

Abstract
Electron microscopy (EM) is an important tool for determining the composition, arrangement and structure of biological macromolecules. When studying structurally heterogeneous samples using EM, classification is a critical step toward achieving higher resolution and identifying biologically significant conformations. We have developed an interactive, web-based tool, called Maskiton, for creating custom masks and performing 2D classifications on aligned single-particle EM images. The Maskiton interface makes it considerably easier and faster to explore the significance of heterogeneity in single-particle datasets. Maskiton features include: resumable uploads to facilitate transfer of large datasets to the server, custom mask creation in the browser, continual progress updates, and interactive viewing of classification results. To demonstrate the value of this tool, we provide examples of its use on several experimental datasets and include analyses of the independent terminus mobility within the Ltn1 E3 ubiquitin ligase, the in-vitro assembly of 30S ribosomal subunits, and classification complexity reduction within Immunoglobulin M. This work also serves as a proof-of-concept for the development of future cross-platform, interactive user interfaces for electron microscopy data processing.
Keywords: electron microscopy, single-particle classification, software, IgM, 30S, Ltn1
Introduction
Transmission electron microscopy (TEM) is a useful tool for determining the composition, arrangement and structure of biological macromolecules. A standard EM methodology is to image samples in a mono-disperse state so that individual particles of the sample can be distinguished and analyzed. Since TEM images of these single-particles are typically extremely noisy, the particles are usually interpreted only after being aligned and averaged to improve the signal-to-noise ratio(Frank, 2006; Ruprecht and Nield, 2001). In many cases, the raw data is also heterogeneous as it can consist of different views of particles in random orientations and structural states. This greatly complicates the task of aligning and sorting particles, but also provides the opportunity for creating three-dimensional reconstructions or observing distinct conformational and compositional states. Characterizing these states, even at relatively low resolution, has provided significant insights into functional mechanisms and relationships. Examples include the flexibility of dynein(Burgess et al., 2003), the assembly of the small 30S subunit of the E coli ribosome(Mulder et al., 2010), domains movements of fatty-acid synthase(Brignole et al., 2009), and flexibility and motions of the Ltn1 complex(Lyumkis et al., 2013).
In TEM, classification broadly refers to the act of sorting heterogeneous datasets into less-heterogeneous subsets so that further processing and interpretation are possible. Classification can be used to improve the resolution of a dataset by identifying and removing ‘bad’ particles, or for distinguishing between distinct structural states present in a dataset. It is common to attempt to exclude portions of datasets from classification if they are known to be unimportant and possibly deleterious to a satisfactory result. The most common example of this is excluding the background around particles using simple circular masks. It is also possible to use customized masks that restrict classifications to specific regions of particles(Penczek et al., 2006), though this is less common.
Examples of algorithms employed in TEM for classification include principal-component analysis(Frank and van Heel, 1982), neural networks(Pascual-Montano, 2001), maximum-likelihood(Scheres et al., 2005), and others. Considerable effort has been put into improving the quality and reliability of classification algorithms so that they can be used with less a priori knowledge or user input(Frey and Dueck, 2007; Scheres et al., 2005; Sorzano et al., 2010). Nevertheless, classification often remains highly dependent on user feedback, since it is an involved, iterative process that requires the investigator to simultaneously judge the validity, and interpret the meaning, of results. Unfortunately, while tools focused on allowing users to interact more seamlessly with their data are of great potential value, they have received far less attention.
In this work we describe a new classification tool, Maskiton, which allows users to create custom masks, launch 2D classifications, and examine their results interactively using a web-based interface. Through the use of custom masks, Maskiton allows focused-classification on specific regions of datasets and direct testing of possible dependencies between different areas of structural heterogeneity. The benefits of Maskiton are demonstrated by its application to three biological case studies: the flexibility of Ltn1 ubiquitin ligase, the assembly of the 30S ribosomal subunit, and the domain movements of Immunoglobulin M.
Methods
Overall Description of Maskiton
Maskiton is a web-based tool for performing 2D classifications of aligned single-particle TEM data. Its primary design goal was to allow custom masks to be easily created and used for focused classification, and to provide fast feedback and results to the user. It consists of a client-side portion that runs locally on the user’s web browser, and server-side components that can be run on one or more machines while serving multiple clients. The client-side portion is responsible for handling user input and displaying results, while the server-side component stores, manages and processes data.
Maskiton User Interface
Users begin by selecting an aligned stack of particles, in IMAGIC format(van Heel and Keegstra, 1981), on their local machine in the web-interface (Figure 1a). Users may also select from a list of datasets that have been made publicly available for demonstration and testing purposes. Selecting an image stack prompts the server to check for the equivalent file in its storage, and if the file is incomplete or missing, requests that the client send the remaining data. This means large uploads can be resumed if stopped or interrupted, a critical feature when the files being uploaded are gigabytes in size. Resumable uploads also obviates the need for server-side infrastructure to annotate and browse previously uploaded data, since the user is left to managing this themselves. While not an explicit security measure, this implementation also has the side-benefit of obfuscating and anonymizing the content of uploaded data, since the server is never given any identifying metadata, such as sample type, etc.
Figure 1.
