

Abstract
Objective:
To highlight the clinical and radiologic features and management of craniofacial fibrous dysplasia with review of literature.
Materials and Methods:
A retrospective review of 6 patients who underwent surgical treatment in a tertiary healthcare centre was done using the parameters of patients' details, clinical features, radiological findings, management and postoperative review.
Results:
Of the six patients, 3 females and 2 males were in the 2nd decade of life and 1 male in the 1st decade of life. The disease was restricted to maxilla in 3 patients, involved the temporal and frontal bones in addition to maxilla in one, involved the frontal bone in one patient and involved frontal and parietal bones in one patient. The primary reason for seeking treatment in all the 6 cases was facial deformity. There was absence of pain in all 6 cases. For surgical treatment in all three cases involving the maxilla, the approach was intraoral while bicoronal approach was used for the other three cases. Treatment consisted of surgical contouring and reshaping the area. All cases were followed up over a period of 2 years with no signs of recurrence.
Conclusion:
Treatment of craniofacial fibro-osseous lesions is highly individualized. Most cases of craniofacial fibrous dysplasia manifest as swellings that cause facial deformity and surgical recontouring after cessation of growth seems to provide the best results.
Keywords: Craniofacial, fibrous dysplasia, monostotic, polyostotic
INTRODUCTION
Fibrous dysplasia (FD) is a non-neoplastic developmental hamartomatous disease of the bone, characterised by a blend of fibrous and osseous elements in the region. With an incidence of 1:4000-1:10,000 it seems to be a rare disease.[1] It represents approximately 2.5% of all bone lesions and about 7% of all benign bone tumors.[2] Initially described as “osteitis fibrosa generalisata” by von Recklinghausen in 1891 in a patient with skeletal deformities due to fibrotic bone changes, the disorder became known as “fibrous dysplasia” in 1938 when Lichtenstein introduced the term.[3] The three subtypes of FD are monostotic, polyostotic and craniofacial. The term “craniofacial fibrous dysplasia” (CFD) is used to describe fibrous dysplasia where the lesions are confined to contiguous bones of the craniofacial skeleton. Most cases of craniofacial fibrous dysplasia cannot be truly categorized as monostotic because of the involvement of multiple adjacent bones of the craniofacial skeleton. They are also not truly polyostotic because bones outside the craniofacial complex are spared. These conditions have a slight female predilection. They are seen in the first 3 decades of life and usually stabilize when the patient reaches skeletal maturity. However, there have also been reports of persistence at later periods underlining their variable clinical behaviour. Fibrous dysplasia involves the maxilla almost twice as often as the mandible, frequenting the posterior region and is usually unilateral in nature.
Diagnosis
The most common presenting symptom in fibrous dysplasia is a gradual, painless enlargement of the involved bone or bones in the craniofacial region, clinically seen as facial asymmetry. In long bones there may be malformed extremity, limb pain, or pathologic fracture. If constriction of foramina or obliteration of bony cavities occurs, orbital dystopia, diplopia, proptosis, blindness, epiphora, strabismus, facial paralysis, loss of hearing, tinnitus, nasal obstruction, etc., may also be evident. In addition to clinical findings, diagnosis is based on results of the radiographic examination and histopathological findings.
Radiological findings
The radiological signs of CFD are very distinctive, visualised as a thin cortex with well defined borders and ground-glass appearance. Radiographically, the appearance varies with the stage of development and amount of bony matrix within the lesion. The radiographic picture is more radiolucent and well defined in the early stages and becomes mottled and more radio opaque as the disease progresses.
MATERIALS AND METHODS
The records of 6 patients of craniofacial fibrous dysplasia were evaluated in terms of patients' details like age, sex, clinical features, radiological picture, management and postoperative review.
