

Abstract
The autism spectrum disorders (ASDs) are a complex group of neuropsychiatric conditions involving language, social communication, and mental flexibility. Here, we attempt to place recent genetic advances within a developmental and anatomical context. Recent progress in identifying ASD candidate genes supports involvement of multiple brain regions, including the frontal lobes, anterior temporal lobes, caudate, and cerebellum. Understanding genetic data within an anatomical context will be critical to explain how individual risk factors operate to shape phenotypic presentation in patients.
The understanding and treatment of neurodevelopmental disorders pose many challenges to biomedicine, not the least of which is that many of the most common clinical diagnoses such as dyslexia, attention-deficit/hyperactivity disorder, or generalized intellectual disability are not defined on the basis of etiology but rather with regard to behavior and cognition. Perhaps nowhere is this more salient than in the ASDs, defined by clinical impairment in language, social interaction, and behavioral flexibility, all prior to 3 years of age. Variation in an ever-growing number of genes appears to modulate risk and presentation,1 and thus it is perhaps not surprising that multiple brain structures have been implicated in these disorders. Stepping back, however, emerging data support the notion that the ASDs can be conceptualized in terms of multiple genetic etiologies that disrupt the development and function of brain circuits mediating social cognition and language.2
The identification of genetic risk factors for ASDs has proceeded by multiple parallel and complementary approaches (Figure). Early insights came from the recognition that individuals with a handful of single-gene syndromes (eg, fragile X syndrome, tuberous sclerosis, and Joubert syndrome) show features of the ASDs at frequencies much higher than expected. Cytogenetic studies identified recurrent, maternally inherited duplications of chromosome 15q11–13, which along with other rare chromosomal abnormalities remains an important cause of the ASDs.3 More recently, individual genes of major effect (eg, NLGN4, NRXN1, and SHANK3) have been identified by resequencing or array-based methods. Although collectively accounting for an estimated 15% of cases, variants at these and other loci are present in no more than 1% to 2% of children with an ASD. Specificity is also an issue here, with such variants having been observed not only in individuals with an ASD diagnosis but also in patients with related conditions such as nonsyndromic mental retardation.4
Figure.
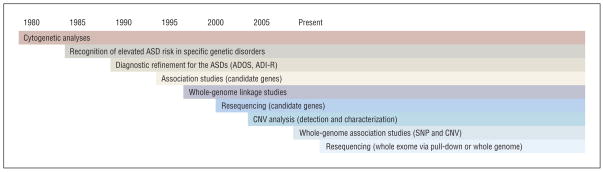
Methodological changes have accelerated progress in autism spectrum disorder (ASD) genetics. The collection of large cohorts via international collaboration together with array-based technologies permitting genome-wide interrogation of genetic variation has resulted in major advances. Similar progress will come from massively parallel sequencing of partial and whole genomes. Although such experiments are soon likely to become routine, interpretation of results, particularly in the context of diverse phenotype data, will require massive computational infrastructure. ADOS indicates Autism Diagnostic Observation Schedule; ADI-R, Autism Diagnostic Interview–Revised; CNV, copy number variation; and SNP, single-nucleotide polymorphism.
In addition to rare alleles of major effect, association studies have demonstrated involvement of numerous common variants with smaller effects.5 Although independently replicated linkage findings are the exception rather than the rule, such studies have proven helpful in the establishment of the veracity of key loci, including those on chromosomes 7q and 17q. Together, these studies underscore the commonly held notion that ASD risk is complex and is likely to involve many different genes and even distinct modes of inheritance among individuals. Large cohorts with 10 000 or more well-phenotyped cases, family members, and unrelated control subjects will be required to fully explore the genetic architecture of the ASDs.
FROM GENES TO PATHOPHYSIOLOGY?