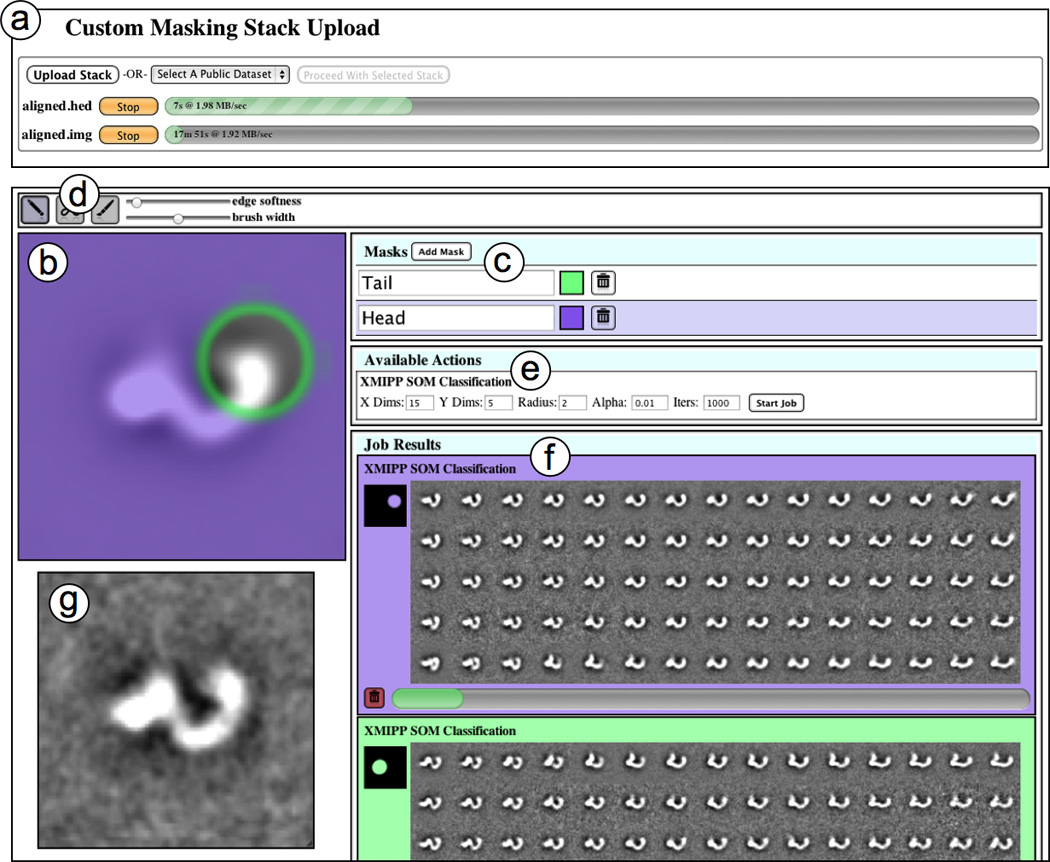
An overview of the Maskiton web interface. This interface allows custom masks to be drawn so that classifications can be focused on specific areas of a dataset. (a) Users can upload new stacks by selecting them on their local machine, or they can select one of the public datasets. Since uploads are resumable, selecting a stack that has already been uploaded is equivalent to selecting the same stack on the server. (b) The stack average is displayed and the mask currently being edited is superimposed on it. The edges of the masked areas are highlighted in green. (c) New masks are created, and can be given labels and colors to keep them organized. The mask superimposed above the stack average is selected by clicking on its entry in the list. (d) Simple drawing tools for editing masks are available. These include a brush, eraser, and sliders for changing width and softness. The softness of mask edges is reflected by the width of the green outline on the mask. (e) The classification parameters are set and new jobs are started on the server by pressing the ‘Start Job’ button. (f) Results for classification jobs started in the current session are displayed, and the results and progress bar are updated as the classification progresses. (g) A zoom-in viewer can be opened by clicking on any of the class images in the results. This viewer displays the class currently beneath the cursor, and allows for movie-like browsing and comparison. This zoom-in view can be moved and resized for convenience.
As soon as a particle stack is uploaded/selected, the server begins calculating the stack’s average so that it can be displayed in the user interface (Figure 1b). The displayed average is updated in the client interface as it is being calculated on the server. Users may begin designing custom masks, or even start classification jobs while the stack average is still being calculated, and results from these classification jobs will be similarly updated as they progress on the server. A variety of drawing tools (Figure 1d) are provided to create the custom masks, and the masks can be assigned labels and colors so that results can be more easily tracked in the interface (Figure 1c). The drawing tools include brushes for painting or erasing portions of masks, and sliders for changing brush width and edge softness. New classification jobs are started by setting classification parameters, described in more detail later, and pressing the ‘Start Job’ button (Figure 1e) in the interface. The class averages produced by classification jobs are then displayed and updated as the classification progresses on the server (Figure 1f). Clicking on a class average in the results panel toggles a zoomed viewer (Figure 1g) that can be moved and resized. While the zoomed viewer is visible, passing the mouse cursor over classes creates ‘movie-like’ transitions in the zoomed view that make it easy to compare them. URLs for the associated jobs are also provided so that they can be saved or shared with collaborators. A video demonstration of this entire process is available in the supplemental materials.
Infrastructure
The Maskiton user-interface is built using HTML5, CSS3 and Javascript. Drawing of custom masks is implemented using the canvas element of HTML5. The client-side code communicates with the server–side component through a RESTful(Fielding, 2000) interface that is used for saving, retrieving, and manipulating masks, images, and jobs. The server-side component of Maskiton is currently composed of three different servers that each handles specific types of resources. An nginx(Reese, 2008) server handles the serving of static content, such as images, a node.js(Tilkov and Vinoski, 2010) server handles file uploads, and a python server handles job submission and progress updates. All three servers can be run on the same machine, or split across separate machines if needed. It should also be possible to run multiple instances from behind HTTP load-balancers if the user load becomes too large for a single server to handle.
