

Abstract
Background
Despite their species abundance and primary economic importance, genomic information about copepods is still limited. In particular, genomic resources are lacking for the copepod Calanus sinicus, which is a dominant species in the coastal waters of East Asia. In this study, we performed de novo transcriptome sequencing to produce a large number of expressed sequence tags for the copepod C. sinicus.
Results
Copepodid larvae and adults were used as the basic material for transcriptome sequencing. Using 454 pyrosequencing, a total of 1,470,799 reads were obtained, which were assembled into 56,809 high quality expressed sequence tags. Based on their sequence similarity to known proteins, about 14,000 different genes were identified, including members of all major conserved signaling pathways. Transcripts that were putatively involved with growth, lipid metabolism, molting, and diapause were also identified among these genes. Differentially expressed genes related to several processes were found in C. sinicus copepodid larvae and adults. We detected 284,154 single nucleotide polymorphisms (SNPs) that provide a resource for gene function studies.
Conclusion
Our data provide the most comprehensive transcriptome resource available for C. sinicus. This resource allowed us to identify genes associated with primary physiological processes and SNPs in coding regions, which facilitated the quantitative analysis of differential gene expression. These data should provide foundation for future genetic and genomic studies of this and related species.
Introduction
Copepods are more abundant than any other multicellular animal group, including the hyper-abundant insects and nematodes [1], [2]. Over 12,000 validated species of copepods have been recognized, which inhabit a domain that extends from the nutrient-rich black oozes of the abyssal ocean depths to the nutrient-poor waters of the highest mountain tarns. As the dominant secondary producers of the marine ecology, copepods play important roles in aquatic food webs. They consume microorganisms and are the main food source for many commercial fishes [3], such as anchovy, cod, herring and salmon. Therefore, copepods are the linchpin of aquatic food webs and they critically support marine fish production. Copepods play important roles in the global carbon budget. Copepods transfer carbon into the deep sea via their vertical migrations between surface and deeper waters [4]. In addition, copepods are sensitive indicators of the climate because ocean warming affects the abundance, distribution and community structure of copepod [5]. Despite their species abundance, diverse geographical distribution, and global importance, limited biological information is available at the molecular level and no model species exist. To date, sequencing efforts and the application of genomic techniques have been limited to a small number of species [2], [6]. Genetic or genomic studies of a broader range of copepods would obviously facilitate future developmental, distributional, and ecological research into copepods.
Recent advances in next-generation sequencing (NGS) and bioinformatics have generated genome-level information for model and non-model organisms [7]–[9]. The increased throughput of NGS platforms, such as massively parallel 454 pyrosequencing, facilitate the rapid and cost-effective generation of massive amounts of sequence data [7], [10]. However, even with high-throughput sequencing technologies, the sequencing of complex genomes remains expensive. Transcriptome sequencing provides an attractive alternative for whole-genome sequencing because it only analyzes the transcribed portions of the genome. This strategy reduces the sequencing cost and experimental complexity, as well as improving the transcript coverage [11], [12]. In addition, this sequencing method allows the de novo assembly and annotation of expressed genes [13], which makes it highly suitable for non-model organisms. Indeed, transcriptome sequencing has been applied successfully to several non-model organisms, which has facilitated the detection of single nucleotide polymorphisms (SNPs), discovery and annotation of genes, analysis of differential gene expression, and functional studies of gene expression [14]–[20].
Calanus sinicus is a dominant copepod in the coastal waters of East Asia including China, Korea, and Japan [21]. C. sinicus may account for 80% of the total zooplankton abundance in the Yellow Sea [22], where it links primary production of the Yellow Sea to fish larvae and juveniles [23]. Given its ecological importance, many researchers have investigated this species in biological and ecological studies [21], [23]–[27]. However, limited genomic resources are available for C. sinicus so several key mechanisms remain unknown in C. sinicus, such as the mechanism of diapause and the population genetics of C. sinicus. During its life cycle, many C. sinicus oversummer in a diapause phase (a dormant over-summering phase where development is suppressed to adapt to the high temperature and seasonal food supply) in the Yellow Sea Cold Water Mass (YSCWM) [25], [26]. However, little is known about the triggers that initiate and terminate the diapause, or the internal processes associated with these triggers. Understanding these processes is important given that subtle changes in environmental conditions, which may affect diapause, may have consequences for the entire copepod-based ecosystem. C. sinicus is the dominant species in the continental shelf waters of the Northwest Pacific Ocean, such as the Yellow Sea population [26]–[28], the Bohai Sea population [28], and the Nanhai Sea population [29]. However, there is no effective molecular marker, leaving the phylogenetic relationships among populations of the species still elusive. Therefore, there is an urgent need to design more genetic tools for C. sinicus.
In this study, we performed de novo transcriptome sequencing of C. sinicus using the 454 GS FLX platform. The ultimate goal of this study was to produce whole-transcriptome sequences, which would provide an invaluable resource for future genetic and genomic studies of this and related species. We identified a wide diversity of candidate genes involved in all major signaling pathways and developmental processes. Given the fact that diapause occurs in copepodite stages IV and V (C4 and C5) and that these copepodites develop into adults at the end of the diapause [25], [27], we also hypothesized that key regulation genes must be involved in diapause even during the transition between active copepodites and adults. We used active copepodites (C4 and C5) and adults (males and females) as the sequencing materials, and we aimed to identify potential candidate genes that were differentially expressed between the C. sinicus copepodites and adults, which included genes involved in the regulation of diapause.