RESULTS
The mean age of the 6 patients was 15.5 years (range, 8-19 years). 3 females and 2 males were in the 2nd decade of life and 1 male in the 1st decade of life. The male to female ratio was 1:1. The universal clinical feature in all six patients was the presence of facial deformity. In addition, two of the patients complained of nasal obstruction due to obliteration of the nasal cavities. The average duration of the condition in the six patients was 8 months. Three of the patients reported with swelling of the maxillary region [Figure 1a]. One of these patients also complained of nasal obstruction of the involved side due to obliteration of the nasal cavity by the lesion. Three of the patients had obvious swellings on the frontal region causing aesthetic concerns [Figures 1a and 2a]. Though the supraorbital and medial orbital regions were also involved due to projection of the frontal bones in 2 of these patients, there were no visual disturbances. The classic clinical features in maxillary involvement was a well defined bony hard swelling in the maxillary region, obliterating the nasolabial groove with no sensory impairment of infra orbital nerve. Intraorally the bony overgrowth extended onto the alveolar crest causing a ledge in the posterior region. In one of these patients, the swelling involved the nasomaxillary region with obvious obliteration of the nasal cavity [Figure 1b and c]. The involvement of frontal bone in three of the patients manifested as prominence of the region creating a very unaesthetic appearance.
Figure 1.
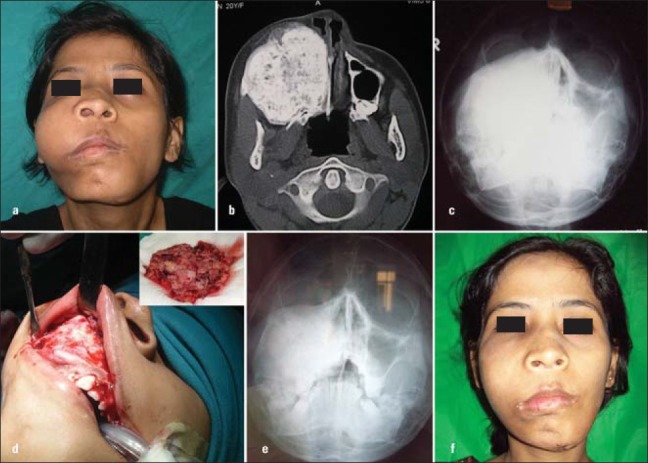
(a) Monostotic maxillary fibrous dysplasia, (b) CT showing extensive maxillary involvement, (c) PNS Radiograph showing maxillary sinus obliteration, (d) Intraoral view of the lesion - Amount of bone removed, (e) Postoperative PNS view, (f) Postoperative view
Figure 2.
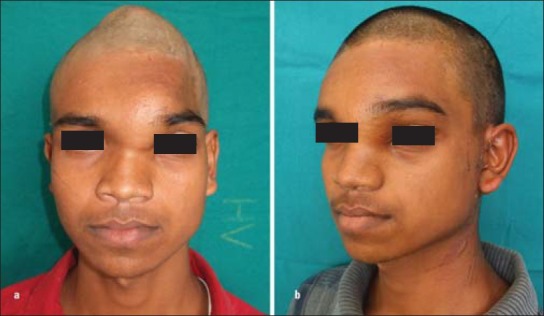
(a) Photograph showing frontal and parietal involvement, (b) Postoperative appearance after recontouring
Routine radiographs were taken in 3 patients and CT scan in 5 of the patients [Figures 1b and c, 3a-c]. Conventional radiographs showed increased radio opaque appearance of the region. The area showed a classic ground glass appearance.
Figure 3.