The server-side software consists largely of Python scripts that perform bookkeeping and coordination of the many concurrent processes spawned while using Maskiton. These processes make use of several programs from third-party EM software packages. The proc2d program from EMAN(Ludtke et al., 1999) is used for converting between file formats, while programs from the Xmipp(Marabini et al., 1996) package are used for image processing, and classification using the self-organizing map (SOM) (Pascual-Montano, 2001) algorithm. Many of the processing steps have been carefully structured to occur in a progressive, incremental manner, so that intermediate results can be forwarded back to the client quickly and regularly (Figure 2). This is largely accomplished by splitting datasets into small subsets and processing them sequentially rather than as single entities. A further speedup is also achieved, when necessary, by starting with pyramidally binned versions of datasets, and moving through progressively less-binned versions until the full, unbinned dataset has been processed. While this does increase the total processing time somewhat, it greatly reduces the latency between progress updates, especially on initial job submission.
Figure 2.
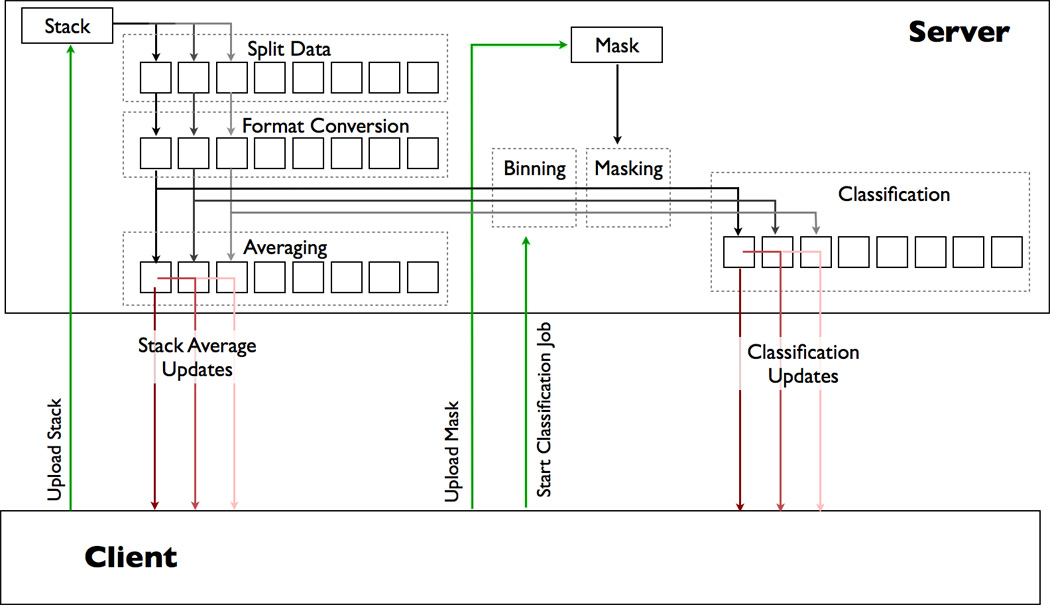
A simplified overview of the client/server interactions and processes used in Maskiton. The client uploads/selects a stack on the server by selecting it in the web browser on the local machine. Once selected, the stack average is displayed in the browser, or if the averaging process is still running, as a series of updates until the full dataset is averaged. This progressive averaging occurs while incrementally splitting and converting the stack from IMAGIC to Xmipp format. The user can submit classification jobs without having to wait for averaging to finish, and the classification processes will reuse as much previously cached data as possible while returning their own incremental progress updates. The server caches a substantial amount of processing to the disk in the event that a future process can reuse a calculation to speed up the return of information to the client.
To make the server-side processing as efficient as possible, processed data is cached at a granular level to make it possible for multiple processes to share computationally expensive steps. This is done using a file-based caching and locking strategy that allows individual Python functions to save processed results to file paths that are uniquely hashed based on inputs. A 128-bit MD5 function is used to generate the file system cache paths making it extremely unlikely for random collisions to occur. File reads and writes are carefully orchestrated using atomic file-system operations, such as linking/renaming operations. This deterministic file-system level caching, along with careful inter-process coordination using atomic file-system operations, allows individual scripts to remain loosely coupled at the code level but still share and reuse a significant amount of processing results.
Availability
A publicly accessible installation of Maskiton is available at http://maskiton.scripps.edu. The source code is available at http://github.com/nramm/maskiton and is released under the Apache license http://www.apache.org/licenses/LICENSE-2.0.html. Development was done under OS X 10.7, and server code has been tested on OS X 10.6, 10.7, 10.8, Fedora 16, and Fedora 17. The client-side browser code has been tested in Safari, Chrome and Firefox.
Public Datasets
Several datasets are made publicly available through the Maskiton interface to allow evaluation of the software without having to upload a dataset. Averages of these datasets are shown in Figure 3, and descriptions of each are as follows:
Ltn1: A dataset of the Ltn1 / Listerin E3 ubiquitin ligase(Bengtson and Joazeiro, 2010) whose structure and extensive flexibility has been analyzed using single particle EM(Lyumkis et al., 2013). This stack consists of 31,297 negatively stained particles, with a box size of 80×80 pixels at 4.36 Å/pixel. Preliminary reference-free alignments of the CTF-corrected particles were generated using the maximum likelihood routine(Scheres et al., 2005) from Xmipp(Marabini et al., 1996). Thereafter, the best classes were input to an iterative procedure consisting of a reference-based alignment in SPIDER(Frank et al., 1996), followed by multivariate statistical analysis and hierarchical ascendant classification in IMAGIC(van Heel and Keegstra, 1981). An iterative procedure consisting of the above steps was used to produce the final aligned stack (Figure 3a). Further details on the data set can be found in Lyumkis et al. (2013).
