

Abstract
Lead is a potent environmental toxin that mimics the effects of divalent metal ions, such as zinc and calcium, in the context of specific molecular targets and signaling processes. The molecular mechanism of lead toxicity remains poorly understood. The objective of this work was to characterize the effect of Pb2+ on the structure and membrane-binding properties of C2α. C2α is a peripheral membrane-binding domain of Protein Kinase Cα (PKCα), which is a well-documented molecular target of lead. Using NMR and isothermal titration calorimetry (ITC) techniques, we established that C2α binds Pb2+ with higher affinity than its natural cofactor, Ca2+. To gain insight into the coordination geometry of protein-bound Pb2+, we determined the crystal structures of apo and Pb2+-bound C2α at 1.9 Å and 1.5 Å resolution, respectively. A comparison of these structures revealed that the metal-binding site is not pre-organized and that rotation of the oxygen-donating sidechains is required for the metal coordination to occur. Remarkably, we found that holodirected and hemidirected coordination geometries for the two Pb2+ ions coexist within a single protein molecule. Using protein-to-membrane Förster resonance energy transfer (FRET) spectroscopy, we demonstrated that Pb2+ displaces Ca2+ from C2α in the presence of lipid membranes through the high-affinity interaction with the membrane-unbound C2α. In addition, Pb2+ associates with phosphatidylserine-containing membranes and thereby competes with C2α for the membrane-binding sites. This process can contribute to the inhibitory effect of Pb2+ on the PKCα activity.
Keywords: Lead toxicity, hemidirected lead, Pb(II), stereochemically active lone pair, C2 domain, peripheral membrane protein, Protein Kinase C, lipid membrane, phosphatidylserine, anionic lipid, NMR, FRET
INTRODUCTION
Lead is a potent environmental toxin that has accumulated in the environment ~1000-fold above its natural level as a result of human activity. According to the Center for Disease Control and Prevention, approximately 250,000 young children in the U.S. have blood levels of lead that exceed 10 micrograms per 1 deciliter of blood.1 Even below the 10 μg/dL “level of concern”, lead levels show significant inverse correlation with IQ in children at 3 and 5 years of age,2 indicating that there is no safe level of exposure. In addition to severe neurocognitive deficiencies,3 lead poisoning can cause anemia, kidney disease, and hypertension.4
Although detrimental effects of lead on human health have been known for decades, the molecular mechanism of lead toxicity remains poorly understood. It is generally accepted that Pb2+ mimics the effects of divalent metal ions, such as Ca2+ and Zn2+, in the context of specific molecular targets.5 One of the Pb2+ targets is the Protein Kinase C (PKC) isoenzymes,6 a family of Ser/Thr kinases that control signaling pathways essential for cell proliferation, differentiation, and survival.7 Ca2+-dependent PKC isoenzymes (α, βI, βII, and γ) have an independently folded C2 domain that anchors the parent enzyme to lipid membranes in response to binding Ca2+ ions.8,9 This membrane-binding step is absolutely essential for the kinase activation. It was hypothesized that at least two Pb2+ binding sites in the PKCα isoenzyme reside on its C2 domain.10
The objective of this work was to understand the effect of Pb2+ on the structure and membrane-binding properties of the C2 domain from PKCα (C2α). Specifically, we wanted to establish if Pb2+ binds to the same sites on the protein as Ca2+; measure the relative binding affinities of Ca2+ and Pb2+ to C2α; determine if the replacement of Ca2+ with Pb2+ has any influence on the structure of C2α and conformation of the Ca2+-binding loops (CBLs); compare the coordination geometries of protein-bound Ca2+ and Pb2+; and finally understand how the change in the chemical environment of C2α brought about by Pb2+ binding affects the downstream reaction, which is the association of C2α with lipid membranes. In addition to reporting on the Pb2+-dependent activity of the parent enzyme, C2α can serve as a paradigm for the Ca2+-dependent C2 domains that are found in other molecular targets of Pb2+, such as synaptotagmin I and phospholipase C.6
The activation of PKC partially purified from rat brain by picomolar concentrations of Pb2+ was first reported in 1988.11 The effect was shown to be specific to Pb2+, as none of the other ten heavy metals that were tested activated PKC to the same extent as Pb2+ at picomolar concentrations. The evidence for multi-site interactions of Pb2+ with PKC was subsequently obtained by both in vivo and in vitro studies.10,12,13 Micromolar concentrations of Pb2+ inhibited the constitutive kinase activity of both Ca2+-dependent and -independent PKCs, suggesting the presence of a low-affinity Pb2+ site in the catalytic domain. Partial activation of PKC at picomolar to nanomolar Pb2+ concentrations was attributed to the presence of the high-affinity Pb2+-binding site in the C2 domain. The second Pb2+-binding site having a lower affinity than the first was suggested to have an inhibitory effect on PKC.10 These experiments established the central role of the C2 domain in the Pb2+-dependent modulation of PKC activity.
Ca2+-responsive C2 domains are independently folded structural domains of ~130 amino acids that are found in a variety of multi-modular proteins.14 The binding of two or more Ca2+ ions drives the association of C2 domains with lipid membranes,15 where they recognize the head group of anionic lipids including phosphatidylserine (PtdSer).16 Computational studies showed that Ca2+ ions alter the electrostatic potential of C2 domains, making the non-specific electrostatic interactions between anionic lipid groups and CBLs a significant contributor to the energetics of the membrane-binding step.17 According to site-directed fluorescence and spin-labeling data, C2 is oriented almost parallel to the membrane surface,18,19 with the first and third CBLs inserted into the head group region of the membrane. In addition to CBLs, an important structural element of C2 is a cationic patch (“β-groove”), which is involved in interactions with phosphoinositides.20,21
Current understanding of how divalent lead interacts with proteins has been shaped by the structural work on the Pb2+-protein and -peptide complexes, mostly having thiolate-rich coordination sites. For example, the best-studied molecular target of Pb2+ is the Zn2+-dependent 5-aminolevulinic acid dehydratase (ALAD), also known as porphobilinogen synthase.22 Structural characterization of the Pb2+-complexed yeast ALAD,23 ab initio studies of the Pb2+ coordination geometry in the human enzyme,24 and biochemical characterization of Pb2+/Zn2+ substitution25 revealed the central role of the stereochemically active 6s2 pair of Pb2+ in altering the geometry of the metal-binding site. The work of Godwin’s laboratory on Pb2+ complexation of cysteine-containing peptides26 showed that Pb2+ adopts a tri-coordinate geometry when replacing structural Zn2+ ions with tetrahedral coordination27 and is thus unable to facilitate proper folding of zinc finger domains. Using the designed cysteine-rich peptides and 207Pb NMR detection, Pecoraro’s group developed sensitive methods for probing the coordination environment of Pb2+ in biological molecules.28
Interactions of Pb2+ with oxygen-rich coordination environment found in Ca2+-binding proteins are not as well characterized as those with sulfur-rich sites. The only high-resolution structure of a Ca2+-binding protein in complex with Pb2+ is that of calmodulin (CaM),38,39 for which the non-EF hand Pb2+ binding sites were hypothesized to be responsible for CaM’s lack of activation at high Pb2+ concentrations.29
In this work, we used C2α as a paradigm for the Ca2+-dependent C2 domains to understand how Pb2+ affects their structure and membrane-binding properties. To our knowledge, this is the first time the interactions within the ternary system comprising a peripheral membrane domain, Pb2+ ions, and lipid bilayers are investigated at the molecular level. Using solution NMR methods and ITC, we established that Pb2+ binds to C2α with high affinity. We report high-resolution structures of apo and Pb2+-complexed C2α, which makes C2α only the second protein after CaM with apo, Ca2+-bound, and Pb2+-bound structures available. Despite being only 4.1 Å apart in the structure, two Pb2+ ions adopt different coordination geometries due to the effect of the stereochemically active 6s2 electron pair. We used FRET spectroscopy to conduct Ca2+- and Pb2+-driven C2α membrane-binding experiments, which revealed that Pb2+ is able to displace Ca2+ from C2α in the presence of PtdSer-containing membranes. In aggregate, our data demonstrate that Pb2+ can potentially act as a concentration-dependent modulator of the C2α-membrane interactions by competing with Ca2+ for the protein metal-binding sites and with C2α for the PtdSer membrane sites.
