

Abstract
Wilson's disease (WD) has varied phenotypic presentations. Here we report the case of a 16-year-old boy who presented with a history of multiple pathological fractures, severe joint deformities, hepatic dysfunction, cognitive decline and limb dystonia. On examination, the patient had pinched out facies, pallor and leukonychia totalis. Bilateral Kayser Fleischer (KF) ring was present. Musculoskeletal examination revealed pectus carinatum, bilateral genu valgus and gun-stock deformity of the left elbow joint. Splenomegaly and moderate ascites were present. Neurological examination revealed mild rigidity and intermittent episodes of dystonic posturing of all four limbs. On this basis a diagnosis of WD with dystonia with cirrhosis of liver with portal hypertension with renal tubular acidosis with renal rickets was thought likely. Investigations confirmed the diagnosis. The patient was started on treatment but he did not improve. He suffered aspiration pneumonia during his hospital stay and succumbed to the illness.
Background
There is a wide variety of phenotypic presentations of Wilson's disease (WD). Generalised osteoporosis or renal rickets could be presenting features of WD. WD is rarely considered as an important differential diagnosis of rickets especially in developing countries like India where nutritional rickets is very common. This case highlights the point that WD should be considered as one of the aetiology in rickets especially when other manifestations like hepatic or neurological manifestations are present. It should also be noted that rarely WD may present as resistant renal rickets without any obvious neurological or hepatic problem.
Case presentation
A 16-year-old previously healthy male presented to our tertiary care hospital with complaints of abnormal posturing of all four limbs for 7 days. There was no history of tongue bite, loss of consciousness or urinary or stool incontinence associated with it. Further the parents gave a history of three episodes of fractures of long bones of limbs with trivial trauma over the last 6 years. The patient developed severe joint deformities of both knees causing him to be bed-ridden for the last 6 months. There was history suggestive of cognitive decline for the last 3 years leading to the child dropping out of school. There was also history of jaundice 1 year ago which was followed by on and off episodes of malena for the last 6 months. However, there was no history of haematemesis. There was no history of diminution of vision, dysarthria or dysphagia. There was no history of bladder or bowel problems. The parents confirmed that there was no history of psychiatric complaints. The family history was unremarkable.
The patient was thinly built and emaciated. His vitals were normal except for mild tachycardia. He had pallor. There were no icterus or oedema feet. He had leukonychia totalis and pinched out facies suggesting protein malnutrition. Eye examination revealed bilateral complete golden brown ring just inside the limbus which was later on confirmed on slit lamb examination as Kayser Fleischer (KF) ring (figure 1). There was no evidence of cataracts. Musculoskeletal examination also revealed pectus carinatum and bilateral genu valgus along with gun-stock deformity at the left elbow joint due to malunion after previous fracture. Per abdominal examination revealed grade 2 splenomegaly and free fluid. Respiratory and cardiovascular system examination did not reveal any abnormality. Patient was drowsy and did not cooperate to higher mental function examination. There was no obvious cranial nerve palsy. Motor system examination revealed mild rigidity in all four limbs. There was no obvious focal neurological deficit. Sensory examination was normal. Deep tendon reflexes were normal and plantars were flexors. During hospital stay patient had intermittent episodes of dystonic posturing of all four limbs.
Figure 1.