30S Ribosomes: A dataset from a time-point in the assembly of the 30S subunit of the E coli ribosome, as described in Mulder, et al. (2010). This stack is from a time point toward the end of the in-vitro assembly and consists of 52,718 negative-stained particles with a box size of 224×224 pixels at 2.24Å/pixel. Particles were picked using DoGPicker(Voss et al., 2009) and CTF estimated and corrected using ACE2(Mallick et al., 2005). To generate the aligned stack, a coarse Xmipp maximum-likelihood classification(Scheres et al., 2005) with 10 references was used to remove particles that fell into uninterpretable classes, and identify a structurally consistent core that was used as a reference for aligning the remaining particles (Figure 3b).
IgM: A negatively stained dataset of the polyclonal Immunoglobulin M (IgM). This stack contains 3,713 particles with a box size of 336×336 pixels at 2.08Å/pixel. An initial, crude stack of 5,989 particles was processed using a coarse, binned by 4, maximum-likelihood alignment from Xmipp(Marabini et al., 1996), with 5 classes requested. These classes were used to remove bad ‘particles’ from the dataset and to create a reference to which the remaining particles were aligned using an iterative Xmipp reference-based alignment. The initial reference structure was similar in appearance to the final stack average (Figure 3c).
Synthetic: A synthetic dataset used to validate that the classification back-end was functioning during development. This dataset is also of instructional value for testing the effect of the various Maskiton classification parameters on resulting classifications. The dataset was generated by replicating and combining two distinct ‘models’ with added Gaussian noise. Each model has a pair of spots diagonally opposite one another, and the two models are mutually exclusive (Figure 3d).
Figure 3.
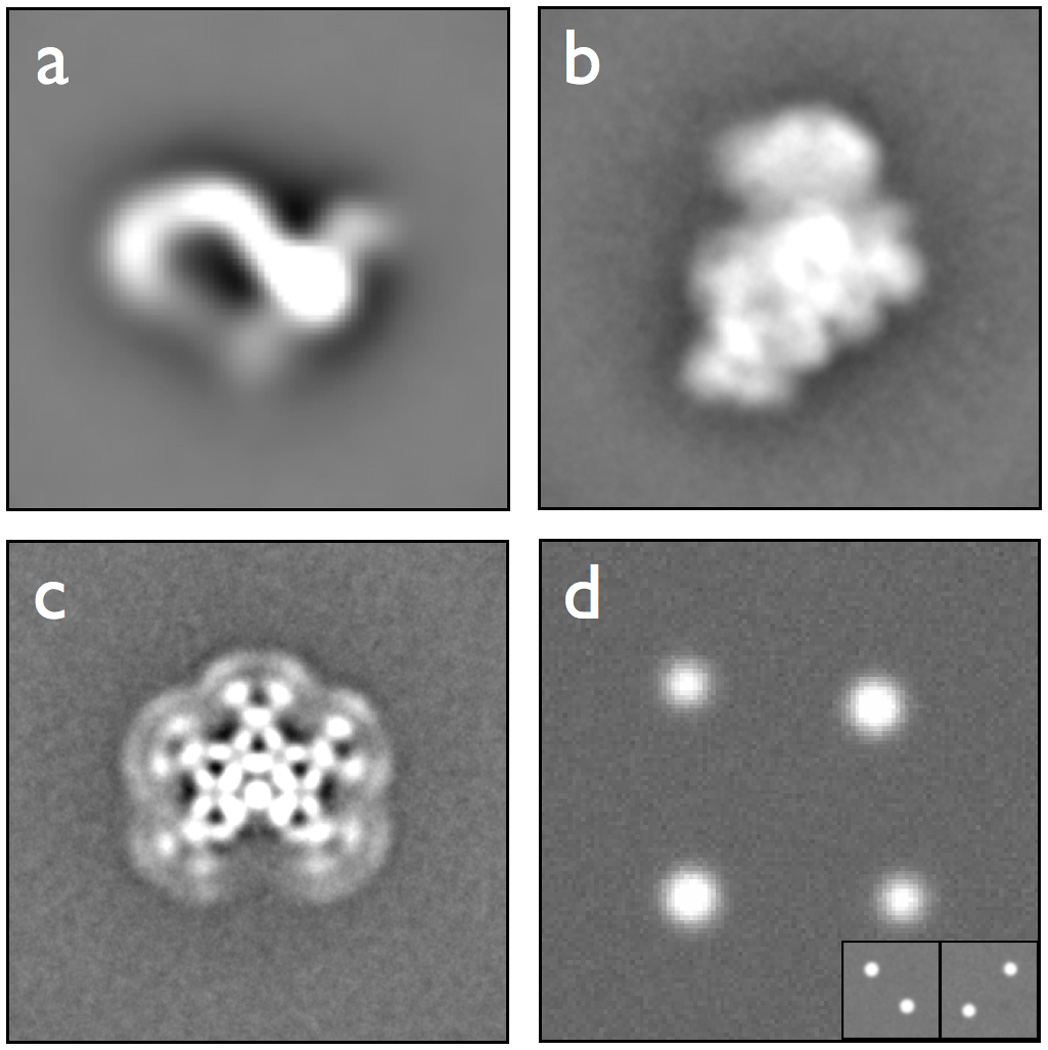
Averages of datasets included in public Maskiton server. (a) Ltn1 (Listerin) dataset. (b) 30S ribosome subunit assembly timepoint. (c) Immunoglobulin M. (d) Synthetic test dataset. The two models that were combined to create the full dataset are shown in inset.