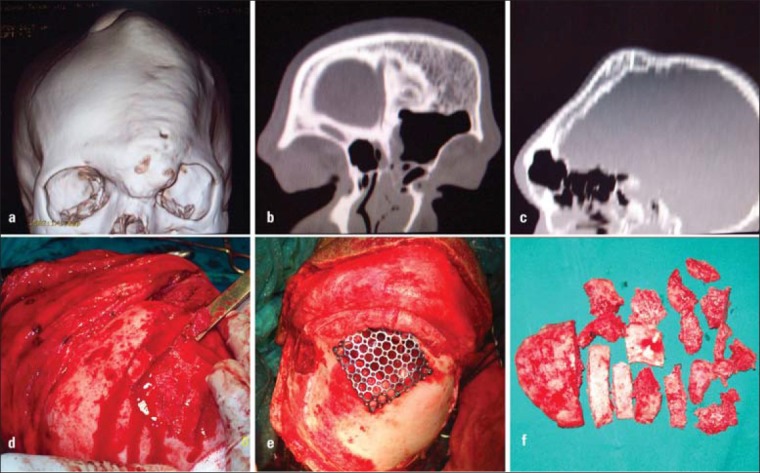
(a) CT showing frontal and parietal involvement, (b) Coronal CT view, (c) Sagittal CT view, (d) Removal of bone using osteotome, (e) Reconstruction of the frontal sinus wall using mesh, (f) The bone chips that were removed during frontal contouring
CT findings
Patient 1 had thickening of the frontal and parietal regions with deposition of bone on the inner aspect, at the expense of the cranial contents [Figures 1b-c]. The frontal area showed thinning and perforation with loss of the anterior wall of the frontal sinus.
Patient 2 showed increased dimensions of the zygoma and partial obliteration of the right maxillary sinus. In patient 3, there was dense ossification of the maxilla, its antrum and slight raising of the orbital floor. Patient 5 and 6 showed gross thickening of the frontal and sphenoid regions with a bilateral frontal bossing including involvement of roof of the orbit. The bone had a mottled appearance with partial obliteration of the anterior cranial fossa.
Lab investigations
Routine investigations including haematology, serum Calcium and serum Alakaline Phosphatase (ALP) were performed. All parameters were within normal limits.
Management
Incision biopsy was done for 4 patients in whom the maxilla was involved. A vestibular approach was used to remove the tissue and histopathological picture of the 4 patients showed loosely arranged curvilinear trabeculae with fibrous elements leading to a diagnosis of fibrous dysplasia.
In 3 patients where the condition was restricted to maxilla, after naso-endotracheal intubation, a vestibular approach was used to deglove the maxilla on the involved side exposing the anterior aspect up to the infraorbital rim. The lesion was removed using rotary instruments and osteotomes. The contouring was checked visually for satisfactory appearance. In one of these patients, the nasal cavity was partially obliterated. The piriform cavity was reshaped by removal of the osseous extension in the nasal cavity.
The approach to the frontal region in 3 patients was the bicoronal approach. After exposing the calvarium, bone was removed in small instalments using micro saws, rotary instruments and osteotomes [Figures 1d, 3d-f]. The contour and aesthetics of the region was evaluated from time to time by putting back the scalp flap and reviewing the appearance. In one of the patients, the frontal sinus was devoid of anterior wall due to thinning. The sinus lining was removed and the anterior wall reconstructed with titanium mesh [Figure 2b].
No attempt was made to remove the whole lesion till the intracranial part, considering the large thickness of the bone in this region and the absence of any signs or symptoms. In one of these patients, since maxillary involvement was also present, an additional intraoral approach was used to recontour this region. The aesthetics was restored in these patients as assessed clinically and radiologically.
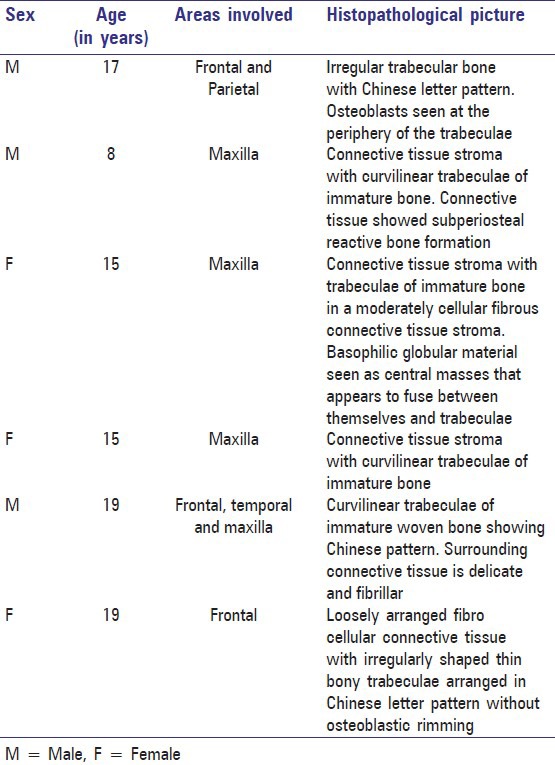
The specimens were subjected to histopathological examination. The reports of the 6 individual cases are given in Table 1.
