NIC+DOCK1wt/wt mice (Fig. 3B). Notably, 100% of the lung metastases in the NIC+DOCK1flx/flx mice were confined to the lung vasculature, whereas close to half the metastases were extravascular in control animals, suggesting a role for DOCK1 in lung invasion (Fig. 3C). In NIC+DOCK1wt/wt animals, lung metastases expressed high levels of DOCK1 (Fig. S8) with respect to the expression detected in the primary tumors (Fig. 2D). Notably, we also noted residual expression of DOCK1 in metastases from NIC+DOCK1flx/flx animals, suggesting that lung lesions are enriched in a small population of cells that resisted Cre-mediated gene inactivation (Fig. S8). We also examined whether immune cells are differentially recruited to breast tumors in the mutant mouse. Equal amounts of CD45+ hematopoietic cells were present in tumors from all genotypes (Fig. S9), ruling out a major contribution of the immune stroma to the observed metastatic phenotype. We conclude that the loss of DOCK1 is likely to have a cell intrinsic effect in NIC transformed cells.
Fig. 3.

DOCK1 regulates HER2-mediated lung metastasis. (A) Incidence of lung metastases is reduced in NIC+DOCK1flx/flx animals. Quantification of the percentage of mice that develop lung metastases for the indicated genotypes. (B) NIC+DOCK1flx/flx mice have a reduction in the amount of lung lesions. (C) H&E staining showing that metastases in NIC+DOCK1wt/wt mice can be found residing inside blood vessels (Left) and invading the lung parenchyma (Center). Metastases in NIC+DOCK1flx/flx were found inside blood vessels (Right). (Scale bar: 250 µm, 10×.) (D) Western blot analysis demonstrating two independent and stable shRNA-mediated knockdown of DOCK1 in NMuMG-NeuNT cells. (E) Representative pictures of the lungs in F. (F) Average lung lesions from animals injected with NMuMG-NeuNT, NMuMG-NeuNTsh1 DOCK1, and NMuMG-NeuNTsh2 DOCK1 cells. (G) DOCK1 regulates STAT3 phosphorylation in HER2-driven MINs. IHC analyses showing pSTAT3 staining in mice lesions. (Scale bar: Upper, 500 µm, 5×; Lower, 100 µm, 20×.) One-way ANOVA followed by a Bonferroni or a Student t test was used to calculate the indicated P values; *P ≤ 0.05 and **P ≤ 0.01.
Because a decrease in the primary tumor volume in NIC+DOCK1flx/flx mice could alter metastasis efficiency, an experimental metastasis assay was performed to validate our in vivo findings. NeuNT-transformed Normal Murine Mammary Gland epithelial (NMuMG) cells form lung metastases upon tail vein injection (19) and they were modified to express two independent shRNAs targeting DOCK1 (Fig. 3D). An equal number of NMuMG-NeuNT, NMuMG-NeuNTsh1 DOCK1, and NMuMG-NeuNTsh2 DOCK1 cells were injected in nude mice and metastasis to lungs was quantified. We observed a 9.6- and 17.3-fold decrease in metastases in mice injected with cells where DOCK1 was down-regulated by shRNAs in comparison with control cells (Fig. 3 E and F and Fig. S10). Collectively, these results demonstrate that DOCK1 is a central mediator of HER2-driven breast cancer metastasis.
As DOCK1 and Rac act in the integrin-signaling pathway, we verified whether upstream components were activated normally in DOCK1-null tumors. In agreement with the position of DOCK1 in the signaling cascade, we tested whether intermediates were activated correctly (Fig. S11). An important mediator of metastasis downstream of HER2, and a partner of activated Rac, is STAT3 (17). We investigated the phosphorylation status of STAT3 (pSTAT3) in MINs and found that it is highly phosphorylated and distributed in a polarized manner in control MINs, whereas only a faint and evenly distributed pSTAT3 signal is detectable in DOCK1-deficient MINs (Fig. 3G and Fig. S6). This difference in STAT3 activation was only noted in MINs because a similar pSTAT3 signal was observed in all fully developed tumors (Figs. S6 and S7). These data identify DOCK1 as a critical component to promote STAT3 phosphorylation during the early stages of HER2 tumorigenesis.
Identification of a Gene Signature Associated with DOCK1 Expression in HER2 Mammary Tumors.