Dataset Alignment
Classification in Maskiton currently requires that particles be aligned. For some structures this process can be quite challenging, as it may be difficult to identify a structurally consistent core to use as a reference. Each heterogeneous data set will exhibit unique characteristics that present different opportunities for customized alignments. The general strategy employed here is to identify and align to the largest homogeneous region (LHR) of the data. Once aligned, the LHR represents a frame of reference outside of which 2D heterogeneity can be analyzed in greater detail using Maskiton.
The general protocol used for the 2D alignment of the sample datasets was:
Identify the predominant orientations within a dataset (aligned 2D classes) using any ab initio 2D alignment and classification algorithm. In order to maximize the benefits of 2D averaging and noise reduction, while minimizing contamination from heterogeneous particle populations within each class, a general starting point is to create enough classes to accommodate ~100 particles within each class. Samples with preferred orientation may tolerate higher particle numbers, while highly heterogeneous samples with multiple orientations may require fewer particle numbers and greater alignment and classification accuracy. A binned version of the dataset is also often used for faster processing.
Align the classes to each other and identify regions of homogeneity and heterogeneity. This step is facilitated by preferred specimen orientation and will be more challenging without prior 3D information to discriminate heterogeneity from orientation. Careful inspection is required.
Particles sharing orientation should be aligned using their LHR as a reference. This reference can consist of using an unmodified class from step 1, if the LHR is large, or by masking to create a reference that focuses on the LHR. Alternatively, one can align the classes themselves and then later apply the alignment transformations to the raw particles of each class. This is often better in cases where the LHR is not as well defined.
Use Maskiton on the aligned dataset and interpret the results. These results will indicate not only interesting observations of the heterogeneity, but also possible misalignments or better LHRs for re-alignment.
If the Maskiton results from step 4 make alignment problems obvious, or provide indication of a better LHR, steps 2 or 3 can be repeated.
Results and Discussion
Motivation and Interface Overview
Our goal was to facilitate focused 2D classification by allowing users to create custom masks without the use of conventional desktop painting applications or explicit file format conversions. Another primary goal was to greatly improve the interactivity of the classification process so that users could more quickly assess the progress of classification jobs and adapt their strategy as required. Performing 2D classification using custom masks is useful for examining specific areas in greater depth, especially in situations where global variations would otherwise dominate subtle differences. Another use of custom masks is to explicitly test if variations within masked areas relate to variations seen outside masked areas. Such can occur, for example, in strutures that have conformational changes propagated between distant regions. We note, however, that since masked 2D classification uses stacks of pre-aligned particles, the user must carefully consider the effect of the global alignment when interpreting the significance of such correlations.
It is easiest to appreciate the benefit of interactive masking and classification by using the interface available at the publicly hosted Maskiton installation: http://maskiton.scripps.edu. A quick video tour is provided in the supplemental materials. The viewing tools provided by Maskiton have also proved a useful aid to gaining an intuitive understanding of variations revealed by classifications. Maskiton thus provides for greater productivity, and by extension, more thorough explorations and interpretations of complex datasets.
A key component of the interactive experience is the emphasis on generating frequent and continual feedback from computational processes that would normally require a substantial amount of time to complete. A major consideration in using the Xmipp SOM routine within Maskiton is that it was relatively easy to integrate it in such a manner that intermediate results were returned quickly and regularly. Another advantage of the SOM algorithm is that it naturally arranges classes in a manner convenient for viewing and comparison by users. We note, however, that the incremental processing strategy employed by Maskiton does theoretically alter the behavior of the underlying classification algorithm so that it is not equivalent to the original. Whether these changes are noticeable in practice, or even detrimental, is highly dependent on the algorithm being used, the data being analyzed, and if any measures have been taken to counteract these effects. For example, binning of the datasets causes the SOM algorithm to behave like a low-resolution biased counterpart of the origial, a side effect that is arguably beneficial. Conversely, one effect of the incremental processing is that it can cause the SOM algorithm to converge much faster than it otherwise would when given the full dataset all at once. We counteracted this effect by significantly modifying some of the default parameter values from the command-line program. We believe other classification algorithms could be similarly modified into ‘low-latency’ versions, as long as the side effects of any required changes were reasoned out carefully and dealt with as needed.
Creating a low-latency interface for processing large datasets is a challenging task. One major obstacle in creating the existing functionality was structuring the processing so that data would not be duplicated while allowing multiple concurrent processes to work with it. Unfortunately, current popular EM data formats are not well suited for this type of environment, as they favor substantial data duplication or modification of existing data, such as image headers, making it impossible to reliably share data between concurrent processes. Since devising a new data format or re-implementing existing algorithms was not part of the original Maskiton goals, we worked around these issues as best we could, but we believe that a more suitable data format is the next step before the incorporation of future features.
The classification parameters exposed by Maskiton map fairly directly to those used in the underlying Xmipp SOM routine(Pascual-Montano, 2001), and readers are referred to the original publication and Xmipp documentation for further details and quantitative explanations of the parameters. Here, we will only describe the general effect these parameters have on Maskiton classifications.
The SOM algorithm works by classifying particles into a two-dimensional grid of classes. The size of this grid, and thus the number of classes, is set by changing the x dim and y dim parameters. While larger grid sizes may reveal more subtle variations in a dataset, they also tend to require a greater number of particles so that each potential class has a better chance of being populated by a statistically significant number of particles. Large grid sizes may also bias Maskiton classification toward interpolating intermediate states from noise. The risk of generating these kinds of artifacts is strongly dependent on the quality of the alignment and the SNR of the data, but Maskiton makes it easy for users to explore different grid sizes to find one appropriate for their data.
