

Abstract
Readily available 1-mesyl-1,2,3-triazoles are efficiently converted into a variety of imidazolones and thiazoles by Rh(II)-catalyzed denitrogenative reactions with isocyanates and isothiocyanates, respectively. The proposed triazolediazoimine equilibrium results in the formation of highly reactive azavinyl metal-carbenes, which react with heterocumulenes causing an apparent swap of 1,2,3-triazole core for another heterocycle.
Drug discovery research constantly calls for new synthetic methods for the rapid and reliable construction of heterocyclic frameworks from easily available precursors. Synthetic approaches that enable straightforward syntheses of several different heterocyclic cores from a common intermediate are especially attractive. Recently discovered denitrogenative transformations of readily accessible 1,2,3-triazoles have led to a new family of synthetic methods towards a variety of valuable organic molecules,1 including nitrogen-containing heterocycles.2 For example, imidazoles1c,3 and pyrroles3c,4 can now be accessed through transannulation reactions of 1-sulfonyl triazoles with nitriles and alkynes, wherein the 1,2,3-triazole core is transformed into another heterocycle. Here, we would like to report an efficient synthesis of imidazolones 2 and thiazole derivatives 3 by a Rh(II)-catalyzed denitrogenative transannulation of 1-sulfonyl-1,2,3-triazoles 1 with isocyanates and isothiocyanates, respectively (eq 1).
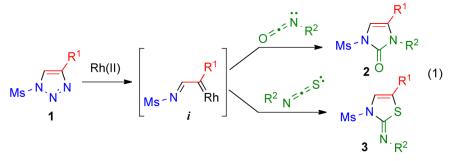 |
A formal [3+2] cycloaddition of heterocumulenes with metalcarbenes derived from diazo compounds has not been extensively studied.5 Moreover, existing literature reports brand isocyanates,5c isothiocyanates5c and carbodiimides5a,b as incompetent reaction partners for Rh(II)-carbenes generated from diazoesters and diazoketones. Only moderate yields of the corresponding heterocyclic products have been reported, and the heterocumulene was used in large excess (typically as a solvent). We and others have recently been successful in employment of Rh(II) azavinyl carbenes i that are conveniently generated from 1,2,3-triazoles.1-4,6 Unlike typical diazo compounds, these readily available7 carbene precursors do not require a slow introduction into the reaction mixture, and a reaction partner is normally used in only a slight excess.8 Given the experimental simplicity of this approach, we envisioned that the addition of heterocumulenes to azavinyl carbenes i could be a promising method for the synthesis of different nitrogen-containing heterocyclic molecules.
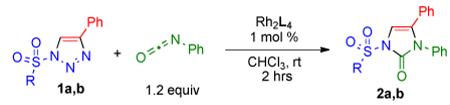
Accordingly, we attempted the reaction of 1-tosyl-4-phenyl-1,2,3-triazole 1a with 1.2 equiv of phenyl isocyanate in the presence of various Rh(II) carboxylate complexes (1 mol %) at ambient temperature (Table 1 and Figure 1). It was anticipated that the attack of the nitrogen lone pair of isocyanate on the azavinyl carbene i would form an “ylide-type” intermediate;3 subsequent cyclization9 would lead to imidazolone 2a. To our delight, the expected heterocyclic product 2a10 was indeed formed in the reaction mixture in moderate yield under these conditions, although the reaction proceeded rather sluggishly. It was found that Rh2(S-PTV)411 6 and Rh2(S-NTV)412 8 catalysts (Figure 1) gave most promising results, although yields did not exceed 60% (Table 1).13
Table 1.
Catalyst Screening for Transannulation with Phenyl Isocyanatea
| L= | Oct | Piv | PTV | PTTL | NTTL | NTV |
|---|---|---|---|---|---|---|
| R = p-Tol (1a) | <5% | 35% | 55% | 51% | 14% | 59% |
| R = Me (1b) | - | 53% | 56% | 89% | 95% | 86% |
Performed on 0.2 mmol scale; NMR yields are shown.
Figure 1.