Table 1.
Histopathological report of the cases
All 6 patients were periodically reviewed every 3 months for 2 years [Figures 1f and 2b]. All three patients having only maxillary involvement had to be subjected to another surgical procedure to remove the remnant overgrowth of the condition in the alveolar crestal region after 3 months. Subsequent reviews did not show any recurrence.
DISCUSSION
Von Recklinghausen first described the condition in 1891 in a patient with skeletal deformities and coined the term “osteitis fibrosa generalisata”. It was renamed “fibrous dysplasia” in 1938 by Lichtenstein. Perhaps the most accurate term to describe fibrous dysplasia is “fibro-osseous dysplasia” or “fibrous osteodysplasia.” Reed,[4] has defined the condition as an “arrest of bone maturation in woven bone with ossification resulting from metaplasia of a nonspecific fibro-osseous type”.
Schlumberger,[5] first reported single bone involvement by the disease process and described it as “monostotic fibrous dysplasia”. Presently, the terms “monostotic” (single bone) and “polyostotic” (multiple bones) are used to describe the extent of this condition in terms of number of bones involved. Monostotic fibrous dysplasia (MFD) constitutes about 70-80% of FD patients and is mostly seen in the second and third decade.[6,7]
The most commonly involved bones are femur, tibia, ribs and facial bones. The involvement of facial and cranial bones in FD occurs in nearly 50% of patients with the polyostotic form and in 10-27% of patients with MFD.[8]
The polyostotic form is more frequently seen in female patients, and about 25% of all patients with polyostotic fibrous dysplasia (PFD) exhibit the disease in more than half of the skeleton.
FD can occur in both types of bones, endochondral and membranous. In addition to these two entities, another presentation is “craniofacial fibrous dysplasia” where the lesions are confined to contiguous bones of the craniofacial skeleton.[9] CFD cannot be truly categorized as monostotic because of the involvement of multiple adjacent bones of the craniofacial skeleton but are not truly polyostotic either because bones outside the craniofacial complex are usually not involved. Between 50 to 100% of patients with polyostotic disease will have craniofacial involvement, whereas only 10% with monostotic lesions will have involvement of these structures.[10]
In the maxillofacial region fibrous dysplasia involves the maxilla almost twice as often as the mandible and is usually seen in the posterior region. Most of these lesions are unilateral. Eversole et al.,[11] classified the craniofacial type as polyostotic because many bones of the craniofacial complex are involved that are separated from each other only by sutures. The polyostotic type may be divided into 3 types: (1) craniofacial FD, in which only the bones of the craniofacial complex are affected; (2) Lichtenstein-Jaffe type, in which multiple bones of the skeleton are involved with café au lait pigmentations on the skin and rare endocrinopathies in a few of these patients; and (3) Albright's syndrome, characterized by the triad of polyostotic FD (mostly unilateral), café au lait pigmentations on the skin, and various endocrinopathies.[12] Another very rare and special form is the Mazabraud syndrome, describing an association of the FD with soft tissue myxomas.
Diagnosis of polyostotic FD is generally based on clinical symptoms and radiological images. In contrast, the monostotic FD requires bone biopsy.
Most authors state that fibrous dysplasia in all forms occurs equally in male and female patients. However, some authors[13] have noted the slight female predilection. The condition manifests in the first 3 decades of life and it is generally believed that many of the cases would stabilize when the patients reach skeletal maturity.[14] The variable clinical behaviour of these lesions can however be illustrated by the series of fibrous dysplasia patients in the study by Harris et al.,[15] where one patient presented with pain at 68 years of age.