DOCK1 is generally assumed to exert its function at the membrane via Rac activation and cytoskeleton remodeling (9). To further characterize the defects in tumor growth and metastasis occurring in DOCK1-deficient HER2 tumors, we established the transcriptome of NIC+DOCK1wt/wt (n = 4) and NIC+DOCK1flx/flx (n = 4) mammary tumors by next generation RNA sequencing and uncovered significant variations in the expression of 45 genes between our experimental conditions (more than twofold change; P value <0.05; of ∼16,000 transcripts sequenced) (Fig. 4A and Table S1). Globally, this represents a constrained change of 0.28% in gene expression. These differential expression data were confirmed by quantitative (Q)-PCR for a subset of the genes (Fig. S12 and Table S2). Among up-regulated genes in the DOCK1-knockout tumors were Casein α/β (CSN1S1 and CSN2), Lactalbumin (LALB1), and Estrogen receptor 1 (ESR1) (Fig. 4A), characteristic markers of differentiated mammary epithelial cells, suggesting that DOCK1-mutant tumors are less dedifferentiated.
Fig. 4.

Identification of a DOCK1-null gene signature enriched in IFN response genes. (A) Heat map of the 45 genes differentially expressed between NIC+DOCK1wt/wt and NIC+DOCK1flx/flx tumors. Red, elevated; green, decreased; intensity of color represents relative change. Genes in red are bona fide players of IFN signaling according to the literature. (B) Gene ontology analyses. (C) Expression levels of a subset of genes from the NIC+DOCK1flx/flx signature correlate with disease-free survival in human HER2+ breast cancer patients. Correlation coefficient >1 means high expression of the gene correlate with a poor prognostic and <1 with a good prognostic. (D) Expression levels of RTP4 and STAT1 independently predict disease-free survival in HER2+ cancer patients.
We performed gene ontology (GO) analyses to gain insights into the biological processes regulated by the differentially expressed genes. Unexpectedly, 34/45 differentially expressed genes, and specifically 34/37 of the down-regulated genes in DOCK1-null tumors, are enriched for IFN response genes (Fig. 4 A and B; genes shown in red). These genes can be broken down further into four functional categories (Fig. 4B). A number of the identified genes in our DOCK1-null tumors have previously been linked to a STAT1-governed cancer gene signature that is predictive of risk of resistance to radiotherapy (20) (Fig. 4A). Likewise, we identified STAT1 as a down-regulated gene in our DOCK1-null gene signature and we could confirm lower levels of STAT1 protein in our DOCK1-null tumors both by IHC and Western blot (Figs. S6 and S7). As all of the other down-regulated genes are targets of STAT1, we envision that our DOCK1-null gene signature could reflect variation in STAT1 activity.
STAT1 is known to regulate transcription of a large set of response genes when IFN signaling is engaged. Unexpectedly, oncogenic transformation by Ras was shown to induce the expression of IFN-responsive genes including Interferon Stimulated Gene 15 (ISG15) that is also linked to poor prognosis and increased cell migration (21–23). We investigated whether such a gene regulation profile could be reproduced ex vivo upon NeuNT transformation. For the genes tested (RTP4, ISG15, STAT1, and UBA7), higher expression could be detected in NMuMG-NeuNT in comparison with parental NMuMG-EV explants (Fig. S13). These genes are also regulated by DOCK1 as they were less expressed in NMuMG-NeuNTsh1 DOCK1 cells (Fig. S13).
We examined the importance of our DOCK1-null gene signature in breast cancer and found that expression levels of five genes (UBA7, RTP4, CSN1S1, DDX60, and IFI44) are individually predictive of disease-free survival in HER2+ patients (Fig. 4C). Furthermore, we examined the survival curves and identified six genes (STAT1, RTP4, OASL, PARP12, LGALS9, and LGALS3BP) for which high levels of expression correlated with a worse survival prognostic in HER2+ patients (Fig. 4D and Fig. S14A). We tested whether the DOCK1-associated gene signature could predict breast cancer outcome in a cohort of patients irrespective of the molecular subtype and found that our DOCK1flx/flx-like signature correlates with good patient survival (Fig. S14B). These data demonstrate that DOCK1 controls the expression of a gene signature that predicts the outcome of breast cancer patients.