Results and Discussion
Sequencing analysis and assembly
Two types of cDNA samples, which represented different developmental stages and adult tissues of C. sinicus, were prepared and sequenced using the 454 GS FLX platform. The two sequencing runs produced a total of 1,470,799 reads with an average length of 355 bases (Table 1). Newbler v2.6 was used with the default parameters to screen for adapter sequences and eliminate poor quality reads. After quality trimming and removal of adapter sequences, 1,368,381 (93% of the raw reads) reads remained in the assembly. Of these, 1,123,512 (76.4%) reads assembled wholly or partially into contigs and 244,869 (16.5%) reads remained as singletons. The remaining reads were excluded because they originated from repeat regions (1,365 reads; 0.09%), were outliers (59,125 reads; 4.0%), or were too short (<40 base pairs: 41,928 reads; 2.9%). All of the raw tag data produced in this study has been deposited in the NCBI SRA database (accession number: SRA064006).
Table 1. Summary of the sequencing and assembly of the C. sinicus transcriptome.
| Raw reads (bade pairs) | 1,470,799(523,341,132) |
| Clean reads | 1,368,381 |
| Isogroup | 19,149 |
| Isotigs | 31,591 |
| Isotig N50 | 873 |
| Mean # isotigs per isogroup | 1.6 |
| Contigs | 56,809 |
| Mean # contigs per isotig | 2.7 |
| Singletons | 244,869 |
Newbler's terminology for assembled reads comprised three elements: contigs (stretches of assembled reads that were free of branching conflicts), isotigs (continuous path through a set of contigs), and isogroups (groups of isotigs arising from the same set of contigs). For consistency, we use this terminology throughout this paper. Our data were assembled into 56,809 contigs, which grouped into 31,591 isotigs. Of these isotigs, 14,791 (46.8%) contained only one contig, and the average number of contigs per isotig was 2.7. The isotig N50 length was 873 bp, which means that 50% of the bases were incorporated into isotigs ≥873 bp. The 31,591 isotigs fell into 19,149 isogroups, where 14,819 (77.4%) contained only one isotig (the average number of isotigs per isogoup was 1.6, Table 1).
The average coverage among contigs was 32.1 reads/bp (median coverage = 7.0 reads/bp, Figure 1B), which means that every base pair in the transcriptome was sequenced 32.1 times on average. This coverage value was much higher than that in previous studies [16], [30], [31] and it should be helpful for distinguishing SNPs from potential sequencing errors in raw reads [32].
Figure 1. Overview of C. sinicus transcriptome sequencing and assembly.

(A) Size distribution of 454 sequencing after removal of adapter and short reads (<40 bases). (B) Sequence coverage. (C) Size distribution of isotigs.
Transcriptome annotation
Several complementary approaches were used to annotate the assembled sequences. First, we used BLASTX to map the 276,460 assembled sequences (31,591 isotigs + 244,869 singletons) against the entire RefSeq protein database with an E-value cut-off of 1e -3. To simplify the statistics, we only reported the results for the longest isotig per isogroup. Of the 19,149 isotigs, 9,497(49.5%) had at least one hit and 8,340 (43.5%) were matched to proteins with known functions. Of the 244,869 singletons, 71,562 (29.2%) had hits and 62,451 (25.6%) were matched to known proteins. These values were higher than those in the comparable BLAST results from most other published studies using 454-generated de novo transcriptomes [16], [20], [31]. This may be attributed to the deeper sequencing, which increased the length of the assembled sequences and accordingly made the sequences more likely to be identified using BLAST. The unidentifiable sequences may originate from untranslated regions (UTRs) or non-conserved portions of protein-coding sequences.
Gene Ontology (GO) analysis was carried out to explore and summarize the functional categories of the genes sequenced in this study. Of the annotated sequences, 25,676 were assigned GO terms. In total, 175,835 GO terms were obtained, which included 41.3% related to biological processes, 32.0% to molecular functions, and 26.7% to cellular components (Figure 2). The percentages of annotated C. sinicus sequences assigned to GO terms were compared with those from Daphnia pulex, the only crustacean with a sequenced genome. We found no significant differences in the percentages of genes with examined GO term categories between the C. sinicus transcriptome and the D. pulex genome, which suggests that there was a similar distribution of genes in the different functional categories, while the sequenced C. sinicus transcriptome did not lack major functional categories of genes.
Figure 2. GO term distribution of BLAST hits from the C. sinicus transcriptome.

Selected GO categories are shown within the top-level divisions of Biological Process, Molecular Function, and Cellular Component. The relative percentages of genes falling into GO categories are comparable between our C. sinicus transcriptome (white) and the D. pulex genome (black).
Final functional classification and pathway assignment were performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The annotated sequences were compared using bi-directional BLAST with an E-value of 1e -3 against the KEGG database. Of these sequences, 8,962 had significant matches in the database. Among the matched sequences, 2,866 sequences with enzyme commission (EC) numbers were assigned to metabolic pathways (Table 2). The metabolic pathways that were well represented in the C. sinicus sequences were carbohydrate metabolism, energy metabolism, amino acid metabolism, and lipid metabolism. Given the important roles of lipids in the lifecycle of copepod [33], especially during diapause, we focused greater attention on lipid metabolism. Genes were found in several pathways involved with fatty acid biosynthesis, such as fatty acid elongation, steroid biosynthesis and ether lipid metabolism (Table S1).
Table 2. KEGG biochemical mappings for C. sinicus.