EXPERIMENTAL SECTION
Materials
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(5-dimethylamino-1-naphthalenesulfonyl) (dansyl-PE) were obtained from Avanti Polar Lipids (Alabaster, AL). The C2α construct comprising residues 155-293 of PKCα (Rattus norvegicus) was cloned into a pET-SUMO vector (Invitrogen),30 over-expressed, and purified as described in Section 1 of the Supporting Information. All buffer solutions used in this study were treated with Chelex 100 (Sigma-Aldrich) to remove residual divalent metal ions. Decalcification of C2α was accomplished by incubating the protein with 0.1 mM EDTA, followed by extensive exchange of the protein solution into the EDTA-free buffer. For the NMR assignment experiments, the buffer composition for the final exchange step was 10 mM MES (pH 6.0), 8% D2O, and 0.02% NaN3. For the NMR-detected Ca2+ and Pb2+ binding experiments, 100 mM KCl was included in the buffer solution.
Site-specific resonance assignments and NMR-monitored binding of divalent metal ions to C2α
All NMR experiments were carried out at 25 °C on Varian Inova spectrometers operating at 1H Larmor frequencies of 500 MHz (11.7 Tesla) and 600 MHz (14.1 Tesla). The temperature was calibrated using methanol. Sequence-specific assignments of the 1HN, 13Cα, 13Cβ, and 15N resonances for the apo C2α were obtained using gradient-enhanced triple-resonance NMR experiments HNCACB, CBCA(CO)NH,31 and C(CO)NH.32 2H-decoupled three-dimensional HNCACB and HN(CO)CACB experiments33 were used to assign the [U-13C,15N; 55%-2H] C2α in complex with Ca2+ and Pb2+. NMR data were processed with nmrPipe34 and assigned with Sparky.35
Binding of Ca2+ and Pb2+ to C2α was monitored using 15N-1H Heteronuclear Single Quantum Coherence (HSQC) spectra of [U-15N]-enriched 160 μM C2α domain. The desired concentration of divalent metal ions in the NMR sample was achieved using concentrated stock solutions of Ca(II) chloride and Pb(II) acetate. The Pb(II) acetate stock solution was prepared in the absence of chloride ions (10 mM MES, 8% D2O, 0.02% NaN3) at pH 6.0 to avoid the formation of Pb(Cl)2 species. The binding curves for Ca2+ and Pb2+ were constructed by plotting the absolute value of the change in 1HN and/or 15N chemical shifts as a function of total metal concentration. The dissociation constant, Kd, for all single-site binding equilibria described in this work, was determined by globally fitting the binding curves using the following equation:
| (1) |
where Δδ is the residue-specific absolute value of the observed change in the chemical shift for 15N or 1HN at total ligand concentration L0, ΔδPL is the absolute value of the residue-specific chemical shift difference between the bound and apo forms of the protein, and P0 is the total protein concentration.36
For the chemical shift perturbation analysis, the normalized change in the chemical shift was determined according to the following equation:
| (2) |
where ΔδH and ΔδN are the residue-specific chemical shift changes, and γH and γN are the gyromagnetic ratios of 1H and 15N nuclei, respectively.
Isothermal Titration Calorimetry
ITC was used to determine the affinity of Pb2+ to the first metal-binding site of C2α. The experiments were carried out at 25 °C using a VP-ITC calorimeter (Microcal Inc., Northampton, MA). Prior to the measurements, decalcified C2α was extensively dialyzed into 10 mM MES buffer (pH 6.0) containing 10 mM KCl. The concentration of protein in the sample cell was 35.4 μM. The Pb2+ titration solution was prepared in the same buffer from a 10 mM stock of Pb(II) acetate that was standardized using Inductively Coupled Plasma (ICP) measurements. The volume of the titration solution injected into the sample cell varied from 2 to 10 μl per injection. Dilution effects were taken into account by injecting the titrant into the reference cell containing pure buffer. The molar ratio of Pb2+ to C2α was 2.5 at the end of the titration. The data were fit to the independent binding sites model involving two sites using the Origin 5.0 software provided by the VP-ITC manufacturer. The presence of two Pb2+ binding sites was independently demonstrated using NMR experiments. The errors reported for the thermodynamic parameters are the errors obtained in the fitting procedures.
Crystallization of apo and Pb2+-bound C2α
The crystals of C2α in the presence and absence of Pb2+ were grown using the hanging-drop vapor diffusion method. Apo C2α (24 mg/mL) was mixed 1:1 with the crystallization buffer comprising 100 mM HEPES (pH 7.5), 0.25 M lithium sulfate monohydrate, and 20% PEG 3350. For the Pb2+ complex, C2α (24 mg/mL) was incubated with 7.5 mM Pb(II) acetate solution and mixed 1:1 with the crystallization buffer comprising 100 mM bis-tris (pH 5.5), 0.25 M lithium sulfate monohydrate, and 20% PEG 3350. The crystals appeared after 4-5 days and were allowed to grow for 3 weeks at 25°C. Data analysis and the structure determination protocol are described in Section 2 of the Supporting Information; the statistics of the crystallographic analysis is given in Table S1. The coordinates have been submitted to the Protein Data Bank (http://www.pdb.org/) and assigned the ID codes of 3RDJ and 3RDL for the apo and Pb2+-bound structures, respectively.