As the list of established genetic risk factors grows, an important challenge will be to understand the manner by which individual genes shape the development and function of regions and circuits affected in disease. Until recently, disorders like fragile X syndrome, Timothy syndrome, tuberous sclerosis, and Smith-Lemli-Opitz syndrome were not considered by many to be bona fide causes of autism but rather clinical peculiarities only tangentially related to the true issues at hand. Recent work demonstrating that no individual cause of autism accounts for more than 1% to 2% of cases, however, has made the importance of these and other ASD-related syndromes more apparent. Moreover, because these syndromic cases are genetically more homogeneous than corresponding “idiopathic” ASDs, it is reasonable to posit that study of these genetic syndromes, where careful contrasts can be made between individuals with defined genotypes, will provide important insights. As summarized here, study of different ASD-related syndromes and monogenic risk factors supports shared involvement of frontal and temporal neocortex, caudate, and cerebellum.
Accelerated early head growth has also been observed among individuals with fragile X syndrome,6 similar to results from idiopathic ASD cases (see later). Although such effects were observed in all mutation carriers relative to control subjects, they were seen a full year earlier in individuals with fragile X syndrome who also carry an ASD diagnosis. Structural imaging of fragile X syndrome cases and controls has also found abnormalities in the caudate (increased), lateral ventricles (increased), and posterior vermis of the cerebellum (reduced).7 Functional magnetic resonance imaging evidence also provides support for involvement of frontostriatal circuitry among cases.8 Dendritic spine maturation is also abnormal in cases, appearing long and thin in cases compared with controls, consistent with an immature phenotype.9,10
Similar results emerge from an analysis of Rett syndrome cases, despite being complicated by behavioral regression, progressive atrophy, and a global (as opposed to regional) hypoplasia.11 A positive relationship between patient age and cerebellar atrophy, however, points to this structure as an important mediator of disease progression. Similarly, frontal and temporal cortices—as well as the caudate—appear subject to the greatest regional reductions in gray matter volume.12 Frontal, motor, and temporal cortices are also implicated by neuropathological investigations, as pyramidal neurons within these regions show reduced dendritic arborization.13,14
Although less is known about Joubert syndrome, which unlike fragile X syndrome can result from mutations in at least 9 identified genes at 10 or more loci, available data suggest that pathological findings in cases are not without parallels. Affected individuals present with hypoplasia of the cerebellar vermis and the molar tooth sign, visible in transverse section as an enlargement of the cerebellar peduncles and associated interpeduncular cistern along with a reduction in the diameter of the midbrain. Examination of phenotypically discordant monozygotic twins makes an important contribution to the understanding of these features—although the molar tooth sign was present in both girls, only the severely affected sibling showed cerebellar hypoplasia.15 Observation of polymicrogyria in some cases with mutations in the AHI1 gene16 suggests that cortical dysgenesis may also be involved in disease pathogenesis, although this feature is not consistent among all families even when considering only mutations in this gene.17
Cortical dysplasia and abnormalities in neuronal migration in patients homozygous for a frameshift mutation within the CNTNAP2 gene18 likewise provide support for an early developmental insult, particularly in the frontal and temporal neocortex, in this ASD-related syndrome. All subjects homozygous for the frameshift mutation had seizures and language regression, and two-thirds of these also met Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition)19 criteria for an ASD. Examination of resected tissue from temporal cortex showed cortical thickening, abnormal neuronal organization and morphology, and ectopic neurons in subcortical white matter. Of particular interest and potentially complicating the use of model systems to study involvement of this gene in disease, the human transcript is highly restricted to corticostriato-thalamic circuitry known to subserve executive function and language, whereas expression in mouse and rat is relatively unremarkable within the telencephalon.20 Consistent with the use of zebra finch as a model system, recent work demonstrates enrichment of Cntnap2 in song nuclei essential for vocal learning.21 Like the sexual dimorphic behavior itself, enrichment of messenger RNA in song nuclei was male specific.