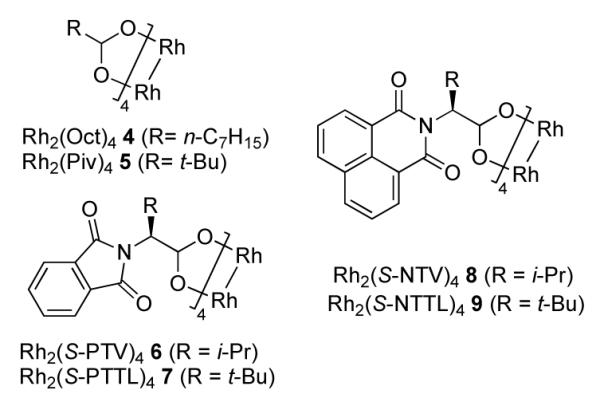
Rhodium(II) carboxylates used in this study.
1-Mesyl-substituted triazoles have previously been recognized as excellent azavinyl carbene precursors in Rh(II)-catalyzed reactions.1a,f,k Therefore, we examined their reactivity in the transannulation with isocyanates. We were pleased to find that triazole 1b underwent the reaction with phenyl isocyanate more efficiently than its 1-tosyl-substituted congener 1a, providing generally higher yields of the corresponding mesyl-substituted product 2b in the presence of various Rh(II) carboxylates (Table 1, Figure 1).13 It was found that complexes 6-7 bearing amino acid derived ligands were superior catalysts compared to simple alkane carboxylates 4-5 (Figure 1). Employment of the bulky Rh2(S-NTTL)412 complex 9 in this reaction allowed for the clean and rapid (1 h) conversion of triazole 1b to the imidazolone 2b in 95% yield (Table 1).
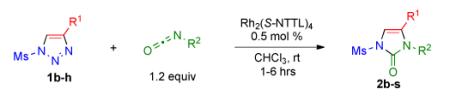
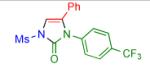
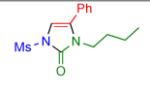
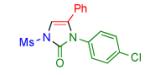
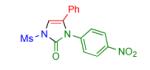
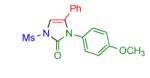
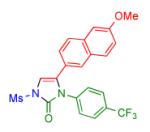
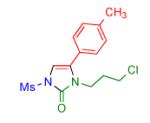
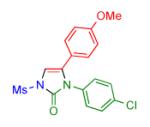
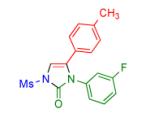
Having identified the optimal substrate-catalyst combination, we examined the scope of the reaction (Table 2). 4-Phenyl triazole 1b reacted smoothly with a wide range of aryl-substituted isocyanates14 to afford the corresponding imidazolones 2 in good to excellent yields (Table 2, entries 1-3 and 7-9). Moreover, 1b underwent this reaction efficiently with primary and secondary alkyl isocyanates, furnishing N-alkyl-substituted heterocyclic products 2n-p in high yields (entries 13-15). Notably, allyl isocyanate reacted with complete chemoselectivity favoring transannulation over cyclopropanation of the double bond (entry 14, Table 2). Next, we examined the scope of this reaction with respect to various aryl substituents at the C-4 position of the triazole. Gratifyingly, substrates possessing electron-rich (entries 6, 12, 17 and 18) and electron-deficient (entries 4, 5, 10 and 16) aryl groups showed similar efficiency in this transannulation reaction and afforded the corresponding imidazolones 2 in high yield (Table 2).
Table 2.
Transannulation of 1-Mesyl-1,2,3-triazoles with Isocyanatesa
| entry | product | yield, %b |
entry | product | yield, %b |
entry | product | yield, %b |
|||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |

|
2b | 95 | 7 |
|
2h | 83 | 13 |
|
2n | 97 |
| 2 |
|
2c | 94 | 8 |
|
2i | 53 | 14 |
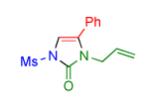
|
2o | 82 |
| 3 |
|
2d | 94 | 9 |
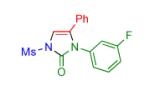
|
2j | 89 | 15 |
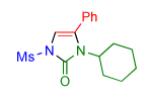
|
2p | 95 |
| 4 |
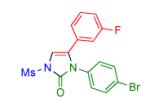
|
2e | 94 | 10 |
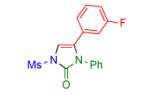
|
2k | 92 | 16 |
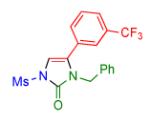
|
2q | 93 |
| 5 |
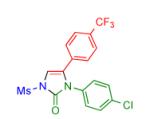
|
2f | 80 | 11 |
|
2l | 69 | 17 |
|
2r | 90 |
| 6 |
|
2g | 85 | 12 |
|
2m | 85 | 18 |
|
2s | 92 |
Procedure: triazole 1 (1.0 mmol), isocyanate (1.2 mmol), Rh2(S-NTTL)4 (0.005 mmol) were stirred in 5 mL of dry chloroform at room temperature for 1-6 hours.