Metal-dependent membrane association of C2α by Surface Plasmon Resonance (SPR)
All SPR measurements were performed at 24 °C using a lipid-coated Pioneer L1 chip (Biacore AB, Piscataway, NJ) in the BIACORE X system as described previously.37 Briefly, after washing the sensor chip surface with the running buffer (20 mM HEPES, pH 7.4, containing 0.16 M KCl), the active surface and the control surface of the sensor chip were coated with POPC/POPS (80:20) and POPC (100%) vesicles, respectively, to give the same resonance unit (RU) values. The level of lipid coating for both surfaces was kept at a minimum, which is necessary for preventing the non-specific adsorption to the sensor chips. This low surface coverage minimized the mass transport effect and kept the total protein concentration above the total concentration of protein binding sites on the vesicles. Equilibrium SPR measurements were carried out at the flow rate of 5 μl/min to allow sufficient time for the response values of the association phase to reach near-equilibrium values Req. Each of the sensorgrams was corrected for a refractive index change by subtraction of the control surface response. We assumed a Langmuir-type binding between the protein (P) and protein-binding sites (L) on vesicles (i.e. P + L ⇔ PL). The Kd was determined by fitting the dependence of Req on the total protein concentration, P0, with the following equation:
| (3) |
Metal-dependent membrane association of C2α by ultracentrifugation
Sucrose-loaded large unilamellar vesicles (LUVs) were prepared as described by Giorgione and Newton.38 In brief, chloroform solutions of the lipid components were mixed at their desired molar ratios. The solvent was removed under N2 followed by two hours under vacuum. The lipid film was hydrated with the sucrose buffer (10 mM MES at pH 6.0 and 170 mM sucrose), vortexed, and subjected to five freeze/thaw cycles. An Avanti Polar Lipids mini-extruder with a 100 nm polycarbonate filter was used to prepare LUVs. Phosphate quantification assays39 were used to determine the lipid concentration after extrusion and the pelleting efficiency of the LUVs, which was >98% for all experiments. 5 μM C2α was incubated with 1.5 mM or 10 mM total lipid LUVs (POPC/POPS, 67:33 mole percent) for one hour, followed by the addition of the divalent metal ion to the desired concentration. After a 20-minute incubation period, LUVs were centrifuged in a Beckman TL-100 tabletop ultracentrifuge for 30 minutes at 50,000 rpm and 25°C. The amount of C2α in the supernatant was quantified using the bicinchoninic acid (BCA) assay kit (Thermo Scientific). The fraction of membrane-bound C2α was calculated as:
| (4) |
where P0 is the total amount of protein in the sample, and Psup is the amount of protein present in the supernatant at a given metal concentration. Metal-free ultracentrifugation experiments were used to quantify the non-specific binding of C2α to membranes, which was <5% in all experiments.
Metal-dependent membrane association of C2α by FRET
Ca2+- and Pb2+-dependent membrane association of C2α was monitored by FRET between the tryptophan residues of C2α and the fluorescent lipid, dansyl-PE.40 Pb2+ or Ca2+ from the concentrated stock solutions was added into a mixture of 0.5 μM C2α in 10 mM MES (pH 6.0) and 100 mM KCl, and LUVs with a total lipid concentration of 150 μM. Pb(Cl)2 remains soluble at these concentrations of Pb2+ and Cl− ions. The LUVs’ composition in the binding experiments was POPC/POPS/dansyl-PE (60:33:7). POPC/dansyl-PE (93:7) LUVs were used to test the PtdSer specificity of C2α in the presence of Pb2+.
The change in dansyl-PE fluorescence at 25°C was monitored using an ISS Koala fluorometer (ISS, Champaign, IL). Excitation and emission wavelengths were 295 and 494 nm, respectively. The “blank” sample contained all reactants but C2α; its signal was subtracted from that of the C2α-containing sample. The change in fluorescence upon metal binding, ΔF, relative to the maximal change, ΔFmax, was plotted as a function of the total metal concentration. The binding curves were fit with the Hill equation:
| (5) |
where H is the Hill coefficient and [M2+]1/2 is the metal concentration required to achieve the half-maximal fluorescence change.
RESULTS
In this section, we describe the experiments designed to probe (i) the affinity of C2α to Pb2+, (ii) the influence of Pb2+ on the structure of C2α and its mode of interaction with lipid bilayers; (iii) the coordination geometry of Pb2+ in complex with C2α; and (iv) the ability of Pb2+ to compete for protein and membrane-binding sites.
C2α binds Pb2+ with high affinity
Early studies of the effect of Pb2+ on the activity of full-length PKCα led to a hypothesis that at least two of the Pb2+-binding sites reside on the C2 domain.10 To establish if C2α is indeed capable of binding Pb2+ and if the modes of protein-metal interaction are different for Pb2+ and Ca2+, we characterized the interaction of [U-15N]-enriched apo C2α with both metal ions by NMR. Expansions of the protein 15N-1H HSQC spectra in the presence of varying concentrations of Ca2+ (Column A) and Pb2+ (Column B) are shown in Figure 1 for four representative residues: Gly190, Asp248, Ile215, and Asp254. Two of these residues, 248 and 254, are involved in direct coordination with Ca2+.41 The arrows in the spectra point towards the increasing concentration of metal ions, which ranges from 0 to 3.2 mM for Ca2+ and from 0 to 1.28 mM for Pb2+.
Figure 1.
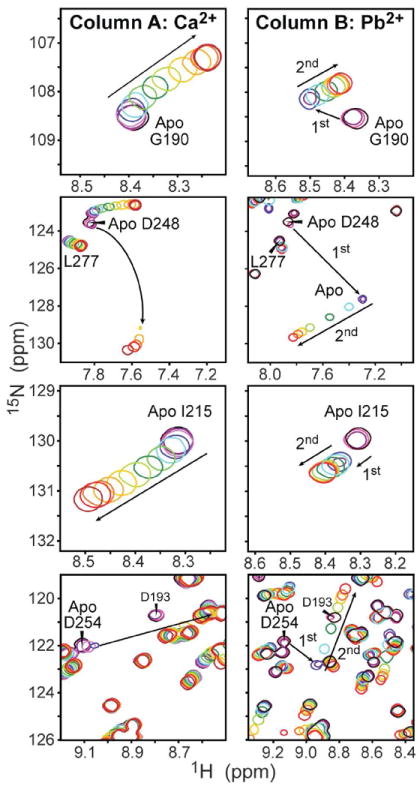
C2α binds Pb2+ with higher affinity than Ca2+. Expansions of the 15N-1H HSQC spectra illustrate the differences in the response of Gly190, Asp248, Ile215, and Asp254 to Ca2+ (column A) and Pb2+ (column B). The protein concentration was 160 μM. The arrows point in the direction of increasing concentration of metal ions, which varies from 0 to 3.2 (1.28) mM for Ca2+ (Pb2+).
The binding regime of Ca2+ to C2α is intermediate-to-fast on the chemical-shift timescale. For residues that are in the fast exchange regime, such as Gly190 and Ile215, the cross-peaks follow a smooth curved trajectory. This behavior is analogous to what has been observed previously for C2 domains from synaptotagmin and PKCβ,42 and is indicative of C2α binding two Ca2+ ions with comparable affinities. As shown in Column B, the spectra of C2α in the presence of Pb2+ exhibit a very different pattern. The NMR spectra clearly show the presence of two Pb2+ binding sites with different binding affinities. The first Pb2+-binding event is slow on the chemical-shift timescale, which manifests itself in the appearance of cross-peaks corresponding to single-Pb2+ bound species in addition to the apo form. The binding of the second Pb2+ is in fast exchange, with the cross-peaks following a linear trajectory.