The radius, alpha and iter parameters all have subtly different effects on Maskiton’s ability to recover distinctly heterogeneous features within the data. Careful specification of these parameters will ensure that a balance can be attained between the recovery of classes that are overly influenced by noise(Stewart and Grigorieff, 2004) vs. those that describe true structural variations within the data. Of the three parameters, radius has the most pronounced effect on classifications. Larger radii may result in less biased and more linearly distributed classes, but smaller radii allow the classification to explore less linear separations, and hence may reveal subtler changes and result in more equally populated classes. Alpha and iter also have a similar effect, but the default values generally work well and do not need to be changed. Alpha controls the ‘learning rate’ of the algorithm, and a low value, such as the default, is generally preferred since it reduces bias by allowing the algorithm to more slowly approach convergence. A higher iter parameter allows each particle in the classification more opportunities to find the best fitting class, and while a higher value may theoretically improve the classification, diminishing returns quickly become apparent.
The importance of classification parameters, their interdependence, and their dependence on the data itself, further underscores the benefit of a highly interactive interface so that users can quickly explore parameter-space. This benefit is not just applicable to SOM classification, but to any data processing procedure with non-trivial effects. Faster, more direct, feedback allows users to more quickly come to their own conclusion as to the behavior of settings on their data, more fairly evaluate whether a method is appropriate for their data, and more thoroughly assess the validity of their results.
Maskiton application: analysis of mobility independence in Ltn1 E3 ligase
Ltn1 is an E3 ubiquitin ligase that plays a critical role in eukaryotic translational surveillance(Bengtson and Joazeiro, 2010). Like all E3s, Ltn1 is a bisubstrate enzyme and is responsible for (1) substrate recognition and (2) recruitment of an E2-Ubiquitin conjugate(Deshaies and Joazeiro, 2009). Recently, EM single particle analysis of Ltn1 revealed that the protein adopts an elongated form and exhibits continuous flexibility about two hinge regions. Using an iterative approach for alignment and classification, Lyumkis et al. (2013) were able to identify multiple conformational snapshots of the protein that can be arranged according to mobility within the N- and C-termini. With a simple 2D arrangement of the conformers, the movements within the termini appeared distinct and independent of one another, which led to a model by which Ltn1 carries out its biological functions.
Maskiton is capable of assessing mobility independence (or dependence) without the need to arrange multiple conformers manually, as was necessary in the analysis in Lyumkis et al. (2013). For example, when the Ltn1 data set is roughly aligned to its central region, heterogeneity is apparent in both the N- (left region) and C- (right region) termini (Figure 4a–e, leftmost panel). A Maskiton classification using a mask on the full protein resolves much of this heterogeneity, leaving only a baseline amount corresponding to the C-terminus (Figure 4a) (this baseline remains present even with iterative sub-classifications). In contrast, the application of a mask to Ltn1’s N-terminus elicits increased heterogeneity within its C-terminus (Figure 4b), indicating that the mobility of Ltn1’s C-terminus does not depend on a single conformation of its N-terminus. Similarly, application of a small mask either to a single (Figure 4c–d) or to multiple (Figure 4e) regions of its C-terminus elicit increased heterogeneity within the N-terminus, likewise indicating mobility independence between the two termini. Had the regions been dependent, it would be possible to resolve all the observed conformations using only a single mask on any one of them.
Figure 4.

Mobility independence within Ltn1’s termini in Maskiton. (a–e) For all masking procedures, the aligned average of the full data set is shown at left, followed by the masked region, and 8 distinct class averages resulting from the masked classification at right. Arrows indicate heterogeneity identified within the outlined/boxed region in the class averages. The aligned dataset was masked with (a) a circular mask to show overall global heterogeneity, (b) an N-terminus mask (left region) to show increased heterogeneity within the C-terminus (right region) of Ltn1, and (c–e) three different C-terminus masks to show increased heterogeneity within the N-terminus of the protein.
Maskiton application: analysis of 30S ribosome subunit assembly
Ribosomes are macromolecular machines that convert messenger RNA into proteins, and an understanding of ribosome biogenesis is central to cellular physiology. The bacterial ribosome contains two subunits (50S and 30S) that can self assemble in vitro. The 30S ribosomal subunit is composed of a single ~1500-nucleotide 16S RNA component and 20 ribosomal proteins (“r-proteins”). Previously, we used single-particle reference-free alignment and classification to identify subpopulations of assembly intermediates in an in-vitro assembly time course (Mulder et al., 2010).
One of the major transitions observed in the 30S assembly pathway occurs when the RNA region corresponding to the ‘head’ begins to fold and become visible in 2D classifications (Figure 5a). Throughout this event, there are subtle changes that occur in parallel, whose precise temporal order can vary (for details, see Mulder et al. (2010)). These include binding of the ribosomal-associated proteins S3, S2, S21, S10, and S14. Maskiton is useful in assessing the temporal dependencies of subunit assembly within the 30S ribosomal subunit. For example, after masking on an area that is expected to cover the S2 ribosomal protein, it is possible to recover classes with and without density in this region, (Figure 5b) as well as classes that show the folding and unfolding of the 30S head. Careful examination of these classes indicates that a folded 30S head may be required for S2 binding, but not vice versa. In contrast, masking on an area covering the ribosomal proteins S11/S21 no longer shows any strong correlation with the folding/unfolding of the 30S head, despite the clear presence and absence of density in that region. Alongside previous work (Williamson, 2005), this indicates that the observed density change is likely due to S21 binding and not S11, since of the two, S21 is believed to bind only after the head is at least partially folded. Using custom masks makes it easier to analyze the potential presence or absence of specific proteins, and test for their interdependence (Figure 5b/c). Maskiton is an exceedingly useful tool for such a discovery-based approach since datasets are easier to interpret, and subsequent experiments faster to devise. As it is, further analysis of the 30S ribosomal subunit dataset using Maskiton has provided additional insights and avenues for experimentation.