Discussion
DOCK1 is established to promote Rac-dependent cell migration/invasion in a variety of in vitro cellular models (11). A key clinical finding of this report is that expression of DOCK1 correlates with poor survival for HER2+ and basal breast cancer patients (1, 2). These breast cancer subtypes present the worst clinical prognosis and remain challenging to treat due to their propensity to metastasize. Our findings that DOCK1 is activated by, and interacts with, HER2 to promote cell migration suggest that it is a critical and therapeutically targetable GEF during breast cancer metastasis. The DOCK1 inhibitor CPYPP (15) blocked Rac activation and migration and it is conceivable that a next generation drug could be used in the clinic in combination with anti-HER2 regimens to limit metastasis. Further studies on the role of DOCK1 in basal breast cancer are also warranted. GEFs responsible for driving migration downstream of HER2 remain poorly defined in vivo. We provide a unique in vivo analysis supporting a role for DOCK1 in cancer progression by demonstrating an essential contribution of this GEF to tumor growth and metastasis in a HER2 breast cancer mouse model. Interestingly, systemic inactivation of the RacGEF TIAM1 was reported to protect mice from Neu-induced tumors, suggesting a role in cell survival in vivo (24). P-REX1 was identified as a GEF mediating Rac activation in luminal breast cancer cells upon HRG stimulation (8). DOCK1 and P-REX1 share similarities as they are recruited to the membrane by Phosphatidylinositol 3,4,5-trisphosphate and can act downstream of G protein coupled receptors to mediate Rac activation in migrating cells (8, 25). P-REX1 is likely to be involved in luminal and estrogen receptor positive breast cancer progression according to its expression profile (8). In our in vivo tumor model, we found P-REX1, in contrast to DOCK1, to be minimally expressed in NIC tumors. Whereas DOCK1 is also highly expressed in luminal breast cancers, high levels of DOCK1 correlated with poor prognosis only in HER2+ and basal breast cancers. These findings suggest that these two GEFs are likely to contribute to the progression of different subtypes of breast cancers.
STAT3 is frequently activated in HER2+ and phospho-SRC+ human invasive breast carcinomas (26). Additionally, joint HER2/Itgβ4 signaling promotes activation of STAT3 for efficient metastasis (17). Inactivation of STAT3 in HER2 tumors supports a role for this protein in metastasis and in tumor growth (27). We report a decrease in pSTAT3 in DOCK1-null MINs and observed similar phenotypes between DOCK1- and STAT3-null mice in growth and metastasis, suggesting that DOCK1 could act upstream of STAT3 activation. It will be important to address whether HER2/Itgβ4 cosignaling relies on DOCK1/Rac for cell proliferation and migration. Deletion of DOCK1 also phenocopies loss of Itgβ1 in HER2 tumors, suggesting that this GEF may signal at the crossroad of HER2/Itgβ1 signaling (28). DOCK1 therefore appears to be a GEF integrating cosignaling of HER2 with integrins (Fig. S15).
We identified a gene signature composed of IFN response genes under the control of HER2 and DOCK1. The signaling events whereby HER2 promotes the expression of these genes are not understood. One possibility would be that HER2 overexpression could increase IFN production by the tumor cells through the IKK/NF-κB pathway (Fig. S15, arrow 1). However, it is unlikely that DOCK1/Rac could be promoting transcription of IFNs because their mRNAs are not detectable in NIC tumors. Alternatively, stromal cells could be providing IFN for the tumor (Fig. S15, arrow 2). Because we could recapitulate HER2-induced expression of IFN response genes ex vivo, our data instead support a model where transcription of these genes is tumor cell autonomous. Our current working model is that DOCK1/Rac could be promoting the expression of IFN response genes by acting at the level of the STAT1/Interferon Regulatory Factor 9 (IRF9) module (Fig. S15, arrow 3). We suggest that the observed down-regulation of STAT1 expression and activation in our DOCK1-mutant tumors contribute to the reduction in transcription of IFN response genes. How DOCK1/Rac regulates STAT1 expression and whether it could directly contribute to activating the transcriptional activity of the STAT1/IRF9 unit remains to be fully investigated.
We identified at least two genes, STAT1 and RTP4, which are individually predictive of patient outcome in the HER2+ breast cancer subtype. Recent genetic studies suggest that STAT1 is a tumor suppressor during HER2 tumorigenesis (29). Similar to our findings here, others link a STAT1 gene network to poor patient outcomes (20). Collectively, these results suggest that STAT1 could be acting in a different manner at stages of tumor initiation, resistance and dissemination; temporal deletion of STAT1 during HER2 oncogenesis would be informative. RTP4, a chaperone escorting GPCRs at the membrane (30), could be a regulator of HER2. We also found a robust modulation of genes regulating ISGylation (ISG15, HERC6, USP18, and UBA7). Recent findings demonstrated that oncogene transformation could induce the expression of ISG15, in a cell autonomous manner, and increase its conjugation on cytoskeletal proteins to modify their activity and promote cell migration (22, 23). Probing the function of this ubiquitin-like posttranslational modification system in HER2 tumorigenesis will be important.