The slow exchange behavior of the first Pb2+ binding event precluded the determination of the dissociation constant, Kd1Pb, based on the NMR spectra. Instead, we used ITC to characterize the thermodynamics of the first binding step. A typical ITC titration profile is shown in Figure 2A. The ITC data were fit as described in the Experimental Section to obtain the following parameters for the first site: n1=0.57±0.01, Kd1Pb=67±1 nM, ΔH1=−5.6±0.1 kcal/mol, and ΔS1=14.1 cal/mol·K. In separate ITC experiments, the stoichiometry of binding n1 ranged from 0.6 to 1.1 for the first site. The deviation of the binding stoichiometry from 1 in C2α has been reported previously.43 In our case, this behavior is likely caused by variable amounts of binding-competent apo C2α, which is extremely sensitive to stirring and other types of mechanical agitation. The apparent stoichiometry of binding had no effect on the obtained values of the thermodynamic parameters.
Figure 2.
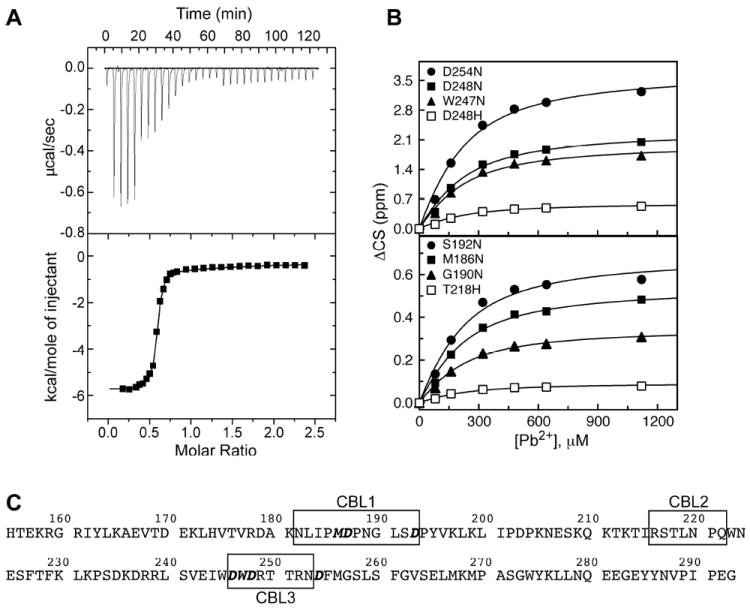
Differential affinities of the two metal-binding sites of C2α to Pb2+. (A) The ITC profile for the titration of apo C2α with Pb2+. (B) NMR binding curves constructed using Pb2+ titration experiments for site (2). (C) Primary structure of C2α highlighting the loop regions (boxed) and coordinating residues (boldface/italicized).
Because of the low affinity of Pb2+ to the second site of C2α, the isotherm for this binding event is extremely shallow with no clear transition. The isotherm shape parameter c is estimated at ~0.4, whereas it needs to be between 5 and 500 to enable reliable determination of the second site parameters. To circumvent this problem, we reduced the total number of fitting parameters by holding n2 constant at 1 and obtained the following values for the second site: Kd2,ITCPb=91±1 μM, ΔH2=−1.8±0.6 kcal/mol, and ΔS2=12.4 cal/mol·K. These values should be treated as estimates only. We used solution NMR experiments to obtain an accurate value of Kd2Pb.
To determine Kd2Pb, we constructed binding curves for the second site by plotting the absolute values of 15N and/or 1H chemical shift changes as a function of the total Pb2+ concentration. Several representative binding curves for residues that belong to CBL3 (top), and CBL1/CBL2 (bottom) are shown in Figure 2B. The relevant regions of the primary structure are indicated in Figure 2C, with the metal-coordinating residues shown in boldface/italics. Fitting 34 residue-specific 1H and 15N binding curves with Eq. (1) produced Kd2Pb=129±4 μM. Analysis of NMR-detected Ca2+-binding curves of C2α is described in Section 3 of the Supporting Information. The estimated value of KdCa is ~270 μM.
From this set of experiments, we concluded that C2α binds Pb2+ with significantly higher affinity than Ca2+. In addition, there is a 2000-fold difference in the Kd values for the two sites of Pb2+. We will hereafter refer to these two sites as high- and low-affinity.
Identification of the high-affinity Pb2+ binding site
Chemical shifts are exquisitely sensitive to the changes in the electronic environment of nuclei caused by binding events. We used chemical shift perturbation analysis to identify the high-affinity Pb2+ site of C2α and determine which regions of the protein are selectively affected by the binding of the first and second Pb2+ ions. To probe individual Pb2+-binding events, the chemical shift perturbations Δ were calculated using Eq. (2) for the C2α·Pb complex relative to the apo form, and for the C2α·Pb2 complex relative to the C2α·Pb.
The results are plotted as a function of primary structure in Figure 3A, and mapped onto the three-dimensional structure of apo C2α (vide infra) in Figure 3B. It is evident that filling the high-affinity Pb2+ site affects both CBL1 and CBL3 regions, while filling the low-affinity site mainly affects CBL3. Based on our NMR data, the crystal structure of the ternary C2α·Ca2·PtdSer complex,41 and the crystal structure of the Pb2+-bound C2α reported in this paper (vide infra), we could immediately assign the high-affinity site to site (1) and low-affinity site to site (2), in the nomenclature used for Ca2+.41
Figure 3.
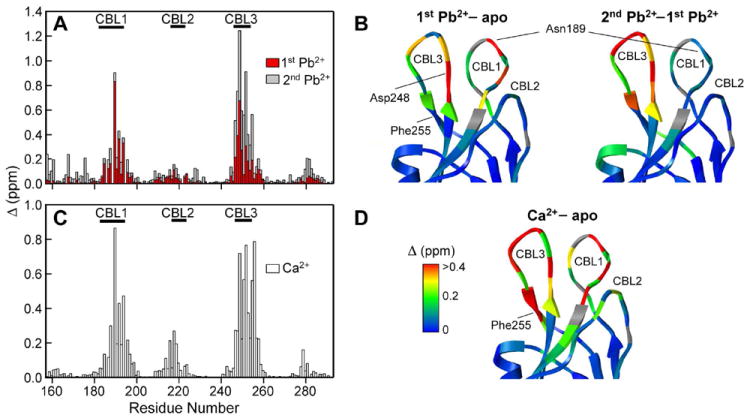
Δ values for the first (red) and second (gray) Pb2+ binding events plotted as a function of primary structure in (A), and mapped onto the CBL region of apo C2α in (B). Δ values for the Ca2+-bound C2α plotted as a function of primary structure in (C), and mapped onto the CBL region of apo C2α in (D). Proline residues are shown in gray.
To compare the effects of Pb2+ and Ca2+ on C2α, we also carried out the chemical shift perturbation analysis of C2α·Ca2 relative to the apo form. The results are shown in Figures 3C and 3D. Overall, the perturbation pattern is similar to that observed for C2α·Pb2, with the CBL1, CBL2, and CBL3 regions being affected the most. Some differences between Pb2+ and Ca2+ are observed in the region between the N-terminus and CBL1. This region is not responsive to Ca2+ but is perturbed upon binding of the second Pb2+ ion.