Figure 5.

Classifications of a time point from the in vitro assembly of the 30S ribosomal subunit in Maskiton. (a) Classes resulting from using a mask covering the full subunit, showing that the major variation in the dataset is attributable to unfolding of the 30S ‘head’, and some further unfolding of the 30S ‘body’. Classes resulting from using a mask covering (b) the S2 ribosomal protein and (c) the S11/21 ribosomal protein. (b–c) Arrows in the expanded insets indicate presence / absence of the density corresponding to the masked region.
Maskiton application: reducing classification complexity in Immunoglobulin M
Immunoglobulin M (IgM), produced by B-cells on initial exposure to an antigen, is a polymer commonly composed of five individual antibodies joined by disulfide bonds. Though the pentameric form of IgM appears to predominate, there is also biochemical evidence of hexameric instances (Gautam and Loh, 2011). The polymeric nature of IgM makes it capable of polyvalent cooperative binding, and thus improved avidity. Current structural knowledge of IgM indicates that the pentameric macromolecule is largely planar, but has a short central stalk, and outside edges that can likely curl toward the stalk as part of its functional role in binding antigens (Czajkowsky and Shao, 2009; Volkov et al., 2003).
Our examination of IgM by EM reveals a star-shaped planar structure of 5 antibody subunits with a missing gap that creates the appearance of an incomplete six-fold symmetric structure (Figure 6a). In this average, the antibodies of IgM radiate outward from a well-defined inner core, becoming less defined at increasing radial distance from the center. The outer blurriness indicates that these regions are likely structurally flexible and have been blurred by averaging. Since the structure contains ten antibody Fab ends, and each is flexible and can likely move somewhat independently, the number of possible structural variations is immense and would be difficult to resolve without using masked classification or the careful analysis of segmented antibody subunits. Using Maskiton, it is straightforward to create a custom mask to observe the range of motions adopted by a single selected Fab end (Figure 6b). The observation that the other, unmasked regions remain poorly defined supports the hypothesis that the Fabs move independent of one another. Similar masks on the other Fab ends have equivalent results, indicating that they all experience the same range of motion. Maskiton also makes it possible to quickly test whether the missing gap in the average is hiding a small population having an antibody at that position (Figure 6c). After extensive exploration using Maskiton, it became apparent that there is no hexameric sub-population in the dataset. Careful measurement of the spacing between the subunits shows that the gap is ~10% smaller than the space occupied by the other antibodies, thus indicating that steric hindrance may prevent the incorporation of a sixth antibody, despite the deceptive appearance.
Figure 6.

Classifications of IgM in Maskiton. (a) An average of the full dataset showing the overall structure of the IgM with an approximate six-fold symmetry broken by a missing gap. (b) Masking only a single antibody arm shows the back-and-forth rotation of the Fab, highlighted by the red arrow. (c) Masking on the missing wedge indicates that this dataset does not contain a small sub-population of IgMs with a sixth antibody. Closer examination of the spacing of this wedge shows that it is ~10% smaller than the spacing occupied by the other antibody subunits.
Conclusions
We have developed a tool for rapid, interactive masking and classification that greatly enhances the process of understanding highly complex and heterogene ous single-particle datasets. Faster, incremental progress updates translate into more rapid data exploration, more extensive interpretation of classification results, and improve a user’s ability to manage and track multiple lines of inquiry. Interactive web-based viewing tools are also useful for quickly sharing results and interpretations with collaborators in a way that is superior to static images and text. These benefits extend to both novice and experienced users, since novice users can more quickly gain an intuitive understanding of how classification behaves, while experienced users can more rapidly explore their data.
Maskiton currently requires aligned stacks, but could be extended in the future to integrate alignment so that classification becomes an iterative, interactive process where users can manually tweak or correct misalignments as they become apparent. In this regard, Maskiton is also a prototype for the incorporation of other steps in EM processing that could benefit from low-latency interfaces. One example is particle-picking, for which there exists many highly-capable particle picking algorithms (Sorzano et al., 2009; Voss et al., 2009; Zhu et al., 2004) that would integrate nicely in an interactive interface. An additional benefit to the Maskiton approach is that it can help alleviate other mundane obstacles to successful software use such as installation, configuration, and parameter determination.
Finally, to demonstrate the utility of Maskiton for focused classification, we applied it to the analysis of several experimental datasets that have been used in previously published results, and compared these results to the ones obtained using Maskiton. The interpretations and results thus obtained were broadly comparable, but the overall effort and time put into obtaining the new Maskiton results was greatly reduced. We have also shown specific examples of how focused classification in Maskiton may allow for a more thorough interpretation of results that serve as a better guide for further experiments. Altogether, these results indicate the value of making focused classification, even in 2D, much easier and approachable for both novice and experienced users.
Supplementary Material
Acknowledgments
This project was supported by a grant from National Institute of General Medical Sciences (9 P41 GM103310-11) from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Materials
S1. A video demonstration of Maskiton features and use.