The manner by which tuberous sclerosis affects neurodevelopment and brain function is complicated by the variability in localization, number, and size of the disease-associated growths (tubers) that accumulate in the developing brain.22 Consistent with pathological findings in other syndromes, however, a comparison of IQ-matched patients with tuberous sclerosis with and without a diagnosis of autism identified altered energy metabolism in temporal neocortex, caudate, and cerebellum among ASD cases.23 Other related work in tuberous sclerosis cases with an ASD identified a relationship between clinical presentation and tuber localization.24–26 Most consistent here is the finding that patients with cerebellar lesions are more severely affected in terms of ASD symptoms compared with patients with lesions restricted to other brain regions. As with patients with homozygous mutations in CNTNAP2, white matter heterotopias may also contribute to ASD features and/or seizures.18
Together, these results suggest that these different monogenic risk factors for autism share a common involvement of frontal and temporal neocortex, caudate, and cerebellum. In some cases, these mutations may act on distinct brain regions to give rise to overlapping neurobehavioral phenotypes, consistent with the notion of disconnection or circuit disruption.2 The advent of new imaging paradigms, such as the study of structural connectivity between brain regions by diffusion tensor imaging or brain networks using functional magnetic resonance imaging, and the extension of these studies to other rare autism-related syndromes promise to further refine our knowledge of the core circuits shared by monogenic disorders that result in ASD.
BRAIN GROWTH IS CONSISTENTLY ALTERED IN ASDs
Idiopathic ASD cases—diverse with regard to both etiology and presentation—have been most extensively studied. At the same time, because of this extensive variability, a study of 10 or 20 randomly selected autistic patients may be best described as individuals with 10 or more different disorders. It is therefore not surprising that relatively little has emerged as consistent in terms of neuropathological findings among idiopathic cases. Not all is lost, however. The well-replicated finding of accelerated postnatal growth in defined brain regions of cases compared with controls27 suggests that a major subset of known and yet to be identified risk factors must contribute to this shared phenotype. Exceptions exist,28 but similar observations have been made by a number of groups in both simplex and multiplex cohorts.29–31 Evidence of early accelerated head growth is intriguing in the context of other work showing selective increases in late-developing white matter in both autism and developmental language disorder,32 suggestive of disrupted connectivity in both conditions.33 Available data support preferential involvement of specific regions, including frontal and temporal cortex, cerebellum, and amygdala,34 nicely paralleling both results reviewed earlier and histopathological studies in idiopathic cases summarized later.
As noted elsewhere,27 such regional effects need not point to differential involvement between brain regions but may simply reflect asynchronous development between them. Consistent with this notion, it is well established that frontal gray matter and the white matter tracts projecting to it show protracted growth.35 Likewise, not all regions are bigger in cases compared with controls. The corpus callosum, the major white matter tract underlying interhemispheric information transfer, for example, is smaller by about 10% in cases compared with controls.36 Separate work showing that frontal and temporal sulci are abnormally shifted in older affected children37 together with evidence of prominent frontal enrichment in the expression of several established ASD genes (B.S.A. and D.H.G., unpublished data, 2009, and findings by Abrahams et al20), however, suggest that these regional findings are most likely the result of differential involvement. These observations also suggest that other genes with such focal frontal or temporal cortical localization in early human brain development may represent candidates of particular interest. Similarly, the protracted patterning of the frontal and temporal cortex may contribute to both the frequency of the ASDs as well as the observed clinical heterogeneity. Although available evidence is limited, regions implicated by imaging and pathological findings generally fit with what is known about the anatomy of social cognition and language, providing a sense of validation between genetics and brain circuits.20,38 Evidence for abnormal development in the second year of life27 together with deficits in structure and function in adults provide a good entry point into neuropathological studies, which we will now discuss.