Isolated yield.
Encouraged by the successful transannulation of 1,2,3-triazoles with isocyanates, our interest turned to the reactivity of other heterocumulenes15 in this transformation. To this end, we attempted reaction of phenyl isothiocyanate with triazole 1b in the presence of Rh2(S-NTTL)4 catalyst 9. Previous studies reported that metalcarbenes are likely to be intercepted by the sulfur atom (rather than by the nitrogen lone pair) of isothiocyanates; a subsequent cyclization of the putative sulfur-ylide intermediate furnishes products with an exocyclic nitrogen atom.5c Indeed, transannulation of 4-phenyl-1,2,3-triazole 1b with 1.2 equiv of phenyl isothiocyanate at ambient temperature afforded N-phenyl thiazole imine derivative 3a in 87% isolated yield (Table 3). The identity of 3a was unambiguously confirmed by the single crystal X-ray analysis, which additionally labeled this product as Z-imine.16
Table 3.
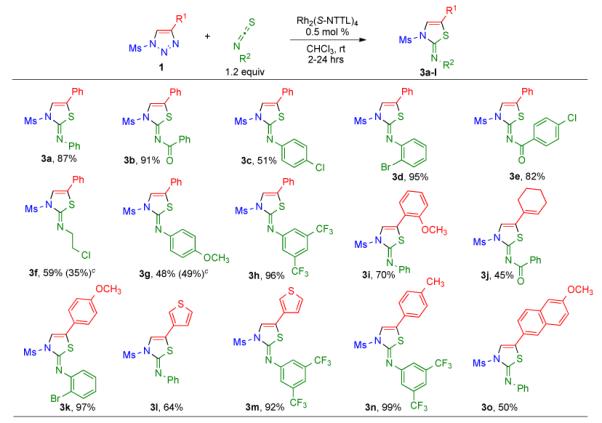
|
Procedure: triazole 1 (1.0 mmol), isothiocyanate (1.2 mmol), Rh2(S-NTTL)4 9 (0.005 mmol) were stirred in 5 mL of dry chloroform at room temperature.
Isolated yields are shown.
Recovered starting material.
Examination of the scope of the Rh(II)-catalyzed transannulation with respect to various 1-mesyl-1,2,3-triazoles 1 and substituted isothiocyanates (Table 3), effectively applying the reaction conditions developed for isocyanates (vide supra), revealed that triazole 1b reacted smoothly with a variety of isothiocyanates under these reaction conditions. In particular, exceptionally good results were obtained with isothiocyanates possessing electron-withdrawing substituents. Thus, reaction with bromo- and trifluoromethyl-substituted aryl isothiocyanates as well as with N-benzoyl-substituted compounds afforded the corresponding thiazole derivatives in excellent yield (e.g. compounds 3b, 3d-e and 3h, Table 3). Electron-rich aryl- and alkyl isothiocyanates reacted more reluctantly, and a significant amount of starting material was recovered even after prolonged (24 hours) reaction times (3f and 3g, Table 3). We tentatively attribute the observed inhibition to the coordination of the formed thiazole product to the rhodium(II) catalyst that blocks the essential reactive site of the latter.