The main presentation of patients with CFD is a diffuse swelling in the affected region. This affects the calvaria, the skull base, the zygoma, the maxilla and the mandible. The maxilla is more often involved than the mandible. The progression of the lesion may cause aesthetic impairment and deformities and clinical symptoms such as visual disturbances, proptosis, orbital dystopia, nasal malfunction, dental problems and sensory disturbances in the affected regions.[16]
FD can resemble conditions like cherubism and other giant cell lesions radiographically and histologically.[17] In general, fibrous dysplasia occurs more readily in membranous bones and involvement of ethmoids and sphenoid sinus is uncommon as these ossify in a cartilage.[18]
Etiology
The etiology of FD is not certain and is probably a genetic predisposition. It is assumed that the mutation is sporadic, postzygotic and located on the GNAS1 gene. This gene is found on chromosome 20q13 and is responsible for the formation of the alpha subunit of stimulating G-proteins.[19] This mutation activates adenylate cyclise and consequently increases intracellular concentrations of cAMP resulting in abnormal osteoblast differentiation and production of dysplastic bone. On the other hand it stimulates release of several cytokines (mainly interleukin-6) which cause normal osteoclasts to congregate and increase bone resorption.[20]
Diagnosis
The usual presentation in fibrous dysplasia is a gradual, painless enlargement of the involved bone or bones causing facial asymmetry.
Other symptoms in CFD are seen due to constriction of cranial foramina or obliteration of bony cavities. These include anosmia, orbital dystopia, diplopia, proptosis, blindness, epiphora, strabismus, facial paralysis, hearing loss, tinnitus, etc.
With fibrous dysplasia, the primary goal is to distinguish it from other benign fibro-osseous lesions especially ossifying fibroma. Ossifying fibroma, unlike FD grows centrifugally and is clearly demarcated from the surrounding normal bone.
Radiology
CFD patients can be assessed by plain radiography, magnetic resonance imaging (MRI) or CT-scans. Radiographically, the appearance of fibrous dysplasia will vary depending on the stage of development and quantity of bony matrix within the lesion. Thus the lesion is more radiolucent and well-defined initially and gradually changes to a mottled, ill-defined radiopacity in the later stages. The radiological picture in fibrous dysplasia is very distinctive showing a thin bony cortex with well defined borders and ground glass appearance. Three distinct patterns have been described by Panda et al.,[18] The pagetoid appearance on CT imaging is characterized by bone expansion and scattered islands of bone formation in a low-attenuation field. The sclerotic type has a homogeneous appearance with a ground-glass appearance. The cystic type appears as a well defined low-attenuation lesion with a sclerotic margin.
Histopathology
The histopathological features are similar in all three types of FD, viewed as benign fibroblastic tissue, arranged in a loose, whorled pattern interspersed with spicules of woven bone with typical osteoblastic rimming embedded in fibrous tissue. Areas of cystic degeneration with hemosiderin laden macrophages, haemorrhage and osteoclasts may also be seen.[21]
Management
The aim of the surgical treatment in patients with FD is to prevent pathological fractures, control the pain and to reduce bone deformities.
The surgical treatment of CFD aims at correcting the facial deformity in most cases and restoring the obliterated foramina when they cause problems like visual disturbances, proptosis, orbital dystopia, nasal malfunction, etc.
Recommended treatment options can be divided into 4 categories:
Observation
Medical therapy
Surgical remodelling
Radical excision and reconstruction
Small asymptomatic lesions of the craniomaxillofacial skeleton that are cosmetically acceptable to the patient are best managed with observation.