| KEGG pathway | Number of genes | Number of sequences (Isogroup + singleton) |
| Metabolism | 1034 | 2866 |
| Carbohydrate Metabolism | 182 | 570 |
| Energy Metabolism | 142 | 435 |
| Lipid Metabolism | 131 | 324 |
| Nucleotide Metabolism | 82 | 302 |
| Amino Acid Metabolism | 154 | 389 |
| Metabolism of Other Amino Acids | 58 | 135 |
| Glycan Biosynthesis and Metabolism | 95 | 201 |
| Metabolism of Cofactors and Vitamins | 80 | 163 |
| Metabolism of Terpenoids and Polyketides | 28 | 56 |
| Biosynthesis of Other Secondary Metabolites | 27 | 81 |
| Xenobiotics Biodegradation and Metabolism | 55 | 210 |
| Genetic Information Processing | 527 | 994 |
| Transcription | 93 | 142 |
| Translation | 199 | 409 |
| Folding, Sorting and Degradation | 183 | 299 |
| Replication and Repair | 52 | 144 |
| Environmental Information Processing | 264 | 612 |
| Membrane Transport | 24 | 39 |
| Signal Transduction | 178 | 482 |
| Signaling Molecules and Interaction | 62 | 91 |
| Cellular processes | 372 | 791 |
| Transport and Catabolism | 167 | 348 |
| Cell Motility | 47 | 84 |
| Cell Growth and Death | 81 | 211 |
| Cell Communication | 77 | 148 |
| Organismal Systems | 581 | 1499 |
| Immune System | 102 | 339 |
| Endocrine System | 99 | 254 |
| Circulatory System | 41 | 91 |
| Digestive System | 92 | 234 |
| Excretory System | 50 | 81 |
| Nervous System | 114 | 346 |
| Sensory System | 15 | 31 |
| Development | 51 | 94 |
| Environmental Adaptation | 17 | 29 |
| Total | 2778 | 6762 |
Estimating the number of genes expressed in C. sinicus
One of the primary goals of transcriptome sequencing projects is to determine the number of expressed genes. The exact gene number is indeterminable if there is no fully sequenced genome. However, several alternative strategies are available to estimate the number of genes expressed in non-model organisms. First, gene numbers can be estimated based on genes that are well matched by sequences. Thus, gene names were assigned to assembled sequences based on the gene product and gene name annotation of the best BLAST match for that sequence. This procedure successfully assigned gene names to 70,791 sequences in the total dataset, i.e., 65,147 of the sequences were ≥300 bp in length and 1,862 of the sequences were ≥1000 bp in length. Among the 70,791 annotated best hits, 14,279 different gene names were assigned. This provided a rough estimate of the number of different genes expressed in the C. sinicus transcriptome. Given that many sequences lacked matches in public sequence databases and thus were not assigned gene names, this number was probably an underestimate. The gene number can also be estimated based on the number of isogroups. Isogroups are groups of isotigs that arise from the same set of contigs, where each isotig in the isogroup represents a transcript variant. Therefore, isogroups may represent putative genes. We identified 19,149 isogroups in our transcriptome, which provided another estimate of the gene number in the C. sinicus transcriptome. No specific solutions are available for determining the number of genes without a sequenced genome, but the estimates described above suggest that over 15,000 different genes were expressed in C. sinicus. This estimate was close to that reported in a previous transcriptome study of another marine copepod, Tigriopus californicus [19], which identified 15,402 unique unigenes in the T. californicus transcriptome.
Genes involved with development
The growth and development of many copepods such as C. sinicus, which is an important link in the food chain between phytoplankton and planktivorous fish, are of particular interest to researchers. In this sequencing project, we identified a wide diversity of candidate genes involved with developmental processes based on BLAST, GO, and KEGG annotations. Among these genes, we identified different groups of growth factors and their receptors involved with cell growth (Table 3).
Table 3. Selected development process genes identified in the C. sinicus transcriptome.
| Process | NumIsotig | Length(range) |
| Growth | ||
| ATP-binding cassette | 7 | 415–1579 |
| prosaposin | 1 | 465 |
| saposin | 10 | 388–1856 |
| vitellogenin receptor | 2 | 488–2772 |
| histone-lysine N-methyltransferase | 7 | 588–1583 |
| serine/threonine-protein kinase PAK 2 | 16 | 357–2002 |
| pre-mRNA-processing factor | 3 | 743–1029 |
| calsyntenin | 1 | 1206 |
| 60S ribosomal protein L12 | 1 | 494 |
| DNA methyltransferase 1-associated protein 1 | 1 | 602 |
| chromodomain-helicase-DNA-binding protein | 1 | 895 |
| transcription factor family member | 92 | 403–1285 |
| insulin receptor | 1 | 729 |
| DNA-binding protein A | 1 | 895 |
| elongation factor | 33 | 356–1885 |
| actin | 196 | 331–1342 |
| ubiquitin-conjugating enzyme E2 E3 | 14 | 378–1810 |
| adenosylhomocysteinase | 6 | 200–1665 |
| menin | 2 | 1084–2603 |
| histone acetyltransferase | 3 | 663–1486 |
| Molting | ||
| CYP315A1; ecdysteroid 2-hydroxylase | 1 | 1390 |
| CYP306A1 | 3 | 485–507 |
| CYP302A1 | 5 | 406–490 |
| CYP305A1 | 2 | 138–151 |
| ecdysone-induced protein 75(E75) | 1 | 747 |
| ecdysone receptor | 4 | 305–2056 |
| ecdysoneless | 1 | 2056 |
| CYP15A1 | 10 | 390–1610 |
| farnesoic acid O-methyltransferase(FAMeT) | 6 | 451–1175 |
| juvenile hormone-inducible protein | 2 | 492–1349 |
| juvenile hormone esterase | 6 | 652–1924 |
| juvenile hormone epoxide hydrolase | 8 | 414–513 |
| ferritin | 56 | 247–773 |
| elongation of very long chain fatty acids protein(ELOV) | 10 | 365–1208 |
| fatty acid binding protein(FABP) | 17 | 467–747 |
| short-chain dehydrogenase/reductase (SDR) | 6 | 449–961 |
Molting and diapause are key biological processes in the life cycle of many insects and copepods. In arthropods, ecdysteroids and juvenile hormone (JH) are key molecules that regulated growth (molting), sexual maturation and egg production [34], [35]. To identify genes associated with molting and diapause, the C. sinicus transcriptome was searched for key ecdysteroid- and JH-related genes. We detected the expression of many known ecdysteroid biosynthesis genes, including members of the cytochrome P450 family, such as CYP315A1, CYP306A1, CYP302A1, and CYP305A1 (Table 3). We also identified transcripts for ecdysone-regulated genes, including E75, ecdysone receptor and ecdysoneless, which are known to play key roles during molting [36]–[38]. JH biosynthesis and response genes were also identified. JH is a key hormone in the regulation of the insect's life history, which maintains the larval state during molts and that directs reproductive maturation [39]. Farnesoic acid O-methyltransferase (FAMeT) and CYP15A1 play significant roles in catalysis during JH biosynthesis pathways [40], [41]. Ferritin and several other genes (ELOV, FABP, SDR) involved in fatty acids metabolism were also identified. These genes were differentially expressed in diapausing and active Calanus finmarchicus [42].