Materials and Methods
Bioinformatics, clinical correlations, and tumor microarray methods are described in SI Materials and Methods. Cell lines, antibodies, and methods for coimmunoprecipitation, kinase, and Rac GTP assays, Western blot, immunohistochemistry, and cell migration assays are described in SI Materials and Methods. Generation of the DOCK1 conditional knockout is described in SI Materials and Methods. For tumorigenesis studies, DOCK1 flox mice were crossed with NIC (MMTV-NeuNDL2-5-IRES-Cre) transgenic mice. Large cohorts of female mice were generated and analyzed in detail 5 wk after tumor onset, which was determined by physical palpation. Further details are available in SI Materials and Methods. RNA deep sequencing, bioinformatics analyses, Q-PCR validation, and statistical analysis are described in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank N. Fradet for excellent technical assistance; M. Patel for reading the manuscript; the Cell Migration Consortium authorities Drs. R. Hynes, R. Horvitz, and A. Burds for help with the construction of the conditional DOCK1 mouse; Dr. S. Keezer (Cell Signaling Technology) for antibody gift; Dr. P. Siegel and J. Ranger for reagents and advice; Dr. M. Kazanietz for discussions; F. Robert and O. Neyret for deep sequencing; S. Riverin for mouse colonies maintenance; and the “Next Generation Sequencing” platform from the McGill University and Genome Québec Innovation Center. This work was funded by Grant 019104 from the Canadian Cancer Society (to J.-F.C.). M.L. and J.H. were supported by a Université Montréal and Invitrogen–Institut de Recherches Cliniques de Montréal scholarships, respectively. J.-F.C. is a recipient of a Junior Investigator award from the Fonds de Recherche du Québec-Santé. W.J.M. holds the Canadian Research Chair in Molecular Oncology.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The RNA sequencing data reported in this paper have been deposited in the NCBI Bioproject database (accession no. 189677).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1213050110/-/DCSupplemental.
References
- 1.Eckhardt BL, Francis PA, Parker BS, Anderson RL. Strategies for the discovery and development of therapies for metastatic breast cancer. Nat Rev Drug Discov. 2012;11(6):479–497. doi: 10.1038/nrd2372. [DOI] [PubMed] [Google Scholar]
- 2.Perou CM, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 3.Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21(2):177–184. doi: 10.1016/j.ceb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 4.Slamon DJ, et al. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 5.Ursini-Siegel J, Schade B, Cardiff RD, Muller WJ. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat Rev Cancer. 2007;7(5):389–397. doi: 10.1038/nrc2127. [DOI] [PubMed] [Google Scholar]
- 6.Steeg PS. Tumor metastasis: Mechanistic insights and clinical challenges. Nat Med. 2006;12(8):895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- 7.Sanz-Moreno V, et al. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008;135(3):510–523. doi: 10.1016/j.cell.2008.09.043. [DOI] [PubMed] [Google Scholar]
- 8.Sosa MS, et al. Identification of the Rac-GEF P-Rex1 as an essential mediator of ErbB signaling in breast cancer. Mol Cell. 2010;40(6):877–892. doi: 10.1016/j.molcel.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Côté JF, Vuori K. GEF what? Dock180 and related proteins help Rac to polarize cells in new ways. Trends Cell Biol. 2007;17(8):383–393. doi: 10.1016/j.tcb.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duchek P, Somogyi K, Jékely G, Beccari S, Rørth P. Guidance of cell migration by the Drosophila PDGF/VEGF receptor. Cell. 2001;107(1):17–26. doi: 10.1016/s0092-8674(01)00502-5. [DOI] [PubMed] [Google Scholar]