Medical therapy has not occupied a prominent role in the management of fibrous dysplasia to date. Recognition of pathogenesis of this disease led to treatment with biphosphonates. They control bone erosion by the inhibition of osteoclastic action. They have a high affinity for hydroxyapatite of resorbed bone and remain tied to it for a long period. The drug is incorporated into the cellular cytoplasm, inhibiting acid phosphatase secretion thereby arresting bone resorption.[22] The theory is that the drug stabilizes the disease and improves the patient's pain. Another drug that has been used is pamidronate given intravenously. Pamidronate containing a basic nitrogen atom in its alkyl side chain represents a second generation drug, characterized by increased potency of inhibition of bone resorption and good tolerance.[23] Pamidronate 60 mg/day through an intravenous route on 3 successive days reduces osteoclastic activity. This is repeated every 6 months for 18 months. It has been shown to reduce intensity of bony pain, bony resorption, and filling of lytic lesions. Another drug that is mentioned in literature is calcitonin.[24] Vitamin D and calcium supplements have also been recommended since serum calcium is low in these patients.
Though there are no uniformly accepted protocols for treatment of CFD, surgical therapy remains the mainstay of therapy for this disease and is directed at correcting or preventing functional deficits and achieving normal facial aesthetics.
Conservative surgery involves a remodelling procedure aimed at achieving reasonably acceptable aesthetics. This can however result in recurrence, especially during the growth period[25] and can range from 15-20%.[16] A more personalised approach is ideal in these conditions and the decision to intervene is based on a careful assessment of the disease on the patient functionally and aesthetically.
The presence of the lesion in the craniofacial region is unique in that the presence of important structures in the vicinity can hamper complete removal of the pathology. Treatment for CFD aims at achieving cosmetic or functional goals. The surgical procedure is usually postponed till puberty hoping that the disease will undergo remission. Although conservative surgical resection is ideal, a more radical approach would be mandatory when the skull base is involved with complications of compression around foramina and orbital apex. The importance of a long term follow up following conservative treatment cannot be over emphasized. In general, stabilization usually occurs when bone maturation is completed. Alvares in a 23 year old follow up has shown stabilisation, 13 years after conservative surgery.[26] Some authors recommend monitoring patients their whole life to assess the progress of the disease. One reason for this is the potential for late growth and dysfunction and a small risk of sarcomatous change. The risk of malignant transformation has been estimated to be 0.4% in fibrous dysplasia and 4% in McCune-Albright syndrome, with the craniofacial region most commonly being the site of occurrence.
Serum alkaline phosphatase levels are an important marker in detecting recurrence of FD.[27] ALP has been shown to decline in patients treated with palmidronate.[23] The authors postulate that recurrence is rare if the postoperative serum ALP level does not increase after complete resection. If the patient is older than 17 years and is in a condition that does not allow complete resection, partial resection is recommended because regrowth is not significantly probable. Surgery merely for aesthetic purposes is sufficient in patients older than 17 years, regardless of their preoperative serum ALP levels.
In 1990 Chen and Noordhoff proposed a treatment algorithm for the management of craniomaxillofacial fibrous dysplasia incorporating aggressive, radical surgery for the resection of diseased tissue.[28] For this algorithm, they proposed that the head and face could be divided into 4 zones based on the esthetic and functional consequences of the disease at each of these sites and the unique anatomic considerations for operating in each area.
Zone 1 represents the fronto-orbito-malar regions of the face. These are esthetically critical and can be adequately reconstructed with simple bone grafting techniques after reconstruction. For this region, they recommended radical excision and reconstruction.
Zone 2 refers to the hair bearing scalp. It is not typically an aesthetic concern, and as such, intervention is optional for the patient.
Zone 3 refers to the central skull base including the sphenoid, pterygoid, petrous temporal bone, and mastoid. Given the difficulty in obtaining surgical access to these areas, the authors recommended observation of lesions in this region.
Zone 4 comprises the tooth bearing portions of the skull, the maxilla and mandible. The authors recommended conservative management, given the difficulty in reconstructing defects in this region.
CONCLUSION
The fibrous dysplasia affecting the craniofacial region is a distinctive entity, that can in most cases be treated by conservative recontouring. The procedure is preferably indicated after the active growth phase has ceased. A long term follow up of these patients is mandatory considering the probable flare up of continuous growth of the lesion.