Differentially expressed genes
Of the whole transcriptome sequences, 959,123 reads were generated from the C. sinicus copepodid larval samples and 842,238 reads were from the adults. We found that 3,798 gene sequences were differentially expressed between the two samples i.e., 1,841 sequences were categorized as up-regulated genes (those with higher expression levels in copepodite samples compared with adult samples), and 1,954 sequences were categorized as down-regulated genes (those with significantly higher expression levels in adults compared with copepodite). Of the 3,798 differentially expressed sequences, we predicted annotations for 1,503 sequences and matched about 640 different proteins (Table S2). GO enrichment of these differentially expressed sequences detected several overrepresented GO categories (Table S3), such as transport process, binding, and motor activities.
With the ultimate goal of fully understanding the molecular mechanism of diapause in C. sinicus, we tried to identify genes involved with the regulation of diapause. Several genes, such as JH, ecdysone, heat shock protein (HSP), ferritin, cytochrome P450, are known to play important roles in diapause regulation [42]–[44]. From all of the genes that were differentially expressed in C. sinicus copepodites and adults, we found several genes that may be involved with diapause (Table 4). Elongation of very long chain fatty acids protein (ELOV) and cuticle protein (CP) were up-regulated in copepodites. ELOV facilitates the regulatory step during fatty acid elongation in mammals [45]. The specific role of ELOV is unknown in C. sinicus but it was reported that ELOV was involved with the biosynthesis of JH [46] and the synthesis of storage lipids [42]. High expression of ELOV suggested that more fatty acids were synthesized in copepodid larvae (copepod stages IV and V), some of which were stored and prepared for later diapause. This hypothesis was consistent with the findings of a previous study where the population of C. sinicus on the continental shelf of the Yellow Sea during May to June exhibited features of preparation for diapause in the Yellow Sea Cold Water Mass, such as the accumulation of lipids in the oil sac and the dominance of C5s [47]. Cuticle protein is one of the structural proteins that comprises the stratum corneum together with chitin [48]. Cuticle protein was found to be highly expressed prior to the final molt and prior to the post-molting growth of adult female salmon louse (Lepeophtheirus salmonis) [49].
Table 4. Expression levels of genes that may be invovled with the diapause regulation of C. sinicus.
| Gene | Gene length/bp | RPKM | p-value | Regulation | |
| Copepodite | Adult | ||||
| Cuticle protein | 1568 | 42 | 0 | 8.10E–11 | up |
| Ferritin | 754 | 70 | 505 | 7.35E–38 | down |
| Heat shock protein | 1003 | 15 | 63 | 1.54E–05 | down |
| ELOV | 1006 | 301 | 6 | 1.38E–44 | up |
| Cytochrome P450 | 1763 | 25 | 7 | 0.00399 | up/down* |
| Juvenile hormone esterase | 1924 | 0 | 31 | 6.64E–10 | up/down |
| FAMeT | 1175 | 47 | 77 | 0.02038 | up/down |
| Ecdysteroid receptor | 2056 | 13 | 2 | 0.00343 | up/down |
| Retinoid X receptor | 747 | 25 | 27 | 0.85347 | up/down |
up/down: Both up and down regulated sequences were found in C. sinicus copepodite or adults. ELOV: elongation of very long chain fatty acids protein. FAMeT: farnesoic acid O-methyltransferase. Gene expression level is calculated by using reads per kilobase of the transcript per million mapped reads (RPKM).
The expression levels of ferritin and heat shock protein were high in adults. As an iron storage protein, ferritin plays a key role in iron metabolism [50] and protects proteins against oxidative damage [51]. In C. finmarchicus, ferritin was up-regulated in diapausing copepodites, where it was predicted to protect proteins and help to delay development [42]. Heat shock proteins (HSPs) are a superfamily of molecular chaperones that regulate diapause in some insects and copepods [52]. HSPs prevent irreversible protein denaturation during stress, facilitate protein refolding or destruction, and are required for stress tolerance [53]. HSP22 was found to be highly expressed in diapausing C. finmarchicus and was predicted to protect proteins from degradation during diapause [44].
Other proteins, such as cytochrome P450, FAMeT, ecdysteroid receptor, and retinoid X receptor were also found to have key roles in diapause regulation in other species [42], [52]. However, these proteins were not differentially expressed in the active copepodites and adults of C. sinicus. We will pay close attention to these genes in future expression profile analyses of active and diapause samples, the transcriptomes of which have been sequenced using the Solexa platform.
Real-time PCR
The significant up- and down-regulation of expressed genes involved with diapause were validated by real-time PCR. The materials used in this experiment were the same as those used in 454 sequencing. The sequences of the primers used are listed in Table 5. The expression levels of the six genes validated using real-time PCR confirmed the robustness of the 454 sequencing results (Figure 3).