There are several features that are worth noting at the level of individual amino acids. The most responsive residue to binding of both Ca2+ and Pb2+ is Asp248, whose sidechain carboxyl group serves as a ligand to both metal sites, according to the crystal structure of the ternary C2α·Ca2·PtdSer complex. Another notable residue is Asn189, which belongs to CBL1. Although Asn189 is not involved in metal-ion coordination, the binding of the first Pb2+ and Ca2+ ions significantly perturbs the chemical shifts of its backbone N-H group. This residue has been implicated in the interactions of C2α with the PtdSer component of plasma membrane.21 Phe255 of the CBL3 region showed a strong chemical shift response to Ca2+ binding but none to Pb2+. Phe255 is involved in putative inter-domain interactions within full-length PKCα that get disrupted when the enzyme associates with lipid membranes upon binding Ca2+.44
In summary, solution NMR experiments enabled us to identify the high-affinity Pb2+ site, and revealed that binding of Pb2+ and Ca2+ has a similar effect on the backbone conformation and electrostatic properties of C2α.
Pb(2) adopts a hemidirected coordination geometry in the C2α·Pb2 structure
To gain insight into the conformational preferences of metal-ion ligands and the coordination geometry of protein-bound Pb2+, we determined the crystal structures of C2α in apo and Pb2+-bound forms. The 1.9 Å resolution structure of apo C2α is the first and only ligand-free structure of a C2 domain from a conventional PKC isoenzyme. The 1.5 Å resolution structure of Pb2+-bound C2α has two well-defined Pb2+ ions specifically coordinated by the protein, and is referred to as the C2α·Pb2 complex in this work. Non-specific association of Pb2+ ions with the charged sidechains on the protein surface was not observed. High resolution of both crystal structures enabled us to analyze the conformation of the CBL regions and determine the detailed geometry of both metal-binding sites.
Figure 4A shows the backbone superposition of apo C2α, C2α·Pb2, and C2α·Ca2·PtdSer41 complexes. With the apo form as a reference structure, the pair-wise root-mean-squared deviations (RMSDs) for the backbone atoms are 0.49 Å and 0.37 Å for the C2α·Ca2·PtdSer and C2α·Pb2 complexes, respectively. At least in protein crystals, the binding of divalent metal ions has little effect on the backbone conformation of the protein core and the CBL regions. However, detailed inspection of the metal coordination site in apo and C2α·Pb2 structures revealed significant differences in the positions of metal-coordinating oxygen atoms.
Figure 4.
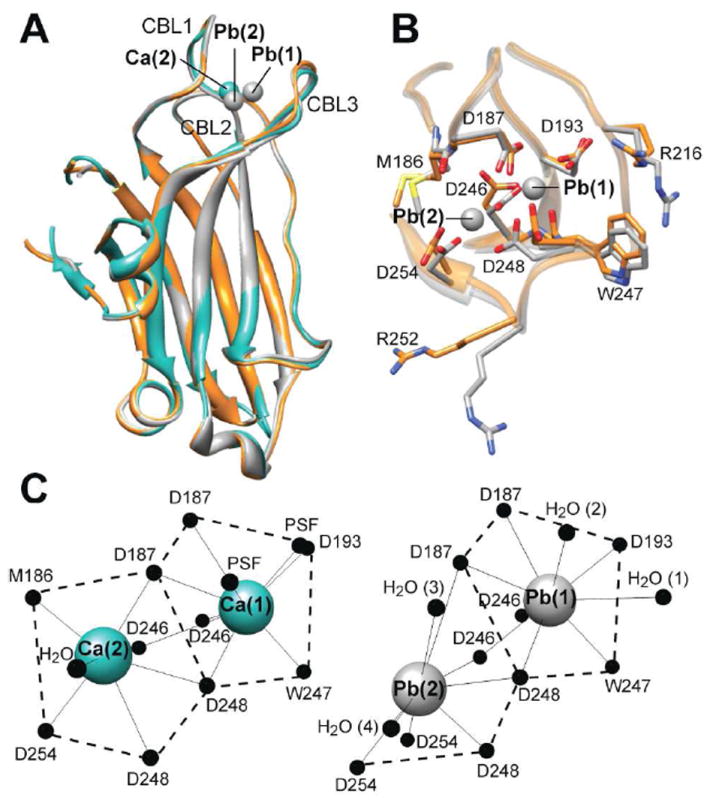
(A) Backbone superposition of apo C2α (orange), C2α·Pb2 (gray), and C2α·Ca2·PtdSer (cyan) complexes. The corresponding PDB ID codes are 3RDJ, 3RDL, and 1DSY. Ca(1) and Pb(1) are superimposable. (B) Detailed view of the metal coordination sites in apo and Pb2+-bound C2α using the same color-coding scheme as in (A). (C) Metal coordination geometries in Ca2+- and Pb2+-bound C2α. PSF stands for 1,2-dicaproyl-sn-phosphatidyl-L-serine.
The metal coordination sites in apo C2α and the C2α·Pb2 complex are contrasted in Figure 4B. Amino acid sidechains that contribute an oxygen ligand to the metal-coordination sphere in either Ca2+ or Pb2+-bound forms are labeled. Notable differences are observed in the sidechain conformations of Asp246, Asp248, and Asp254. As shown in Figure 4C, the carboxyl oxygens of Asp246 and Asp248 sidechains are involved in coordination bonds with both metal sites, while the Asp254 sidechain coordinates only to site (2). In addition to the coordinating residues, we observed a significant change in the conformations of the Arg252 and Arg216 sidechains. Both residues belong to the CBL regions, with Arg216 implicated in the interactions with PtdSer.45
The metal coordination geometries for Ca2+ and Pb2+ are compared in Figure 4C. Pb(1), which we identified as a high-affinity site, has nine ligands that are arranged around the metal center in a manner similar to that of Ca(1). To identify the ligands, we used the 3.5 Å cutoff distance determined in the analysis of the Pb-O bond lengths in protein-Pb2+ complexes46 deposited in the Protein Data Bank. The seven protein ligands are: Asp187 Oδ1 and Oδ2; Asp193 Oδ2; Asp246 Oδ1 and Oδ2; Trp 247 O; and Asp248 Oδ1. There are also two water molecules whose oxygens provide the two top axial ligands. In the Ca(1) coordination sphere, these positions are occupied by the phosphoryl oxygens of PtdSer, although some uncertainty about the ligand conformation in the structure was noted by the authors.41
A striking difference was observed in the metal coordination geometries of site (2). In Pb2+-bound structure, we observed that eight ligands occupy one coordination hemisphere, while the other hemisphere is devoid of any ligands. This is the hallmark of the hemidirected coordination geometry of lead. The hemidirected geometry is characterized by the spatial expansion of the 6s2 lone pair of Pb, forcing all ligands to be accommodated on one side of the coordination sphere. The protein ligands for Pb(2) are: Asp187 Oδ1; Asp246 Oδ2; Asp254 Oδ1 and Oδ2; and Asp248 Oδ2. There are two additional oxygen ligands that are provided by water molecules.