Footnotes
Source of Support: Nil
Conflict of Interest: No.
REFERENCES
- 1.Assaf AT, Benecke AW, Riecke B, Zustin J, Fuhrmann AW, Heiland M, et al. Craniofacial fibrous dysplasia (CFD) of the maxilla in an 11-year old boy: A case report. J Craniomaxillofac Surg. 2012;40:788–92. doi: 10.1016/j.jcms.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 2.Ricalde P, Horswell BB. Craniofacial fibrous dysplasia of the fronto-orbital region: A case series and literature review. J Oral Maxillofac Surg. 2001;59:157–67. doi: 10.1053/joms.2001.20487. [DOI] [PubMed] [Google Scholar]
- 3.Lichtenstein L. Polyostotic fibrous dysplasia. Arch Surg. 1938;36:874–98. [Google Scholar]
- 4.Reed RJ. Fibrous dyplasia of bone. A review of 25 cases. Arch Pathol. 1963;75:480–95. [PubMed] [Google Scholar]
- 5.Schlumberger HG. Fibrous dysplasia of single bones (monostotic fibrous dysplasia) Mil Surg. 1946;99:504–27. [PubMed] [Google Scholar]
- 6.Tabrizi R, Ozkan BT. Craniofacial fibrous dysplasia of orbit. J Craniofac Surg. 2008;19:1532–7. doi: 10.1097/SCS.0b013e31818ac270. [DOI] [PubMed] [Google Scholar]
- 7.Keskin M, Karabekmez FE, Ozkan BT, Tosun Z, Avunduk MC, Savaci N. Simultaneous occurrence of facial fibrous dysplasia and ameloblastoma. J Craniomaxillofac Surg. 2009;37:102–5. doi: 10.1016/j.jcms.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 8.Ben Hadj Hamida F, Jlaiel R, Ben Rayana N, Mahjoub H, Mellouli T, Ghorbel M, et al. Craniofacial fibrous dysplasia: A case report. J Fr Ophtalmol. 2005;28:e6. doi: 10.1016/s0181-5512(05)81011-3. [DOI] [PubMed] [Google Scholar]
- 9.Daves ML, Yardley JH. Fibrous dysplasia of bone. Am J Med Sci. 1957;234:590–606. doi: 10.1097/00000441-195711000-00009. [DOI] [PubMed] [Google Scholar]
- 10.Kim DD, Ghali GE, Wright JM, Edwards SP. Surgical treatment of giant fibrous dysplasia of the mandible with concomitant craniofacial involvement. J Oral Maxillofac Surg. 2012;70:102–18. doi: 10.1016/j.joms.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 11.Eversole LR, Sabes WR, Rovin S. Fibrous dysplasia: A nosologic problem in the diagnosis of fibro osseous lesions of the jaws. J Oral Pathol. 1972;1:189. doi: 10.1111/j.1600-0714.1972.tb01659.x. [DOI] [PubMed] [Google Scholar]
- 12.Albright F, Butler AM, Hampton AO, Smith P. Syndrome characterized by osteitis fibrosa disemminata, areas of pigmentation and endocrine dysfunction, with precocious puberty in females-report of five cases. N Engl J Med. 1937;216:727–46. [Google Scholar]
- 13.Kruse A, Pieles U, Riener MO, Zunker Ch, Bredell MG, Grätz KW. Craniomaxillofacial fibrous dysplasia: A 10 year database 1996-2006. Br J Oral Maxillofac Surg. 2009;47:302–5. doi: 10.1016/j.bjoms.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 14.Rahman AM, Madge SN, Billing K, Anderson PJ, Leibovitch I, Selva D, et al. Craniofacial fibrous dysplasia: Clinical characteristics and long-term outcomes. Eye (Lond) 2009;23:2175–81. doi: 10.1038/eye.2009.6. [DOI] [PubMed] [Google Scholar]
- 15.Harris WH, Dudley HR, Jr, Barry RJ. The natural history of fibrous dysplasia: An orthopedic, pathological and roentgenographic study. J Bone Joint Surg Am. 1962;44-A:207–33. [PubMed] [Google Scholar]