Table 5. Oligonucleotide primer sequences for real time PCR.
| Gene | Primer sequence | Product size(bp) |
| ELOV | F: 5′ ACGGTTCTCATCTATTGCT 3′ | 149 |
| R: 5′ ATCGCAATCAATTTCGGGAC 3′ | ||
| Actin | F: 5′ GAAGATCTGGCATCACACC 3′ | 109 |
| R: 5′ CATCTTCTCTCTGTTTGCCTT 3′ | ||
| Cuticle protein 7 | F: 5′ GAGGATCCAGCACACCAA 3′ | 118 |
| R: 5′ AACCGCATTTCCATATCCAC 3′ | ||
| Cuticle protein 19 | F: 5′ CCCTCAGCCATTTGCCTA 3′ | 63 |
| R: 5′ TTCTTGAAGTTAGCCTTAGAGT 3′ | ||
| Ferritin | F: 5′ AACCGTGATGATCAAGCTC 3′ | 104 |
| R: 5′ CGCTTGGTCTGATATTCCAT 3′ | ||
| HSP 70 | F: 5′ ATCTTCGAGGTCAAGTCCAC 3′ | 116 |
| R: 5′ TTCATGTCCTTCTTGTGCTT 3′ | ||
| HSP 90 | F: 5′ TGGTTTCTACTCCGCCTA 3′ | 78 |
| R: 5′ CAGACATATTGCTCGTCA 3′ |
Figure 3. Real-time PCR validation of differentially expressed genes that may be involved in diapause.

The values were normalized against actin. Significant levels were *P<0.05, **P<0.01, and values indicate the mean ± S.E.
SNP discovery
Potential SNPs were detected using CLC Genomics Workbench software. We identified 284,154 high-quality SNPs and 30,409 indels from 18,891 isotigs (Figure 4). The predicted SNPs included 179,345 transitions and 132,756 transvertions. The overall frequency of all SNP types in the transcriptome, including indels, was 1 per 179 bp. These potential SNPs provide an extensive set of genetic markers for copepods and will facilitate future studies of genetic connectivity and genetic mapping at a previously unprecedented level of detail.
Figure 4. Classification of single nucleotide polymorphisms (SNPs) indentified in the C. sinicus transcriptome.
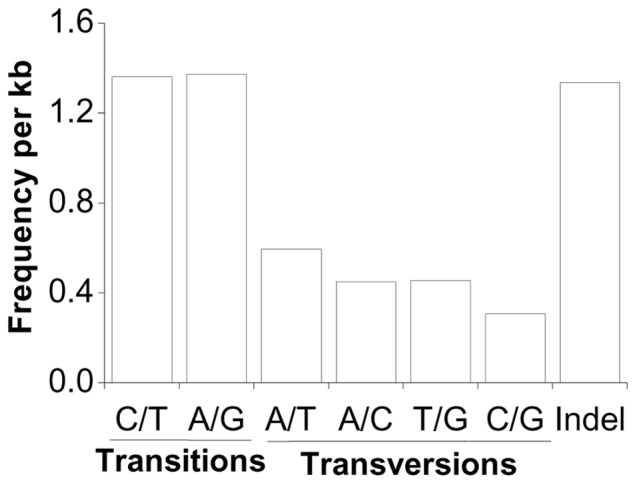
The overall frequency of these SNPs is one per 179 bp.
C. sinicus dominates the continental shelf waters in the Northwest Pacific Ocean. The population structure of C. sinicus is central to understanding the mechanism of its population recruitment. However, there are no effective molecular markers, which make it difficult to determine the phylogenetic relationships among the populations of this species. Wang reported that, compared with other gene markers (COX1 and ITS), more information could be obtained using SNP gene markers in population studies of C. sinicus [54]. Using the abundance of SNPs determined by 454 sequencing, we will select effective SNPs for studying the population structure of C. sinicus.
Conclusions
In this work, we performed de novo transcriptome sequencing of C. sinicus using the 454 GS FLX platform. We identified many candidate genes that were potentially involved with growth, molting, and lipid metabolism. This set of candidate genes is valuable for further research. We produced a large set of Expressed Sequence Tags (ESTs) and SNPs and made them publicly available for marker development. Our comparative sequencing of copepodid larvae and adults has provided information about the gene variations involved with a number of pathways, which might potentially reflect developmental changes.
Materials and Methods
Sample preparation and sequencing
Copepods were collected from Jiaozhou Bay, China, and brought to the laboratory in fresh seawater. The studies were performed according to the Institute of Oceanology, Chinese Academy of Sciences guidelines for the use and care of laboratory animals in research. All necessary permits were obtained for the field studies. Copepods were collected by a 500 µm mesh zooplankton net (mouth opening 0.5 m2) and preserved in fresh seawater temporarily. All tows were carried out vertically from 4 m above the bottom to the sea surface. Adults (females and males) and copepodid larvae (C4 and C5) were collected immediately and stored separately in 4 L of 0.45 μm-filtered seawater at 15°C for 24 hours. Using an anatomical lens copepods were picked up at different developmental stages with adults of different sexes, including copepod stages C4 and C5, and female, and male adults. Copepods were put in 1.5 ml frozen tubes and then seawater was removed using bibulous paper. All samples were flash frozen and stored in liquid nitrogen until analysis.
Total RNA was extracted from each of the four samples (>50 mg wet weight each) using a RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and purified further with a TURBO DNA-free Kit (Ambion, Austin, TX, USA). The quantity and quality of total RNA was analyzed using a 1% agarose gel and an Agilent 2100 bioanalyzer. The RNA from C4 and C5 were pooled in equal proportions to produce the copepodite sample while the adult males and females were pooled equally to produce the adult sample. Equal total RNA were reverse-transcribed into cDNA using a SMART PCR cDNA Synthesis Kit (Clontech Laboratories, Inc. CA, USA). Sample of copepodid stages (C4 and C5) was run on a sequencing plate and adult sample was run on another sequencing plate. The 454 sequencing experiments were performed by the Chinese National Human Genome Center.
Sequence data analysis and assembly
Prior to assembly, adapter sequences and low-quality sequences were trimmed from the raw reads. All reads shorter than 40 bp were removed based on an assumption that small reads would fail to assemble and that they might represent sequencing artifacts. The trimmed and size-selected reads were assembled using the Newbler assembly program (v2.6) where all of the parameters were set to their default values. Singletons, i.e., reads that were not incorporated into any contigs, were retained in the data set because many were likely to be fragments of low-expressed transcripts. Assembled contigs and singletons were pooled for subsequent analyses.