NEUROPATHOLOGICAL STUDIES REVEAL SOME CONSISTENCY BUT ALSO HIGHLIGHT HETEROGENEITY
The most consistent neuropathological finding among the ASDs is the observation of errors in neuronal migration,39 particularly in frontal and temporal lobes. Like findings from syndromic cases discussed earlier, however, these results suggest that a diverse number of missteps can give rise to an ASD. One intriguing observation is based on the examination of cortical minicolumns: organizations of cells, perpendicular to that pial surface, thought to contribute to integration of neuronal information across cortical layers. While cortical thickness at least on average appears similar between cases and controls,40 examination of spacing between minicolumns revealed that relative to typically developing individuals, distance is reduced in the frontal and temporal cortex of individuals with a spectrum condition.41
Like the accelerated frontal growth described earlier, narrow minicolumns in cases may represent a pathological feature able to transcend individual causes. Recent results indicating similarly narrow columnar spacing among distinguished scientists42 fit with the notion that variation here occurs along a continuum that may be related to specific aspects of cognition as opposed to affection status. Because minicolumn findings are predominant in the frontal and temporal cortex, results mesh well with expression patterns for known ASD genes CNTNAP220 and MET (Zohar Mukamel, PhD, and D.H.G., 2009, unpublished data); it is possible that variation in additional genes yet to be identified that have similar patterns of restricted expression may also be contributory to specific aspects of brain patterning modulating core aspects of disease.
The amygdala, involved in the modulation of social behavior, has long been implicated in the ASDs. Early work from Bauman and Kemper43 in which histopathological analyses in a control subject were contrasted against those in an individual with autism and seizures determined that neurons in the amygdala were abnormally small and showed elevated packing density. Subsequent work using modern stereological methods in a larger, seizure-free cohort found different, albeit related, abnormalities with fewer neurons in the amygdala of cases compared with controls.44 The fact that the effects were most prominent in the lateral nucleus of the amygdala is intriguing given that a reduced neuronal number has also been observed in this structure in schizophrenia.45
Finally, no discussion of ASD histopathological findings would be complete without mention of anomalies detected in Purkinje cells of the cerebellum.39,43,46 Although recent stereologically based methods found no significant groupwise differences between cases and controls,47 this work did observe abnormalities in a full half of all probands. Rather than being dismissed as inconsistent, these results highlight the need to pursue such investigations with a focus on individual cases in the context of genetic and environmental factors that contribute toward etiology. Averaging across cases likely to have many different etiologies only serves to obscure observations likely to hold in more homogeneous subgroups. Good neuropathological analyses on large numbers of well-phenotyped patients of known head size and the integration of these data with genetic analyses will permit the determination of how specific risk factors shape developmental brain patterning and subsequently come to influence behavior.
CONCLUSIONS AND A LOOK TO THE FUTURE
Understanding how broadly expressed genes give rise to regionalized deficits in brain anatomy and connectivity represents a major challenge for the field. Although a variety of hypotheses will need to be explored, one possibility is that such molecules may lead to localized findings via interaction with other, more focally expressed molecules. Alternatively, subtle but global disruption of brain circuits may preferentially affect the more vulnerable higher-order association areas that depend heavily on the precise timing of input from other regions. Asymmetrical brain development and function are also critical, particularly given observed abnormalities in the ASD brain.48 Further complicating matters is that at least some of the structures and circuits relevant to known pathophysiology have diverged substantially between humans and rodents. A key hypothesis that integrates these potential mechanisms and accommodates the ever-growing list of candidate genes1 is that connectivity between frontal, temporal, and additional interconnected regions mediating language and social behavior is critical to understanding the ASDs.2 Regardless, the union of genetics and anatomy through concurrent investigation of etiology and pathology within individual cases represents an entry point into disease mechanisms and provides hope for improved patient care.
Acknowledgments
Funding/Support: The work in the Geschwind laboratory is supported by Autism Speaks, the Cure Autism Now Foundation, and grants U54 MH68172, P50 HD055784, R01 MH64547, and R37 MH60233 from the National Institute of Mental Health.
Footnotes
Financial Disclosure: None reported.
Additional Contributions: We thank the families who participate in our genetic studies, especially the Autism Genetic Resource Exchange (http://www.AGRE.org), and our many colleagues and collaborators.
Author Contributions: Study concept and design: Abrahams and Geschwind. Analysis and interpretation of data: Abrahams and Geschwind. Drafting of the manuscript: Abrahams and Geschwind. Critical revision of the manuscript for important intellectual content: Abrahams and Geschwind. Obtained funding: Geschwind. Administrative, technical, and material support: Geschwind. Study supervision: Geschwind.