- 16.Valentini V, Cassoni A, Marianetti TM, Terenzi V, Fadda MT, Iannetti G. Craniomaxillofacial fibrous dysplasia: Conservative treatment or radical surgery? A retrospective study on 68 patients. Plast Reconstr Surg. 2009;123:653–60. doi: 10.1097/PRS.0b013e318196bbbe. [DOI] [PubMed] [Google Scholar]
- 17.Hyckel P, Berndt A, Schleier P, Clement JH, Beensen V, Peters H, et al. Cherubism-New hypotheses on pathogenesis and therapeutic consequences. J Craniomaxillofac Surg. 2005;33:61–8. doi: 10.1016/j.jcms.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 18.Panda NK, Parida PK, Sharma R, Jain A, Bapuraj JR. A clinicoradiologic analysis of symptomatic craniofacial fibro-osseous lesions. Otolaryngol Head Neck Surg. 2007;136:928–33. doi: 10.1016/j.otohns.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 19.Demirdöver C, Sahin B, Ozkan HS, Durmus¸ EU, Oztan HY. Isolated fibrous dysplasia of the zygomatic bone. J Craniofac Surg. 2010;21:1583–4. doi: 10.1097/SCS.0b013e3181edc5af. [DOI] [PubMed] [Google Scholar]
- 20.Mandrioli S, Carinci F, Dallera V, Calura G. Fibrous dysplasia. The clinico-therapeutic picture and new data on its etiology. A review of the literature. Minerva Stomatol. 1998;47:37–44. [PubMed] [Google Scholar]
- 21.Abdelkarim A, Green R, Startzell J, Preece J. Craniofacial polyostotic fibrous dysplasia: a case report and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2008;106:e49–e55. doi: 10.1016/j.tripleo.2008.03.023. [DOI] [PubMed] [Google Scholar]
- 22.Vasikaran SD. Biphosphonates: An overview with special reference to alendronate. Ann Clin Biochem. 2001;38:608–23. doi: 10.1258/0004563011901037. [DOI] [PubMed] [Google Scholar]
- 23.Kos M, Luczak K, Godzinski J, Klempous J. Treatment of monostotic fibrous dysplasia with pamidronate. J Craniomaxillofac Surg. 2004;32:10–5. doi: 10.1016/j.jcms.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 24.Yasuoka T, Takagi N, Hatakeyama D, Yokoyama K. Fibrous dysplasia in the maxilla: Possible mechanism of bone remodeling by calcitonin treatment. Oral Oncol. 2003;39:301–5. doi: 10.1016/s1368-8375(02)00037-4. [DOI] [PubMed] [Google Scholar]
- 25.Feingold RS, Argamaso RV, Strauch B. Free fibula flap mandible reconstruction for oral obstruction secondary to giant fibrous dysplasia. Plast Reconstr Surg. 1996;97:196–201. doi: 10.1097/00006534-199601000-00032. [DOI] [PubMed] [Google Scholar]
- 26.Alvares LC, Capelozza AL, Cardoso CL, Lima MC, Fleury RN, Damante JH. Monostotic fibrous dysplasia: A 23-year follow-up of a patient with spontaneous bone remodelling. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;107:229–34. doi: 10.1016/j.tripleo.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 27.Park BY, Cheon YW, Kim YO, Pae NS, Lee WJ. Prognosis for craniofacial fibrous dysplasia after incomplete resection: AGE and serum alkaline phosphatise. Int J Oral Maxillofac Surg. 2010;39:221–6. doi: 10.1016/j.ijom.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 28.Chen YR, Noordhoff MS. Treatment of craniomaxillofacial fibrous dysplasia: How early and how extensive? Plast Reconstr Surg. 1990;86:835–42. [PubMed] [Google Scholar]