Another signature of the hemidirected geometry is shorter than average metal-ligand distances for ligands opposite of the active lone pair, and longer than average distances for ligands adjacent to the lone pair.47 The Pb-O coordination bond lengths for Pb(1) and Pb(2) are summarized in Table 1. Pb(1) has a holodirected geometry characterized by uniform distribution of ligands in the coordination sphere. As a result, the Pb-O distances are fairly uniform and do not deviate significantly from the average of 2.7±0.4 Å, which was reported in the Protein Data Bank analysis by Kirberger et al.46 In the Pb(2) coordination sphere, we detected <2.4 Å distances for two ligands that are directly opposite to the active lone pair, Asp246 (Oδ2) and Asp248 (Oδ2). In contrast, the two ligands that are flanking the “void” in the coordination sphere, D187 Oδ1 and H2O (3), have coordination bond lengths of 3.4-3.5 Å (see Table 1).
Table 1.
Pb-O Distances in the C2α·Pb2 structure.
| Pb(1), CN=9 Holodirected | Bond length (Å) | Pb(2), CN=8 Hemidirected | Bond length (Å) |
|---|---|---|---|
| D187 (Oδ1) | 2.69 | D187 (Oδ1) | 3.40b |
| D187 (Oδ2) | 2.69 | D246 (Oδ2) | 2.38c |
| D193 (Oδ2) | 2.57 | D248 (Oδ1) | 2.80 |
| D246 (Oδ1) | 2.53 | D248 (Oδ2) | 2.34 |
| D246 (Oδ2) | 2.61 | D254 (Oδ1) | 2.59 |
| W247 (O) | 2.70 | D254 (Oδ2) | 2.55 |
| D248 (Oδ1) | 2.74 | H2O (3) | 3.47 |
| H2O (1)a | 3.28 | H2O (4) | 2.61 |
| H2O (2) | 2.98 | ||
|
| |||
| Average distance | 2.75±0.24 | Average distance | 2.77±0.44 |
Water molecule labels are shown in Figure 4C.
Distances that are longer than average are shown in boldface and underlined.
Distances that are shorter than average are shown in boldface.
In summary, our structural work shows that the apo form of C2α is a rigid scaffold with flexible loops. The metal coordination site that is formed mostly by CBL1 and CBL3 is not pre-organized for metal binding. In contrast to Ca(2), Pb(2) adopts a hemidirected coordination geometry, in which the oxygen-donating ligands occupy a single coordination hemisphere.
Pb(1) reduces the affinity of C2α to calcium ions
Having determined the coordination geometries of Pb(1) and Pb(2), we tested the feasibility of the formation of mixed Pb(1)/Ca(2) C2α species. It has been suggested that binding of Ca2+ to site (2) when Pb2+ is present at high-affinity site (1) results in full activation of PKCα.10 Taking advantage of the differential Pb2+-binding affinities of C2α, we prepared the C2α·Pb complex. The progress of Ca2+ binding to C2α·Pb was monitored by 15N-1H HSQC NMR experiments.
Figure 5 shows the expansion of the NMR spectra illustrating the response of Asp248 to increasing concentration of Ca2+. The Ca2+ concentration ranged from zero (blue) to 20 mM (red). The maximum Ca2+ excess relative to C2α and Pb2+ was 125-fold. The titration behavior can be broken down into four steps that are summarized in Scheme 1 of Figure 5.
Figure 5.
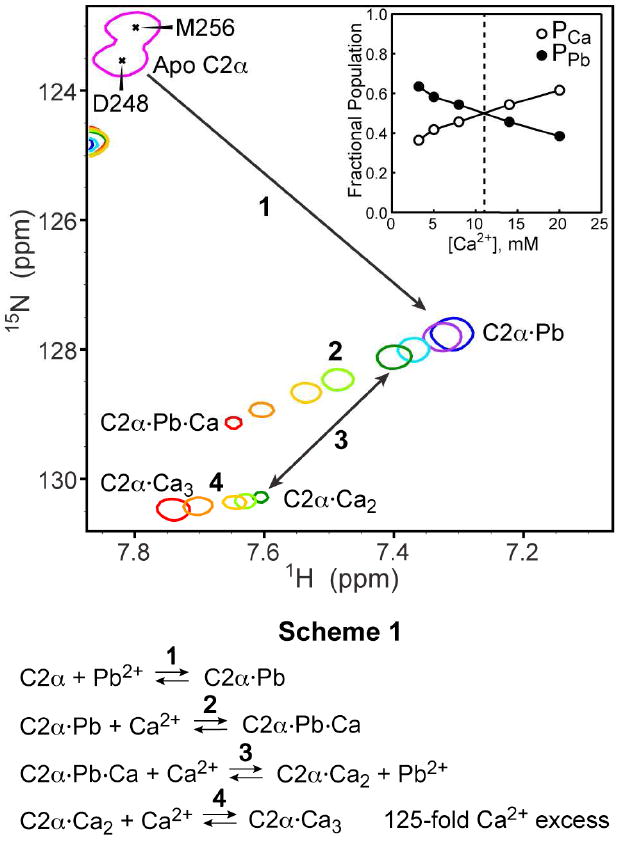
Expansion of the 15N-1H HSQC spectra showing the response of residue Asp248 in the preformed C2α·Pb complex to the addition of Ca2+ ions. Concentration of Ca2+ ions ranged from 0 mM (blue) to 20 mM (red). Inset: fractional populations of Ca-only (PCa) and Pb-containing (PPb) protein species as a function of the total Ca2+ concentration.
Step 1 corresponds to the formation of the high affinity C2α·Pb complex by adding a stoichiometric amount of Pb2+ to a 160 μM solution of C2α. Ca2+ binds to the vacant site (2) of the C2α·Pb in Step 2 to form the C2α·Pb·Ca complex. C2α·Pb and C2α·Pb·Ca are in fast exchange on the chemical shift timescale, as manifested by the smooth trajectory of the exchange-averaged cross-peak in response to increasing Ca2+ concentration. At a total Ca2+ concentration of 3.2 mM, we observed an appearance of another cross-peak whose position is coincident with that of Asp248 in the C2α·Ca2 complex (see Figure 1). The two cross-peaks are shown in dark green and are connected with a double-headed arrow in Figure 5. This process, referred to as Step 3 in our reaction scheme, is the displacement of Pb2+ from the high-affinity site (1) by Ca2+. The exchange of Pb2+ for Ca2+ in the C2α·Pb·Ca complex is a slow process on the chemical shift timescale. This means that the sum of the “on” and “off” kinetic rate constants in Step 3 is much smaller than the Asp248 chemical shift difference in the C2α·Pb·Ca and C2α·Ca2 complexes.48 At Ca2+ concentrations above 3.2 mM, we observed the titration of the newly formed C2α·Ca2 species in what is defined as Step 4. We interpret this process as the binding of a third Ca2+ ion to the C2α·Ca2 with the formation of C2α·Ca3. The formation of the C2α·Ca3 species is supported by the observation that the cross-peaks affected most by high concentrations of Ca2+ belong to Asp248, Asp254, Thr251, and Arg252. These residues coordinate the third Ca2+ ion in the two crystal structures of C2α that were obtained under conditions of large Ca2+ excess with respect to the protein.49,50
Increasing Ca2+ concentration also results in the redistribution of the cross-peak intensities between the Pb2+-bound and Ca2+-only C2α complexes. This is illustrated in the inset of Figure 5, which shows fractional populations of Pb2+-bound (C2α·Pb·Ca and C2α·Pb) and Ca2+-only (C2α·Ca2 and C2α·Ca3) protein species. The populations were calculated using the cross-peak volumes determined in Steps 2 and 4. Equal populations are reached around 11 mM total concentration of Ca2+. Using the results of the titration experiments, we estimated the affinity of Ca2+ to the preformed C2α·Pb complex as described in Section 4 of the Supporting Information. The apparent dissociation constant is 13±1 mM, which represents a ~50-fold reduction in Ca2+-binding affinity compared to the apo C2α. Formation of the mixed C2α·Pb·Ca species as a result of Pb exposure in vivo cannot be completely excluded. However, given the metal-binding affinities that we determined in this work, a more plausible scenario would involve the formation of either C2α·Pb2 complex or fully Ca2+-bound species if Ca2+ were in large excess compared to Pb2+.