Sequence annotation
Before the gene name annotation, the assembled sequences were mapped against Swiss-Prot and the NCBI non-redundant (Nr) protein databases using BLASTX with an E-value threshold of 10−3. Gene names were assigned to each assembled sequence based on the best BLAST hit.
GoPipe [55] was used to annotate the assembled sequences with Gene Ontology (GO) terms describing biological processes, molecular functions, and cellular components. GoPipe is a tool for integrating BLAST results into streamlined GO annotations for batched sequences. We used a precomputed GO annotation from the D. pulex genome [56] to compare sequences with different GO annotations in C. sinicus and D. pulex.
Finally, the assembled sequences were compared to the KEGG database [57]. KEGG pathways were assigned to the assembled sequences using the online KEGG Automatic Annotation Server (KAAS), http://www.genome.jp/kegg/kaas/. The bi-directional best hit (BBH) method was used (with an E-value of 1e -3) to obtain KEGG Orthology (KO) assignment. Using the KEGG database, we further studied the complex biological behaviors of genes and determined pathway annotations for sequences.
Differential gene expression analysis
The expression level of a transcript was quantified in reads per kilobase of the transcript per million mapped reads (RPKM) in the transcriptome [58]. Differentially expressed genes were identified in C. sinicus copepodid larvae (C4 and C5) and adults (male and female) by a DEGseq package using the MARS (MA-plot-based method with Random Sampling model) method [59]. Genes were regarded as differentially expressed if they exhibited two fold or greater change using a 0.1% or less false discovery rate (FDR). Differentially expressed genes were regarded as up-regulated if the expression levels in copepodid larval samples were significantly higher than those in adult samples. Down-regulated genes were those with significantly higher levels of expression in adults compared with larvae.
A Fisher's exact test was used with a threshold of 5% as the false discovery rate to determine the differentially regulated genes in the two samples for each GO term.
Real-time PCR
The significant up- or down- regulated expressed genes associated with diapause were validated and quantified by real-time PCR. Actin, which has been used for the real-time PCR of copepod genes by other workers [42], was chosen to normalize the target gene quantities. Oligonucleotide primers were designed using the 454 sequencing data to target 75–150 bp amplicons (Table 5). The total RNA of copepodite samples and adult samples were prepared and analyzed as RNA used in 454 sequencing. The same amount of total RNA from each sample was reverse transcribed into cDNA using a RevertAid Frist Strand cDNA synthesis kit (Fermentas, Ontario, Canada).
Real-time PCR was performed using the SYBR Green® real-time PCR assay with an Eppendorf Mastercycler® ep realplex S. All samples were run in duplicate wells for each gene on a single plate. The PCR mixture contained 10.0 μL of SYBR Premix Ex Taq (Takara, Dalian, China), 0.4 μL (each) of forward and reverse primer (10 mM), 2 μL of 1∶10 diluted cDNA, and 7.2 μL of RNase-free water. The PCR conditions were as follows: 95°C for 30 s; 40 cycles of 95°C for 5 s and 60°C for 20 s. After 40 cycles, the PCR products from each reaction were subjected to melt-curve analysis to ensure that only a single product was amplified. To confirm that the correct amplification had occurred, the PCR products were analyzed by electrophoresis and PCR product sequencing. To check the amplification efficiencies, four different dilutions (of 5, 5∧2, 5∧3 and 5∧4) were tested using cDNA for the target and internal control genes.
The statistical significance of the data were tested using one-way ANOVA and independent sample t-tests. Significance was accepted at ≤0.05. The statistical analyses were performed using SPSS 18.
SNP discovery
CLC Genomics Workbench software was used to detect potential SNPs in isotigs with sufficient depth of coverage. SNPs were identified using an arbitrary criterion of at least 4 reads supporting the consensus or variant and a similarity of 95%.
Supporting Information
KEGG pathway annotation of genes involed in the lipid metabolism.
(XLSX)
Genes differentially expressed in C. sinicus copepodid larvae and adults.
(XLSX)
Go terms of genes differentially expressed in C. sinicus copepodites and adults.
(DOC)
Acknowledgments
We would like to thank Xiaodong Wang and Yanqing Wang for their assistance with sample collection and the crew of the RV ‘Chuangxin’ for their support during sampling. We would also like to thank Chinese National Human Genome Center who carried out the 454 runs.