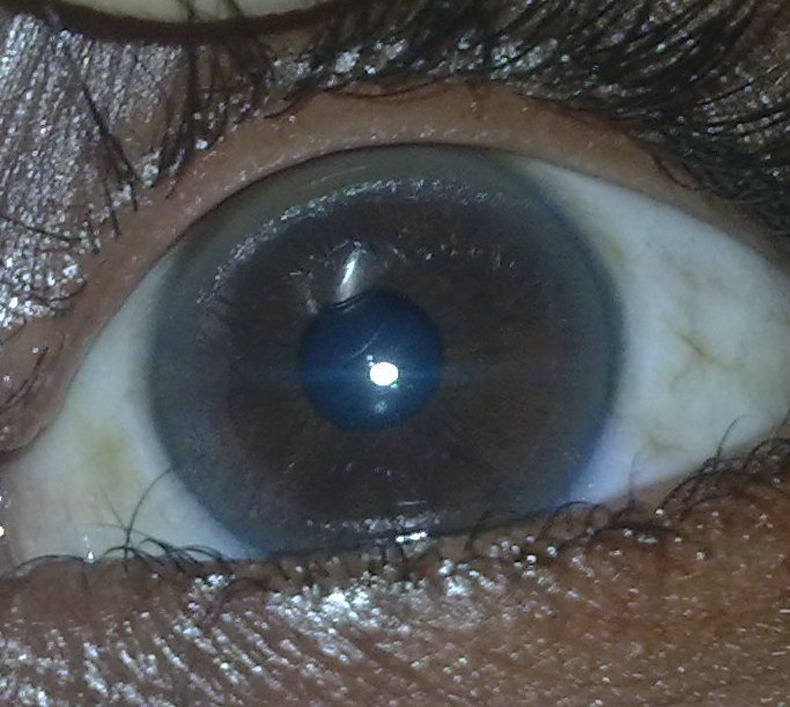
Naked eye appearance of Kayser Fleischer ring.
Investigations
The routine blood investigations of patient revealed mild anaemia and thrombocytopaenia with normal leucocyte count. His serum sodium and potassium and renal functions were normal. His liver functions showed hypoalbuminaemia, mildly raised bilirubin (1.6 mg/dl) along with raised serum glutamic oxaloacetic transaminase (120 IU/l) and serum glutamic pyruvic transaminase (110 IU/l). His coagulation profile was deranged with international normalised ratio of 1.5. His ultrasonography of abdomen was suggestive of cirrhosis of liver, splenomegaly and moderate free fluid. He was subjected to upper gastrointestinal scopy which revealed grade 2 oesophageal varices. His serum ceruloplasmin was found low and 24 h urinary copper was high (541.5 µg). Urine examination revealed abnormally high pH along with high urinary calcium. His serum total and ionic calcium were low while phosphorus was below normal. During hospital stay patient developed pathological fracture of left humerus. The radiogram of left arm revealed not only fracture but also gross osteomalacia (figure 2). The MRI of brain was suggestive of increased signal intensities in basal ganglia and thalami on T2-weighted images along with diffuse age-inappropriate cerebral atrophy (figure 3).
Figure 2.
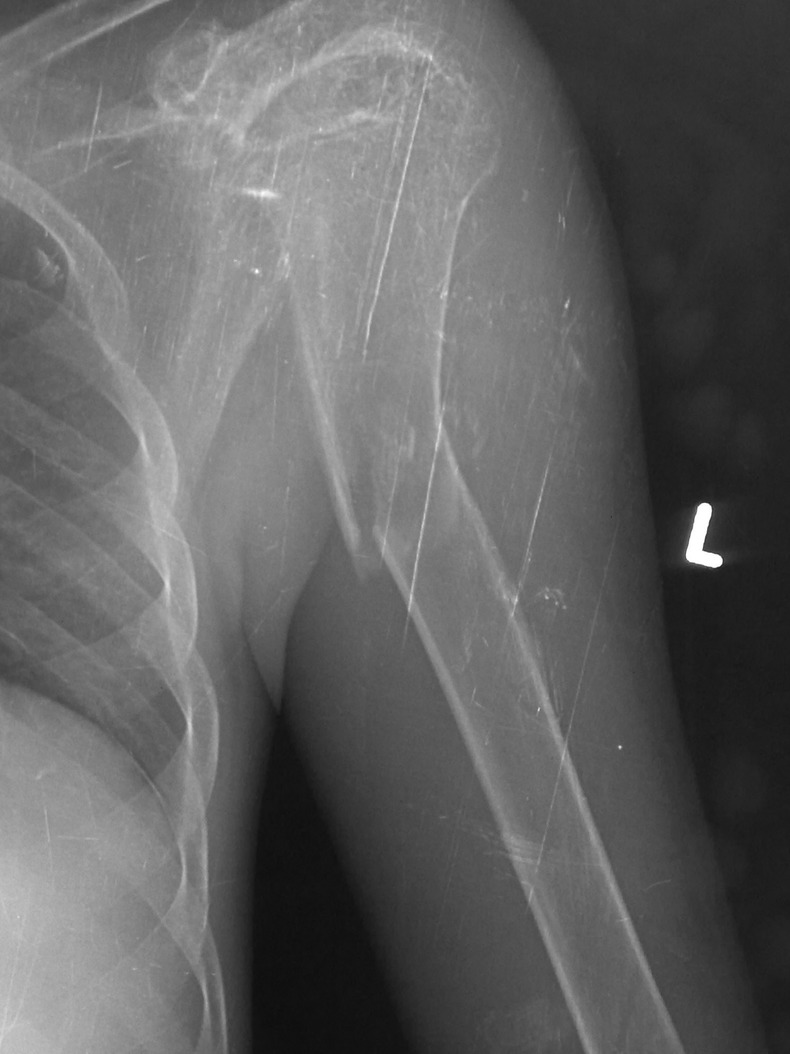
Left humerus radiogram showing gross osteomalacia including fracture.