Examination of the scope of this reaction with respect to various substituents at the C-4 position of the triazole revealed that a range of 1-mesyl-substituted 4-aryl and 4-heteroaryl 1,2,3-triazoles produced the corresponding thiazoles 3 in good to excellent yields (3i and 3k-o, Table 3). Moreover, substrate possessing an alkenyl moiety at the C-4 position also participated in this reaction affording 5-alkenyl thiazole 3j, albeit in a moderate yield.17
Further modification of heterocyclic transannulation products 2 and 3 could offer additional synthetic opportunities expanding the repertoire of useful drug-like molecules that can be accessed starting from 1-sulfonyl-1,2,3-triazoles. A sulfonyl group is often used to protect N-H moiety of nitrogen-containing heterocycles and can easily be removed using a number of methods.18 We found that the mesyl group of the imidazolone 2b can be efficiently removed by treatment with magnesium in refluxing methanol producing the parent diphenyl NH-imidazolone19 10 in good yield (Scheme 1). Similarly, treatment of thiazole imine 3m with 1-hydroxybenzotriazole (HOBt) at room temperature resulted in clean desulfonylation, nearly quantitatively furnishing 2-(arylamino)-thiazole 11 (Scheme 1).
Scheme 1.

Desulfonylation of Mesyl-Protected Transannulation Products.
The new method for the construction of imidazolones and thiazoles via a Rh(II)-catalyzed transannulation reaction of 1-sulfonyl-1,2,3-triazoles with heterocumulenes is practical and reliable. This formal [3+2] cycloaddition reaction features metal-stabilized azavinyl carbenes as reactive intermediates, which are conveniently generated from stable and readily available 1-mesyl-1,2,3-triazoles. Together with a straightforward synthesis of triazoles, this new method offers a modular sequence for the formal addition of “S-C-N” and “N-C-N” fragments to the acetylenic triple bonds leading to useful families of five-membered nitrogen-containing heterocycles.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Institute of General Medical Sciences, National Institutes of Health (GM-087620) and National Science Foundation (CHE-0848982). We also thank Dr. Arnold L. Rheingold and Dr. Curtis E. Moore for X-ray analysis.
Footnotes
ASSOCIATED CONTENT Supporting Information Experimental procedures, characterization data, NMR spectra, and crystallographic data (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
Notes The authors declare no competing financial interest.
REFERENCES
- (1) (a).For cyclopropanation, see: Chuprakov S, Kwok SW, Zhang L, Lercher L, Fokin VV. J. Am. Chem. Soc. 2009;131:18034. doi: 10.1021/ja908075u. Grimster N, Zhang L, Fokin VV. J. Am. Chem. Soc. 2010;132:2510. doi: 10.1021/ja910187s. Zibinsky M, Fokin VV. Org. Lett. 2011;13:4870. doi: 10.1021/ol201949h. Chuprakov S, Gevorgyan V. Org. Lett. 2007;9:4463. doi: 10.1021/ol702084f. Alford JS, Davies HML. Org. Lett. 2012;14:6020. doi: 10.1021/ol3029127. For carbenoid insertions, see: Chuprakov S, Malik JA, Zibinsky M, Fokin VV. J. Am. Chem. Soc. 2011;133:10352. doi: 10.1021/ja202969z. Miura T, Biyajima T, Fujii T, Murakami M. J. Am. Chem. Soc. 2012;134:194. doi: 10.1021/ja2104203. For 1,2-alkyl shift, see: Selander N, Worrell BT, Fokin VV. Angew. Chem. Int. Ed. 2012;51:13054. doi: 10.1002/anie.201207820. Miura T, Funakoshi Y, Morimoto M, Biyajima T, Murakami M. J. Am. Chem. Soc. 2012;134:17440. doi: 10.1021/ja308285r. Gulevich AV, Gevorgyan V. Angew. Chem. Int. Ed. 2013;52:1371. doi: 10.1002/anie.201209338. For arylation with boronic acids, see: Selander N, Worrell BT, Chuprakov S, Velaparthi S, Fokin VV. J. Am. Chem. Soc. 2012;134:14670. doi: 10.1021/ja3062453.
- (2).For review, see: Chattopadhyay B, Gevorgyan V. Angew. Chem. Int. Ed. 2012;51:862. doi: 10.1002/anie.201104807.