Funding Statement
This research was supported by special funds from National Key Basic Research Program of China (2011CB403604), National Natural Science Foundation of China (41076099), and National Marine Public Welfare Project of China (201005018). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Humes AG (1994) How many copepods? Hydrobiologia 292: 1–7. [Google Scholar]
- 2. Bron JE, Frisch D, Goetze E, Johnson SC, Lee CE, et al. (2011) Observing copepods through a genomic lens. Frontiers in Zoology 8: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beaugrand G, Brander KM, Lindley JA, Souissi S, Reid PC (2003) Plankton effect on cod recruitment in the North Sea. Nature 426: 661–664. [DOI] [PubMed] [Google Scholar]
- 4. Frangoulis C, Christou E, Hecq J (2004) Comparison of marine copepod outfluxes: nature, rate, fate and role in the carbon and nitrogen cycles. Advances in Marine Biology 47: 253–309. [DOI] [PubMed] [Google Scholar]
- 5. Richardson AJ (2008) In hot water: zooplankton and climate change. ICES Journal of Marine Science: Journal du Conseil 65: 279–295. [Google Scholar]
- 6. Minxiao W, Song S, Chaolun L, Xin S (2011) Distinctive mitochondrial genome of Calanoid copepod Calanus sinicus with multiple large non-coding regions and reshuffled gene order: Useful molecular markers for phylogenetic and population studies. BMC Genomics 12: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hudson ME (2008) Sequencing breakthroughs for genomic ecology and evolutionary biology. Molecular Ecology Resources 8: 3–17. [DOI] [PubMed] [Google Scholar]
- 8. Collins LJ, Biggs PJ, Voelckel C, Joly S (2008) An approach to transcriptome analysis of non-model organisms using short-read sequences. Genome Informatics 21: 3–14. [PubMed] [Google Scholar]
- 9. Rothberg JM, Leamon JH (2008) The development and impact of 454 sequencing. Nature Biotechnology 26: 1117–1124. [DOI] [PubMed] [Google Scholar]
- 10. Marguerat S, Bähler J (2009) RNA-seq: from technology to biology. Cellular and Molecular Life Sciences 67: 569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morozova O, Hirst M, Marra MA (2009) Applications of new sequencing technologies for transcriptome analysis. Annual Review of Genomics and Human Genetics 10: 135–151. [DOI] [PubMed] [Google Scholar]
- 12. Emrich SJ, Barbazuk WB, Li L, Schnable PS (2007) Gene discovery and annotation using LCM-454 transcriptome sequencing. Genome Research 17: 69–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vera JC, Wheat CW, Fescemyer HW, Frilander MJ, Crawford DL, et al. (2008) Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. Molecular Ecology 17: 1636–1647. [DOI] [PubMed] [Google Scholar]
- 14. Parchman TL, Geist KS, Grahnen JA, Benkman CW, Buerkle CA (2010) Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BMC Genomics 11: 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fraser B, Weadick C, Janowitz I, Rodd H, Hughes K (2011) Sequencing and characterization of the guppy (Poecilia reticulata) transcriptome. BMC Genomics 12: 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ben EC, Nathan S, Kristen P, Yuichiro S, Siegfried R, et al. (2011) The maternal and early embryonic transcriptome of the milkweed bug Oncopeltus fasciatus . BMC Genomics 12: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gregory R, Darby AC, Irving H, Coulibaly MB, Hughes M, et al. (2011) A De Novo Expression Profiling of Anopheles funestus, Malaria Vector in Africa, Using 454 Pyrosequencing. PLoS One 6: e17418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hou R, Bao Z, Wang S, Su H, Li Y, et al. (2011) Transcriptome Sequencing and De Novo Analysis for Yesso Scallop (Patinopecten yessoensis) Using 454 GS FLX. PLoS One 6: e21560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barreto FS, Moy GW, Burton RS (2011) Interpopulation patterns of divergence and selection across the transcriptome of the copepod Tigriopus californicus . Molecular Ecology 20: 560–572. [DOI] [PubMed] [Google Scholar]
- 20. Meyer E, Aglyamova G, Wang S, Buchanan-Carter J, Abrego D, et al. (2009) Sequencing and de novo analysis of a coral larval transcriptome using 454 GSFlx. BMC Genomics 10: 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hulsemann K (1994) Calanus sinicus Brodsky and C. jashmovi, nom. nov.(Copepoda: Calanoida) of the north-western Pacific Ocean: a comparison, with notes on the integumental pore pattern in Calanus s. str. Invertebrate Systematics 8: 1461–1482. [Google Scholar]
- 22. Chen Q (1964) Study on the reproduction, sex ratio and body size of Calanus sinicus (in Chinese with English abstract). Oceanologia et Limnologia Sinica 6: 272–287. [Google Scholar]
- 23. Uye S (2000) Why does Calanus sinicus prosper in the shelf ecosystem of the Northwest Pacific Ocean? ICES Journal of Marine Science: Journal du Conseil 57: 1850–1855. [Google Scholar]
- 24. Kang HK, Lee CR, Choi KH (2011) Egg production rate of the copepod Calanus sinicus off the Korean coast of the Yellow Sea during spring. Ocean Science Journal 46: 133–143. [Google Scholar]
- 25. Wang S, Li C, Sun S, Ning X, Zhang W (2009) Spring and autumn reproduction of Calanus sinicus in the Yellow Sea. Marine Ecology Progress Series 379: 123–133. [Google Scholar]
- 26. Sun S, Zhang G (2005) Over-summering strategy of Calanus sinicus . GLOBEC Int Newsl 11: 34. [Google Scholar]
- 27. Pu XM, Sun S, Yang B, Zhang GT, Zhang F (2004) Life history strategies of Calanus sinicus in the southern Yellow Sea in summer. Journal of Plankton Research 26: 1059–1068. [Google Scholar]
- 28. Bi H, Sun S, Gao S, Zhang G (2001) The ecological characteristics of zooplankton community in the Bo hai Sea II. The distribution of copepoda abundance and seasonal dynamics. Acta Ecologica Einica 21: 177–185. [Google Scholar]
- 29. Zhang W, Tang D, Yang B, Gao S, Sun J, et al. (2009) Onshore–offshore variations of copepod community in northern South China Sea. Hydrobiologia 636: 257–269. [Google Scholar]