- 11.Smith HW, Marra P, Marshall CJ. uPAR promotes formation of the p130Cas-Crk complex to activate Rac through DOCK180. J Cell Biol. 2008;182(4):777–790. doi: 10.1083/jcb.200712050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng H, et al. Activation of Rac1 by Src-dependent phosphorylation of Dock180(Y1811) mediates PDGFRα-stimulated glioma tumorigenesis in mice and humans. J Clin Invest. 2011;121(12):4670–4684. doi: 10.1172/JCI58559. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Feng H, et al. Phosphorylation of dedicator of cytokinesis 1 (Dock180) at tyrosine residue Y722 by Src family kinases mediates EGFRvIII-driven glioblastoma tumorigenesis. Proc Natl Acad Sci USA. 2012;109(8):3018–3023. doi: 10.1073/pnas.1121457109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haibe-Kains B, et al. A three-gene model to robustly identify breast cancer molecular subtypes. J Natl Cancer Inst. 2012;104(4):311–325. doi: 10.1093/jnci/djr545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishikimi A, et al. Blockade of inflammatory responses by a small-molecule inhibitor of the Rac activator DOCK2. Chem Biol. 2012;19(4):488–497. doi: 10.1016/j.chembiol.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 16.Ursini-Siegel J, et al. ShcA signalling is essential for tumour progression in mouse models of human breast cancer. EMBO J. 2008;27(6):910–920. doi: 10.1038/emboj.2008.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo W, et al. Beta 4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell. 2006;126(3):489–502. doi: 10.1016/j.cell.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 18.Kiyokawa E, et al. Activation of Rac1 by a Crk SH3-binding protein, DOCK180. Genes Dev. 1998;12(21):3331–3336. doi: 10.1101/gad.12.21.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Northey JJ, et al. Signaling through ShcA is required for transforming growth factor beta- and Neu/ErbB-2-induced breast cancer cell motility and invasion. Mol Cell Biol. 2008;28(10):3162–3176. doi: 10.1128/MCB.01734-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khodarev NN, et al. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc Natl Acad Sci USA. 2004;101(6):1714–1719. doi: 10.1073/pnas.0308102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bektas N, et al. The ubiquitin-like molecule interferon-stimulated gene 15 (ISG15) is a potential prognostic marker in human breast cancer. Breast Cancer Res. 2008;10(4):R58. doi: 10.1186/bcr2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsai YC, et al. Interferon-β signaling contributes to Ras transformation. PLoS ONE. 2011;6(8):e24291. doi: 10.1371/journal.pone.0024291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Desai SD, et al. ISG15 disrupts cytoskeletal architecture and promotes motility in human breast cancer cells. Exp Biol Med (Maywood) 2012;237(1):38–49. doi: 10.1258/ebm.2011.011236. [DOI] [PubMed] [Google Scholar]
- 24.Strumane K, Rygiel T, van der Valk M, Collard JG. Tiam1-deficiency impairs mammary tumor formation in MMTV-c-neu but not in MMTV-c-myc mice. J Cancer Res Clin Oncol. 2009;135(1):69–80. doi: 10.1007/s00432-008-0437-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanematsu F, et al. DOCK180 is a Rac activator that regulates cardiovascular development by acting downstream of CXCR4. Circ Res. 2010;107(9):1102–1105. doi: 10.1161/CIRCRESAHA.110.223388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diaz N, et al. Activation of stat3 in primary tumors from high-risk breast cancer patients is associated with elevated levels of activated SRC and survivin expression. Clin Cancer Res. 2006;12(1):20–28. doi: 10.1158/1078-0432.CCR-04-1749. [DOI] [PubMed] [Google Scholar]
- 27.Ranger JJ, Levy DE, Shahalizadeh S, Hallett M, Muller WJ. Identification of a Stat3-dependent transcription regulatory network involved in metastatic progression. Cancer Res. 2009;69(17):6823–6830. doi: 10.1158/0008-5472.CAN-09-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huck L, Pontier SM, Zuo DM, Muller WJ. beta1-integrin is dispensable for the induction of ErbB2 mammary tumors but plays a critical role in the metastatic phase of tumor progression. Proc Natl Acad Sci USA. 2010;107(35):15559–15564. doi: 10.1073/pnas.1003034107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raven JF, et al. Stat1 is a suppressor of ErbB2/Neu-mediated cellular transformation and mouse mammary gland tumor formation. Cell Cycle. 2011;10(5):794–804. doi: 10.4161/cc.10.5.14956. [DOI] [PubMed] [Google Scholar]
- 30.Saito H, Kubota M, Roberts RW, Chi Q, Matsunami H. RTP family members induce functional expression of mammalian odorant receptors. Cell. 2004;119(5):679–691. doi: 10.1016/j.cell.2004.11.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.