References
- 1.Abrahams BS, Geschwind DH. Genetics of autism. In: Speicher MR, Antonarakis SE, Motulsky AG, editors. Human Genetics: Problems and Approaches. 4. Heidelberg, Germany: Springer-Verlag; 2010. [Google Scholar]
- 2.Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17(1):103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 3.Vorstman JA, Staal WG, van Daalen E, van Engeland H, Hochstenbach PF, Franke L. Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol Psychiatry. 2006;11(1):1, 18–28. doi: 10.1038/sj.mp.4001781. [DOI] [PubMed] [Google Scholar]
- 4.Laumonnier F, Bonnet-Brilhault F, Gomot M, et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet. 2004;74(3):552–557. doi: 10.1086/382137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9(5):341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiu S, Wegelin JA, Blank J, et al. Early acceleration of head circumference in children with fragile X syndrome and autism. J Dev Behav Pediatr. 2007;28 (1):31–35. doi: 10.1097/01.DBP.0000257518.60083.2d. [DOI] [PubMed] [Google Scholar]
- 7.Reiss AL, Abrams MT, Greenlaw R, Freund L, Denckla MB. Neurodevelopmental effects of the FMR-1 full mutation in humans. Nat Med. 1995;1(2):159–167. doi: 10.1038/nm0295-159. [DOI] [PubMed] [Google Scholar]
- 8.Hoeft F, Hernandez A, Parthasarathy S, Watson CL, Hall SS, Reiss AL. Frontostriatal dysfunction and potential compensatory mechanisms in male adolescents with fragile X syndrome. Hum Brain Mapp. 2007;28(6):543–554. doi: 10.1002/hbm.20406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wisniewski KE, Segan SM, Miezejeski CM, Sersen EA, Rudelli RD. The fra(X) syndrome: neurological, electrophysiological, and neuropathological abnormalities. Am J Med Genet. 1991;38(2–3):476–480. doi: 10.1002/ajmg.1320380267. [DOI] [PubMed] [Google Scholar]
- 10.Irwin SA, Galvez R, Greenough WT. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex. 2000;10(10):1038–1044. doi: 10.1093/cercor/10.10.1038. [DOI] [PubMed] [Google Scholar]
- 11.Murakami JW, Courchesne E, Haas RH, Press GA, Yeung-Courchesne R. Cerebellar and cerebral abnormalities in Rett syndrome: a quantitative MR analysis. AJR Am J Roentgenol. 1992;159(1):177–183. doi: 10.2214/ajr.159.1.1609693. [DOI] [PubMed] [Google Scholar]
- 12.Subramaniam B, Naidu S, Reiss AL. Neuroanatomy in Rett syndrome: cerebral cortex and posterior fossa. Neurology. 1997;48(2):399–407. doi: 10.1212/wnl.48.2.399. [DOI] [PubMed] [Google Scholar]
- 13.Belichenko PV, Oldfors A, Hagberg B, Dahlstrom A. Rett syndrome: 3-D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport. 1994;5(12):1509–1513. [PubMed] [Google Scholar]
- 14.Armstrong D, Dunn JK, Antalffy B, Trivedi R. Selective dendritic alterations in the cortex of Rett syndrome. J Neuropathol Exp Neurol. 1995;54(2):195–201. doi: 10.1097/00005072-199503000-00006. [DOI] [PubMed] [Google Scholar]
- 15.Raynes HR, Shanske A, Goldberg S, Burde R, Rapin I. Joubert syndrome: monozygotic twins with discordant phenotypes. J Child Neurol. 1999;14(10):649–654. doi: 10.1177/088307389901401005. [DOI] [PubMed] [Google Scholar]
- 16.Dixon-Salazar T, Silhavy JL, Marsh SE, et al. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet. 2004;75(6):979–987. doi: 10.1086/425985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valente EM, Brancati F, Silhavy JL, et al. International JSRD Study Group. AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Ann Neurol. 2006;59(3):527–534. doi: 10.1002/ana.20749. [DOI] [PubMed] [Google Scholar]