- 30. O'Neil S, Dzurisin J, Carmichael R, Lobo N, Emrich S, et al. (2010) Population-level transcriptome sequencing of nonmodel organisms Erynnis propertius and Papilio zelicaon . BMC Genomics 11: 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zeng V, Villanueva K, Ewen-Campen B, Alwes F, Browne W, et al. (2011) De novo assembly and characterization of a maternal and developmental transcriptome for the emerging model crustacean Parhyale hawaiensis . BMC Genomics 12: 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brockman W, Alvarez P, Young S, Garber M, Giannoukos G, et al. (2008) Quality scores and SNP detection in sequencing-by-synthesis systems. Genome Research 18: 763–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Irigoien X (2004) Some ideas about the role of lipids in the life cycle of Calanus finmarchicus . Journal of Plankton Research 26: 259–263. [Google Scholar]
- 34. Dubrovsky EB (2005) Hormonal cross talk in insect development. Trends in Endocrinology and Metabolism 16: 6–11. [DOI] [PubMed] [Google Scholar]
- 35. Kidokoro K, Iwata K, Fujiwara Y, Takeda M (2006) Effects of juvenile hormone analogs and 20-hydroxyecdysone on diapause termination in eggs of Locusta migratoria and Oxya yezoensis . Journal of Insect Physiology 52: 473–479. [DOI] [PubMed] [Google Scholar]
- 36. Pierceall WE, Li C, Biran A, Miura K, Raikhel AS, et al. (1999) E75 expression in mosquito ovary and fat body suggests reiterative use of ecdysone-regulated hierarchies in development and reproduction. Molecular and Cellular Endocrinology 150: 73–89. [DOI] [PubMed] [Google Scholar]
- 37. Henrich VC, Livingston L, Gilbert LI (1993) Developmental requirements for the ecdysoneless (ecd) locus in Drosophila melanogaster . Developmental Genetics 14: 369–377. [DOI] [PubMed] [Google Scholar]
- 38. Li T, Bender M (2000) A conditional rescue system reveals essential functions for the ecdysone receptor (EcR) gene during molting and metamorphosis in Drosophila . Development 127: 2897–2905. [DOI] [PubMed] [Google Scholar]
- 39. Riddiford LM (2008) Juvenile hormone action: a 2007 perspective. Journal of Insect Physiology 54: 895–901. [DOI] [PubMed] [Google Scholar]
- 40. Helvig C, Koener J, Unnithan G, Feyereisen R (2004) CYP15A1, the cytochrome P450 that catalyzes epoxidation of methyl farnesoate to juvenile hormone III in cockroach corpora allata. Proceedings of the National Academy of Sciences of the United States of America 101: 4024–4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vannini L, Ciolfi S, Dallai R, Frati F, Hoffmann KH, et al. (2010) Putative-farnesoic acid O-methyltransferase (FAMeT) in medfly reproduction. Archives of Insect Biochemistry and Physiology 75: 92–106. [DOI] [PubMed] [Google Scholar]
- 42. Tarrant AM, Baumgartner MF, Verslycke T, Johnson CL (2008) Differential gene expression in diapausing and active Calanus finmarchicus (Copepoda). Marine Ecology Progress Series 355: 193–207. [Google Scholar]
- 43. Denlinger DL (2002) Regulation of diapause. Annual Review of Entomology 47: 93–122. [DOI] [PubMed] [Google Scholar]
- 44. Aruda AM, Baumgartner MF, Reitzel AM, Tarrant AM (2011) Heat shock protein expression during stress and diapause in the marine copepod Calanus finmarchicus . Journal of Insect Physiology 57: 665–675. [DOI] [PubMed] [Google Scholar]
- 45. Jakobsson A, Westerberg R, Jacobsson A (2006) Fatty acid elongases in mammals: their regulation and roles in metabolism. Progress in Lipid Research 45: 237–249. [DOI] [PubMed] [Google Scholar]
- 46. Tillman JA, Seybold SJ, Jurenka RA, Blomquist GJ (1999) Insect pheromones – an overview of biosynthesis and endocrine regulation. Insect Biochemistry and Molecular biology 29: 481–514. [DOI] [PubMed] [Google Scholar]
- 47.Wang S, (2009) Reproduction, population recruitment and life history of Calanus sinicus in the Yellow Sea (Dissertation, in Chinese with English abstract). Qingdao: Institute of Oceanology, Chinese Academy of Sciences. [Google Scholar]
- 48. Liu Q, Yuan Y, Lin J, Zhong Y (2010) Advance of researches on insect cuticular proteins and the regulation mechanism of their gene expression (in Chinese with English abstract). Chinese Journal of Applied Entomology 002: 247–255. [Google Scholar]
- 49. Eichner C, Frost P, Dysvik B, Jonassen I, Kristiansen B, et al. (2008) Salmon louse (Lepeophtheirus salmonis) transcriptomes during post molting maturation and egg production, revealed using EST-sequencing and microarray analysis. BMC Genomics 9: 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Harrison PM, Arosio P (1996) The ferritins: molecular properties, iron storage function and cellular regulation. Biochimica et Biophysica Acta 1275: 161–203. [DOI] [PubMed] [Google Scholar]
- 51. Reif DW (1992) Ferritin as a source of iron for oxidative damage. Free Radical Biology & Medicine 12: 417–427. [DOI] [PubMed] [Google Scholar]
- 52. MacRae TH (2010) Gene expression, metabolic regulation and stress tolerance during diapause. Cellular and Molecular Life Sciences 67: 2405–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vos MJ, Hageman J, Carra S, Kampinga HH (2008) Structural and Functional Diversities between Members of the Human HSPB, HSPH, HSPA, and DNAJ Chaperone Families. Biochemistry 47: 7001–7011. [DOI] [PubMed] [Google Scholar]
- 54.Wang M (2010) Application of molecular markers to the researches on pelagic copepods in the Chinese coastal regions (Dissertation, in Chinese with English abstract). Qingdao: Institute of Oceanology, Chinese Academy of Sciences. [Google Scholar]
- 55. Chen Z, Xue C, Zhu S, Zhou F, Ling XB, et al. (2005) GoPipe: streamlined gene ontology annotation for batch anonymous sequences with statistics. Progress in Biochemistry and Biophysics 32: 187–191. [Google Scholar]
- 56. Colbourne JK, Pfrender ME, Gilbert D, Thomas WK, Tucker A, et al. (2011) The Ecoresponsive Genome of Daphnia pulex . Science 331: 555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M (2010) KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Research 38: 355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods 5: 621–628. [DOI] [PubMed] [Google Scholar]
- 59. Wang L, Feng Z, Wang X, Zhang X (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26: 136–138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
KEGG pathway annotation of genes involed in the lipid metabolism.
(XLSX)
Genes differentially expressed in C. sinicus copepodid larvae and adults.
(XLSX)
Go terms of genes differentially expressed in C. sinicus copepodites and adults.
(DOC)