Figure 3.
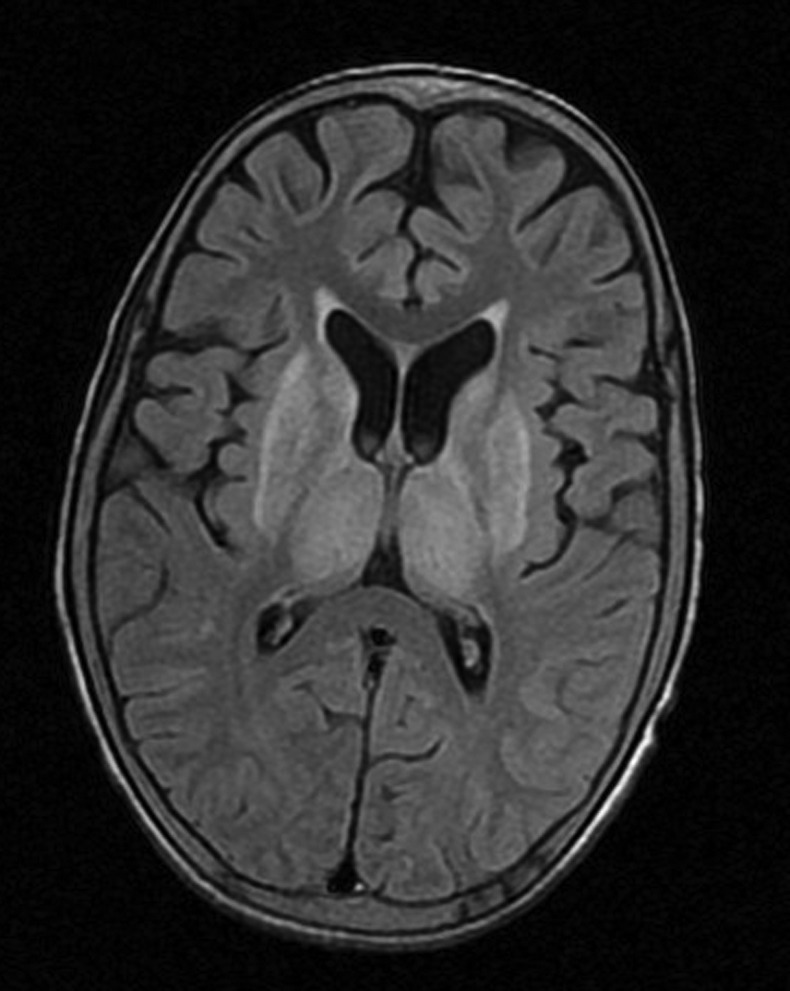
MRI of brain showing increased signal intensities in basal ganglia and thalami on T2-fluid attenuated inversion recovery image.
Differential diagnosis
On this basis a diagnosis of WD with dystonia with cirrhosis of liver with portal hypertension with renal tubular acidosis with renal rickets was kept.
Treatment
The patient was started on zinc and d-penicillmine. He was given trihexyphenydyl for dystonia, propranolol for portal hypertension and oral sodium bicarbonate, calcium and vitamin D3 supplementation for renal tubular acidosis.
Outcome and follow-up
The patient did not show any significant improvement in the first 5 days of his hospital stay. Later he developed bilateral aspiration pneumonia and succumbed to the illness on the next day.