- 18.Strauss KA, Puffenberger EG, Huentelman MJ, et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med. 2006;354(13):1370–1377. doi: 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- 19.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- 20.Abrahams BS, Tentler D, Perederiy JV, Oldham MC, Coppola G, Geschwind DH. Genome-wide analyses of human perisylvian cerebral cortical patterning. Proc Natl Acad Sci U S A. 2007;104(45):17849–17854. doi: 10.1073/pnas.0706128104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Panaitof SC, Abrahams BS, Dong H, Geschwind DH, White SA. Language-related Cntnap2 gene is differentially expressed in sexually dimorphic song nuclei essential for vocal learning in songbirds. J Comp Neurol. doi: 10.1002/cne.22318. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Humphrey A, Higgins JN, Yates JR, Bolton PF. Monozygotic twins with tuberous sclerosis discordant for the severity of developmental deficits. Neurology. 2004;62(5):795–798. doi: 10.1212/01.wnl.0000113745.58425.ef. [DOI] [PubMed] [Google Scholar]
- 23.Asano E, Chugani DC, Muzik O, et al. Autism in tuberous sclerosis complex is related to both cortical and subcortical dysfunction. Neurology. 2001;57(7):1269–1277. doi: 10.1212/wnl.57.7.1269. [DOI] [PubMed] [Google Scholar]
- 24.Bolton PF, Griffiths PD. Association of tuberous sclerosis of temporal lobes with autism and atypical autism. Lancet. 1997;349(9049):392–395. doi: 10.1016/S0140-6736(97)80012-8. [DOI] [PubMed] [Google Scholar]
- 25.Weber AM, Egelhoff JC, McKellop JM, Franz DN. Autism and the cerebellum: evidence from tuberous sclerosis. J Autism Dev Disord. 2000;30(6):511–517. doi: 10.1023/a:1005679108529. [DOI] [PubMed] [Google Scholar]
- 26.Eluvathingal TJ, Behen ME, Chugani HT, et al. Cerebellar lesions in tuberous sclerosis complex: neurobehavioral and neuroimaging correlates. J Child Neurol. 2006;21(10):846–851. doi: 10.1177/08830738060210100301. [DOI] [PubMed] [Google Scholar]
- 27.Courchesne E, Pierce K, Schumann CM, et al. Mapping early brain development in autism. Neuron. 2007;56(2):399–413. doi: 10.1016/j.neuron.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 28.Dissanayake C, Bui QM, Huggins R, Loesch DZ. Growth in stature and head circumference in high-functioning autism and Asperger disorder during the first 3 years of life. Dev Psychopathol. 2006;18(2):381–393. doi: 10.1017/S0954579406060202. [DOI] [PubMed] [Google Scholar]
- 29.Dementieva YA, Vance DD, Donnelly SL, et al. Accelerated head growth in early development of individuals with autism. Pediatr Neurol. 2005;32(2):102–108. doi: 10.1016/j.pediatrneurol.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 30.Hazlett HC, Poe M, Gerig G, et al. Magnetic resonance imaging and head circumference study of brain size in autism: birth through age 2 years. Arch Gen Psychiatry. 2005;62(12):1366–1376. doi: 10.1001/archpsyc.62.12.1366. [DOI] [PubMed] [Google Scholar]
- 31.Dawson G, Munson J, Webb SJ, Nalty T, Abbott R, Toth K. Rate of head growth decelerates and symptoms worsen in the second year of life in autism. Biol Psychiatry. 2007;61(4):458–464. doi: 10.1016/j.biopsych.2006.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herbert MR, Ziegler DA, Makris N, et al. Localization of white matter volume increase in autism and developmental language disorder. Ann Neurol. 2004;55(4):530–540. doi: 10.1002/ana.20032. [DOI] [PubMed] [Google Scholar]