Pb2+ binding drives membrane association of C2α. Pb2+ successfully competes with Ca2+ for C2α binding sites in the presence of lipid membranes
The next step was to understand the influence of Pb2+ on the interactions of C2α with PtdSer-containing membranes. Based on the functional and structural data obtained on multiple C2 domains, it has been hypothesized that one of the functions of metal ions is to serve as a “bridge” between the protein and lipid head-group by forming Ca-O coordination bond(s).14 To understand if the unusual coordination geometry of Pb(2) has any effect on the C2α function, we tested the membrane-binding properties of C2α in the presence of Pb2+ using FRET spectroscopy, ultracentrifugation binding assays, and SPR.
FRET experiments were carried out using LUVs comprising POPC/dansyl-PE and POPC/POPS/dansyl-PE lipids with mole percentages of 93:7 and 60:33:7, respectively. The normalized change in protein-to-membrane FRET as a function of the total metal concentration is shown in Figure 6A for Ca2+ and Pb2+. No membrane binding was observed in the absence of the PtdSer component, irrespective of the nature of the divalent metal ion. In the presence of 33% PtdSer, both Ca2+ and Pb2+ were capable of driving the C2α binding to the lipid membrane.
Figure 6.
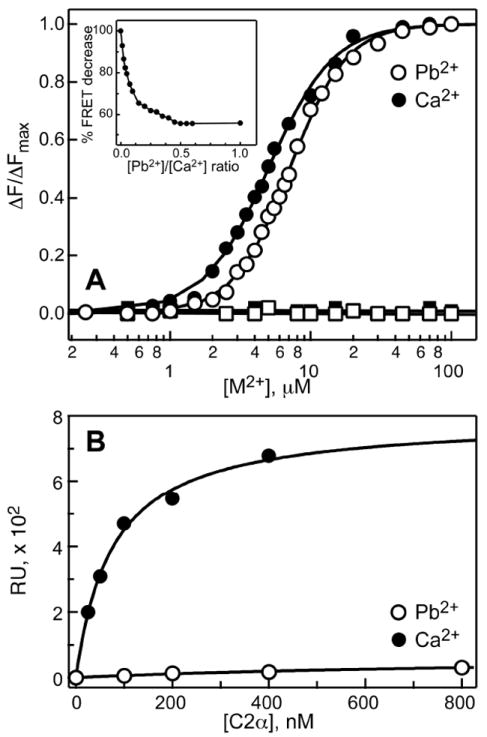
(A) Normalized protein-to-membrane FRET as a function of Pb2+ (empty circles) or Ca2+ (filled circles) concentration. No binding is observed in the absence of PtdSer membrane component (filled and open squares). Inset: Pb2+/Ca2+ competition experiment showing the displacement of Ca2+ with Pb2+. (B) C2α membrane-binding curves detected by SPR in the presence of 100 μM Ca2+ (filled circles) and Pb2+ (open circles). Response units (RUs) are plotted against total protein concentration.
The data were fitted with Eq. (2) to obtain the following parameters: HPb=2.17±0.04, [Pb2+]1/2=7.08±0.05 μM, HCa=1.88±0.05, and [Ca2+]1/2=5.00±0.07 μM. The Ca2+ values are in good agreement with the previous studies of C2α.15 From this set of experiments, we concluded that Pb2+ is almost as effective as Ca2+ in promoting the association of C2α with PtdSer-containing membranes.
To determine if Pb2+ can successfully compete with Ca2+ for C2α metal-binding sites in the presence of lipid membranes, we carried out the Pb2+/Ca2+ competition experiment. C2α was saturated with Ca2+ at 100 μM in the presence of LUVs, and Pb2+ ions were subsequently added into the mixture. The change in FRET intensity was monitored as a function of Pb2+ concentration. The maximum attainable protein-to-membrane FRET intensity in the presence of Pb2+ is ~56% of that in the presence of Ca2+. The results of the competition experiment are shown in the inset of Figure 6A. The FRET intensity steadily decreases with the increase of Pb2+ concentration. A plateau region indicative of complete displacement of Ca2+ by Pb2+ is reached at the Pb2+/Ca2+ ratio of 0.5. Given the comparable membrane affinities of Ca2+- and Pb2+-bound C2α, the displacement of Ca2+ by Pb2+ most likely occurs in the membrane-unbound form of the protein. Pb2+-bound C2α can subsequently undergo membrane insertion and eventually replaces the Ca2+-containing membrane-bound protein species at high Pb2+ concentrations.
Pb2+ competes with C2α for PtdSer binding sites
In addition to FRET, we used SPR experiments to test the effect of Pb2+ on the C2α membrane-binding properties. The experiments were carried out at 100 μM concentration of metal ions, while the C2α concentration varied from 0 to 800 nM. The active and control surfaces of the sensor chip were coated with POPC/POPS and POPC, respectively. The SPR binding curves are presented in Figure 6B. In the presence of Ca2+, C2α showed the expected membrane-binding behavior with an apparent Kd of 76±9 nM, which is in good agreement with the previously reported Kd value of 71 nM.21 Surprisingly, hardly any signal was observed for the C2α in the presence of Pb2+. Two possible explanations for this behavior are either a complete loss of PtdSer selectivity resulting in the equivalent signal from the active and control surfaces, or the absence of C2α binding to the vesicle-coated L1 chip. Either explanation would contradict the results of our FRET experiments, in which we observed both Pb2+-driven binding and PtdSer selectivity.
We used ultracentrifugation-binding assays to gain insight into the apparent discrepancy between the SPR and FRET data. In these assays, apo C2α, LUVs, and metal ions in varying concentrations are mixed, incubated to allow for the membrane binding to occur, and subjected to ultracentrifugation to separate the membrane and aqueous fractions. The advantage of this method compared to FRET and SPR is that it enables direct detection of free protein in the supernatant after pelleting the membrane fraction by ultracentrifugation.