- (3) (a).Horneff T, Chuprakov S, Chernyak N, Gevorgyan V, Fokin VV. J. Am. Chem. Soc. 2008;130:14972. doi: 10.1021/ja805079v. Zibinsky M, Fokin VV. Angew. Chem., Int. Ed. 2013;52:1507. doi: 10.1002/anie.201206388. For N-fused imidazoles, see: Chuprakov S, Hwang FW, Gevorgyan V. Angew. Chem., Int. Ed. 2007;46:4757. doi: 10.1002/anie.200700804.
- (4) (a).Miura T, Yamauchi M, Murakami M. Chem. Commun. 2009:1470. doi: 10.1039/b819162j. [DOI] [PubMed] [Google Scholar]; (b) Chattopadhyay B, Gevorgyan V. Org. Lett. 2011;13:3746. doi: 10.1021/ol2014347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5) (a).For isolated reports, see: Hubert AJ, Feron A, Warin R, Teyssie P. Tetrahedron Lett. 1976:1317. Drapier J, Feron A, Warin R, Hubert AJ, Teyssie P. Tetrahedron Lett. 1979:559. Bien S, Kimmel T, Tirosh N. Acta Chem. Scand. 1993;47:218.
- (6).Selander N, Fokin VV. J. Am. Chem. Soc. 2012;134:2477. doi: 10.1021/ja210180q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7) (a).Raushel J, Fokin VV. Org. Lett. 2010;12:4952. doi: 10.1021/ol102087r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yoo EJ, Ahlquist M, Kim SH, Bae I, Fokin VV, Sharpless KB, Chang S. Angew. Chem. Int. Ed. 2007;46:1730. doi: 10.1002/anie.200604241. [DOI] [PubMed] [Google Scholar]
- (8) (a).Typically, to avoid the carbene dimerization side reactions, a dilute solution of a diazo compound is added over several hours to a mixture of rhodium catalyst and a large excess of another reagent. Certain 1,2,3-triazoles, however, undergo tautomerization in solution to form α-diazoimines in presumably low concentration. See: Dimroth O. Ann. 1909;364:183. Gilchrist TL, Gymer GE. Adv. Heterocycl.Chem. 1974;16:33. See also ref. 3.
- (9).Although unlikely, alternative concerted [3+2] cyclization path and [4+2] cycloaddition, followed by reductive elimination, may also be operating in this transformation.
- (10).Structure of compound 2a was confirmed by X-ray crystallography (CCDC 888986). See Supporting Information for details.
- (11) (a).Hashimoto S, Watanabe N, Sato T, Shiro M, Ikegami S. Tetrahedron Lett. 1993;34:5109. [Google Scholar]; (b) Tsutsui H, Abe T, Nakamura S, Anada M, Hashimoto S. Chem. Pharm. Bull. 2005;53:1366. doi: 10.1248/cpb.53.1366. [DOI] [PubMed] [Google Scholar]
- (12) (a).Müller P, Allenbach YF, Robert E. Tetrahedron: Asymmetry. 2003;14:779. [Google Scholar]; (b) Müller P, Bernardinelli G, Allenbach YF, Ferry M, Flack HD. Org. Lett. 2004;6:1725. doi: 10.1021/ol049554n. [DOI] [PubMed] [Google Scholar]
- (13).See Supporting Information for a complete catalyst screening table.
- (14).One notable exception includes ortho-substituted aryl isocyanates, employment of which resulted in sluggish and low-yielding reactions, presumably due to the increased steric hindrance at the reaction center. Similarly, triazoles possessing sterically demanding groups at the C-4 reacted with isocyanates hesitantly.
- (15).Attempts to utilize carbodiimides in this reaction were unsuccessful.
- (16).The structure has been deposited with the Cambridge Crystallographic Data Centre (CCDC 888987). See SI for details.
- (17).Presumably, the highly reactive 1-alkenyl carbene undergoes a number of side-reactions (e.g. carbene dimerization); this results in the observed diminished yield in the transannulation reaction.
- (18).Wuts PGM, Greene TW. Protective Groups in Organic Synthesis. 4rd Ed WILEY; New York: 2007. pp. 872–894. [Google Scholar]
- (19).Lee SH, Yoshida K, Matsushita H, Clapham B, Koch G, Zimmermann J, Janda KD. J. Org. Chem. 2004;69:8829. doi: 10.1021/jo048353u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.