References
- Bengtson MH, Joazeiro CAP. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature. 2010;467:470–473. doi: 10.1038/nature09371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brignole EJ, Smith S, Asturias FJ. Conformational flexibility of metazoan fatty acid synthase enables catalysis. Nat. Struct. Mol Biol. 2009;16:190–197. doi: 10.1038/nsmb.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess SA, Walker ML, Sakakibara H, Knight PJ, Oiwa K. Dynein structure and power stroke. Nature. 2003;421:715–718. doi: 10.1038/nature01377. [DOI] [PubMed] [Google Scholar]
- Czajkowsky DM, Shao Z. The human IgM pentamer is a mushroom-shaped molecule with a flexural bias. PNAS. 2009;106:14960–14965. doi: 10.1073/pnas.0903805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, Joazeiro CAP. RING domain E3 ubiquitin ligases. Annu. Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- Fielding RT. Architectural styles and the design of network-based software architectures. 2000 [Google Scholar]
- Frank J. Three-Dimensional Electron Microscopy Of Macromolecular Assemblies. USA: Oxford University Press; 2006. [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, et al. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Frank J, van Heel M. Correspondence analysis of aligned images of biological particles. J. Mol Bio. 1982;161:134–137. doi: 10.1016/0022-2836(82)90282-0. [DOI] [PubMed] [Google Scholar]
- Frey BJ, Dueck D. Clustering by Passing Messages Between Data Points. Science. 2007;315:972–976. doi: 10.1126/science.1136800. [DOI] [PubMed] [Google Scholar]
- Gautam S, Loh KC. Immunoglobulin-M purification--challenges and perspectives. Biotechnol Adv. 2011;29:840–849. doi: 10.1016/j.biotechadv.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- Lyumkis D, Doamekpor S, Bengston M, Lee JW, Toro T, et al. Single-Particle Electron Microscopy Reveals Extensive Conformational Variability of the Ltn1 E3 Ligase. PNAS. 2013 doi: 10.1073/pnas.1210041110. 2013 Jan 14 epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick SP, Carragher B, Potter CS, Kriegman DJ. ACE: automated CTF estimation. Ultramicroscopy. 2005;104:8–29. doi: 10.1016/j.ultramic.2005.02.004. [DOI] [PubMed] [Google Scholar]
- MC, Marco S, Fernández JJ, et al. Xmipp: An Image Processing Package for Electron Microscopy. J. Struct Biol. 1996;116:237–240. doi: 10.1006/jsbi.1996.0036. [DOI] [PubMed] [Google Scholar]
- Mulder AM, Yoshioka C, Beck AH, Bunner AE, Milligan RA, et al. Visualizing ribosome biogenesis: parallel assembly pathways for the 30S subunit. Science. 2010;330:673–677. doi: 10.1126/science.1193220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual-Montano A. A Novel Neural Network Technique for Analysis and Classification of EM Single-Particle Images. J. Struct Biol. 2001;133:233–245. doi: 10.1006/jsbi.2001.4369. [DOI] [PubMed] [Google Scholar]
- Penczek PA, Frank J, Spahn CMT. A method of focused classification, based on the bootstrap 3D variance analysis, and its application to EF-G-dependent translocation. J. Struct. Biol. 2006;154:184–194. doi: 10.1016/j.jsb.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Reese W. Nginx: the high-performance web server and reverse proxy. Linux Journal. 2008:2. 2008. [Google Scholar]
- Ruprecht J, Nield J. Determining the structure of biological macromolecules by transmission electron microscopy, single particle analysis and 3D reconstruction. Prog. Biophys. Mol Biol. 2001;75:121–164. doi: 10.1016/s0079-6107(01)00004-9. [DOI] [PubMed] [Google Scholar]
- Scheres SHW, Valle M, Nuñez R, Sorzano COS, Marabini R, et al. Maximum-likelihood multi-reference refinement for electron microscopy images. J. Mol Bio. 2005;348:139–149. doi: 10.1016/j.jmb.2005.02.031. [DOI] [PubMed] [Google Scholar]
- Sorzano COS, Bilbao-Castro JR, Shkolnisky Y, Alcorlo M, Melero R, et al. A clustering approach to multireference alignment of single-particle projections in electron microscopy. J. Struct Biol. 2010;171:197–206. doi: 10.1016/j.jsb.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorzano COS, Recarte E, Alcorlo M, Bilbao-Castro JR, San-Martín C, et al. Automatic particle selection from electron micrographs using machine learning techniques. J. Struct Biol. 2009;167:252–260. doi: 10.1016/j.jsb.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart A, Grigorieff N. Noise bias in the refinement of structures derived from single particles. Ultramicroscopy. 2004;102:67–84. doi: 10.1016/j.ultramic.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Tilkov S, Vinoski S. Node.js: Using JavaScript to Build High-Performance Network Programs. Internet Computing, IEEE. 2010;14:80–83. [Google Scholar]
- van Heel M, Keegstra W. IMAGIC: A fast, flexible and friendly image analysis software system. Ultramicroscopy. 1981;7:113–129. [Google Scholar]
- Volkov VV, Lapuk VA, Kayushina RL, Shtykova EV, Varlamova EY, et al. Low-resolution structure of immunoglobulins IgG1, IgM and rheumatoid factor IgM-RF from solution X-ray scattering data. J Appl Crystallogr. 2003;36:503–508. [Google Scholar]
- Voss NR, Yoshioka CK, Radermacher M, Potter CS, Carragher B. DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J. Struct. Biol. 2009;166:205–213. doi: 10.1016/j.jsb.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson JR. Assembly of the 30S ribosomal subunit. Q Rev Biophys. 2005;38:397–403. doi: 10.1017/S0033583506004264. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Carragher B, Glaeser RM, Fellmann D, Bajaj C, et al. Automatic particle selection: results of a comparative study. J. Struct Biol. 2004;145:3–14. doi: 10.1016/j.jsb.2003.09.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.