Discussion
During the last few decades, inherited metabolic disorders have acquired much importance due to improved biochemical and genetic testing. Among these disorders, WD (hepatolenticular degeneration) is of very great interest to the clinicians because it is a potentially curable illness if recognised early.1
Recently there have been dramatic advances in the characterisation of the genetic basis for WD as well as in its treatment.2 WD is an inborn error of copper metabolism characterised by accumulation of copper in the brain, the liver, cornea and other tissues.3 It is an autosomal-recessive disorder resulting from mutation in the ATP7B gene on chromosome 13. About 300 specific mutations have been identified. There is a wide variety of phenotypic presentations of WD which could be due to genetic heterogeneity and various types of mutations involved.3
Clinical presentation of WD in India is different than that of the Western world. Wadia et al observed that generalised osteoporosis or renal rickets could be prominent manifestations of WD. As reported by a few other Indian authors, rickets and osteoporosis could be presenting features of WD.3 The musculoskeletal syndrome of WD seems to be more common in the Indian subcontinent.4
Osteomalacia, spontaneous fractures, osteoarthritis, osteochondritis dissecans, chondrocalcinosis and subchondral cyst formation have been well described in the literature. A total of 88% of patients with WD have radiographic evidence of osteoporosis.4 Various mechanisms implicated in the pathogenesis of musculoskeletal manifestations of WD are:
A genetic variant of WD;
Secondary to renal tubular acidosis,
Hypoparathyroidism;
Prolonged immobilisation;
Due to hepatic dysfunction.5
WD is rarely considered as an important differential diagnosis of rickets especially in developing countries like India where nutritional rickets is very common. This case highlights the point that WD should be considered as one of the aetiology in rickets especially when other manifestations like hepatic or neurological manifestations are present. It should also be noted that rarely WD may present as resistant renal rickets without any obvious neurological or hepatic problem.6
Renal involvement in the form of renal tubular acidosis (RTA) was proven by non-anion gap hyperchloremic metabolic acidosis, high urinary pH and hypercalciuria. We did not find out the exact type of RTA as it could make little difference in our treatment of the underlying disease. Osteopaenia in these patients is because of acidosis. Acidosis may interrupt metabolic transformation of calcium phosphate to hydroxyapatite. It also causes hyperphosphaturia, hypercalciuria and disturbed 1,25(OH)D3 production.6 WD as a cause of renal tubular dysfunction can be easily missed if not sought for.
The aim of this case report was to highlight the importance of carefully searching for rare causes of intractable rickets like WD. This patient had only recurrent pathological fractures over the last 6 years. He had not had any neurological complaints until 3 years ago. Had the treating doctor suspected and diagnosed the patient with WD as a cause of osteomalacia and recurrent pathological fractures and had treated the patient adequately, the patient might not have developed hepatic and neurological manifestation and possibly could have had a good prognosis.
Learning points.
Wilson's disease (WD) has varied phenotypic presentations. Musculoskeletal syndrome of WD is more common in the Indian subcontinent.
Renal rickets and osteoporosis could be presenting features of WD.
WD should be considered as one of the aetiology in rickets especially when other manifestations like hepatic or neurological manifestations are present.
Footnotes
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Taly AB, Prashanth LK, Sinha S. Wilson's disease: an Indian perspective. Neurol India 2009;2013:528–40 [DOI] [PubMed] [Google Scholar]
- 2.Pfeiffer RF. Wilson's disease. Semin Neurol 2007;2013:123–32 [DOI] [PubMed] [Google Scholar]
- 3.Goyal JP, Kumar N, Rao SS, et al. Wilson's disease presenting as resistant rickets. Gastroenterol Res 2011;2013:34–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palkar AV, Shrivastava MS, Padwal NJ, et al. Renal tubular acidosis due to Wilson's disease presenting as metabolic bone disease. BMJ Case Rep. Published 11 Aug 2011. doi:10.1136/ bcr.04.2011.4121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rais NN, Dalal DD, Bhandarkar SD, et al. Wilson's disease (a case report). J Postgrad Med 1980;2013:149–51 [PubMed] [Google Scholar]
- 6.Olia MB, Haghighi A. Vitamin D-resistant rickets as presenting feature of Wilson's disease. JIACM 2004;2013:277–80 [Google Scholar]