- 33.Just MA, Cherkassky VL, Keller TA, Kana RK, Minshew NJ. Functional and anatomical cortical underconnectivity in autism: evidence from an FMRI study of an executive function task and corpus callosum morphometry. Cereb Cortex. 2007;17(4):951–961. doi: 10.1093/cercor/bhl006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carper RA, Courchesne E. Localized enlargement of the frontal cortex in early autism. Biol Psychiatry. 2005;57(2):126–133. doi: 10.1016/j.biopsych.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 35.Levitt P. Structural and functional maturation of the developing primate brain. J Pediatr. 2003;143(4 suppl):S35–S45. doi: 10.1067/s0022-3476(03)00400-1. [DOI] [PubMed] [Google Scholar]
- 36.Schultz RT, Gauthier I, Klin A, et al. Abnormal ventral temporal cortical activity during face discrimination among individuals with autism and Asperger syndrome. Arch Gen Psychiatry. 2000;57(4):331–340. doi: 10.1001/archpsyc.57.4.331. [DOI] [PubMed] [Google Scholar]
- 37.Levitt JG, Blanton RE, Smalley S, et al. Cortical sulcal maps in autism. Cereb Cortex. 2003;13(7):728–735. doi: 10.1093/cercor/13.7.728. [DOI] [PubMed] [Google Scholar]
- 38.Mundy P. Annotation: the neural basis of social impairments in autism: the role of the dorsal medial-frontal cortex and anterior cingulate system. J Child Psychol Psychiatry. 2003;44(6):793–809. doi: 10.1111/1469-7610.00165. [DOI] [PubMed] [Google Scholar]
- 39.Bailey A, Luthert P, Dean A, et al. A clinicopathological study of autism. Brain. 1998;121(pt 5):889–905. doi: 10.1093/brain/121.5.889. [DOI] [PubMed] [Google Scholar]
- 40.Hutsler JJ, Love T, Zhang H. Histological and magnetic resonance imaging assessment of cortical layering and thickness in autism spectrum disorders. Biol Psychiatry. 2007;61(4):449–457. doi: 10.1016/j.biopsych.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 41.Casanova MF, Buxhoeveden DP, Switala AE, Roy E. Minicolumnar pathology in autism. Neurology. 2002;58(3):428–432. doi: 10.1212/wnl.58.3.428. [DOI] [PubMed] [Google Scholar]
- 42.Casanova MF, Switala AE, Trippe J, Fitzgerald M. Comparative minicolumnar morphometry of three distinguished scientists. Autism. 2007;11(6):557–569. doi: 10.1177/1362361307083261. [DOI] [PubMed] [Google Scholar]
- 43.Bauman M, Kemper TL. Histoanatomic observations of the brain in early infantile autism. Neurology. 1985;35(6):866–874. doi: 10.1212/wnl.35.6.866. [DOI] [PubMed] [Google Scholar]
- 44.Schumann CM, Amaral DG. Stereological analysis of amygdala neuron number in autism. J Neurosci. 2006;26(29):7674–7679. doi: 10.1523/JNEUROSCI.1285-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kreczmanski P, Heinsen H, Mantua V, et al. Volume, neuron density and total neuron number in five subcortical regions in schizophrenia. Brain. 2007;130 (pt 3):678–692. doi: 10.1093/brain/awl386. [DOI] [PubMed] [Google Scholar]
- 46.Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57(1):67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- 47.Whitney ER, Kemper TL, Bauman ML, Rosene DL, Blatt GJ. Cerebellar Purkinje cells are reduced in a subpopulation of autistic brains: a stereological experiment using calbindin-D28k. Cerebellum. 2008;7(3):406–416. doi: 10.1007/s12311-008-0043-y. [DOI] [PubMed] [Google Scholar]
- 48.Herbert MR, Ziegler DA, Deutsch CK, et al. Brain asymmetries in autism and developmental language disorder: a nested whole-brain analysis. Brain. 2005;128(pt 1):213–226. doi: 10.1093/brain/awh330. [DOI] [PubMed] [Google Scholar]