Figure 7A shows a plot of fractional population of the membrane-bound C2α as a function of the metal-to-protein ratio. As expected, addition of Ca2+ ions resulted in full membrane binding of C2α. Similarly, C2α associated with membranes in response to Pb2+, reaching full binding at the metal-to-protein ratio of ~30. However, the Pb2+ binding curve showed a noticeable lag at low Pb2+ concentrations. In addition, at Pb2+ concentrations exceeding 0.5 mM (metal-to-protein ratio >100), we observed dissociation of C2α from the membrane. To rule out the technique-specific factors, we carried out a protein-to-membrane FRET experiment under conditions identical to those used in the ultracentrifugation assays. It is evident from Figure 7B that FRET data are in complete agreement with the ultracentrifugation results of Figure 7A. To determine if the membrane-dissociation behavior of C2α at high Pb2+ concentration can be “rescued” by adding more lipids into the system, we increased the concentration of LUVs 7-fold and carried out an ultracentrifugation binding assay. The “high-lipid” binding curve of Figure 7C demonstrates that full membrane binding of C2α can be achieved by increasing the lipid-to-Pb2+ ratio.
Figure 7.
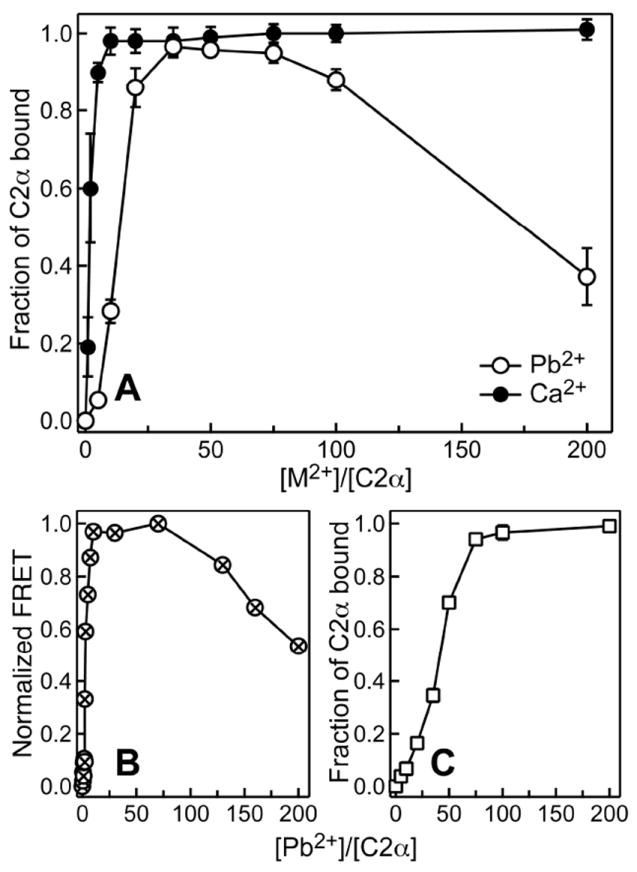
Metal-dependent membrane binding of C2α. (A) Fractional population of membrane-bound C2α obtained in ultracentrifugation binding assays as a function of Ca2+-to-protein (filled circles) and Pb2+-to-protein (open circles) ratios. (B) Pb2+-dependent membrane binding monitored by protein-to-membrane FRET under conditions identical to those used in (A). (C) Ultracentrifugation binding experiments with a 7-fold higher lipid concentration but otherwise identical conditions to those of (A).
In summary, at low concentrations of Pb2+ and high lipid-to-metal ratios, Pb2+-bound C2α readily associated with PtdSer-containing membranes. In contrast, at high concentrations of Pb2+ the fractional population of membrane-bound C2α decreased more than two-fold. No Pb2+-driven C2α binding was observed in the SPR experiments, where the lipid-coated sensor chip was equilibrated with Pb2+-containing buffer prior to the start of the measurements.
To test the integrity of PtdSer-containing LUVs in the presence of Pb2+, we collected the cryoelectron microscopy (cryoEM) images of LUVs under conditions identical to those of the ultracentrifugation binding assays. The concentration of Pb2+ of 1 mM was the highest we ever used in our experiments. The cryoEM images of LUVs in the presence of Pb2+ and Ca2+ are shown in Figure S4 of the Supporting Information. Although high concentrations of Pb2+ promote aggregation of vesicles, the integrity of the lipid bilayers is preserved. The most plausible explanation for the reduced membrane binding of C2α at high Pb2+-to-lipid ratios is then a direct competition of Pb2+ with C2α for the PtdSer binding sites.
To directly test association of Pb2+ with PtdSer-containing LUVs, we carried out ultracentrifugation binding assays in the absence of C2α. The amount of Pb2 in the membrane and supernatant fractions was quantified using inductively coupled plasma (ICP) measurements. Indeed, increasing amounts of Pb2+ were detected in the membrane fraction upon increasing the total Pb2+ concentration. Based on the ICP data, we constructed the binding curve shown in Figure S5 for the association of Pb2+ with the PtdSer component of the LUVs. Fitting the data with a single-site binding model produced a Kd of 119 ± 12 μM. The results of the ICP experiments confirmed that Pb2+ could indeed compete for PtdSer binding with C2α, and hence modulate the protein-membrane interactions in a concentration-dependent manner. The ability of Pb2+ ions to simultaneously associate with both C2α and PtdSer-containing membranes gives rise to complex binding equilibria, where the identity of the dominant species depends on the relative and total concentration of the protein, metal ion, and LUVs.
DISCUSSION
The central objective of this work was to understand the effect of Pb2+ ions on the structure and membrane-binding properties of C2α, a calcium-dependent membrane-binding domain of PKCα. We used NMR, ITC, X-ray crystallography, SPR, and FRET techniques to gain insight into the molecular mechanism of the interactions between C2α, Pb2+, and PtdSer-containing lipid membranes. The picture that emerged from our studies is that of Pb2+ acting as a concentration-dependent modulator of C2α-membrane interactions.
High-affinity binding of Pb2+ to C2α and the Pb2+/Ca2+ competition
Using NMR spectroscopy, we established that there are two Pb2+-binding sites on the C2α domain (Figure 1). There is a 2000-fold difference between the Pb2+ affinities of the two sites, with Kd1Pb of 67±1 nM and Kd2Pb of 129±4 μM that were measured by ITC and NMR, respectively. In contrast, the two sites have comparable affinities to Ca2+ with an estimated KdCa of 270 μM. Our finding that C2α binds Pb2+ tighter than Ca2+ is in agreement with the results of the divalent metal ion-Tb3+ competition studies of synaptotagmin I (a protein with two C2 domains)51 and full-length PKC.52
We determined that Pb2+ is almost as effective as Ca2+ in its ability to promote the association of C2α with PtdSer-containing membranes. Using protein-to-membrane FRET experiments of Figure 6A, we determined the concentrations at which the binding is half-maximal: [Pb2+]1/2=7.08±0.05 μM and [Ca2+]1/2=5.00±0.07 μM. Another C2-domain containing protein, synaptotagmin I, was shown to bind lipid membranes in the presence of Pb2+.51 In that study, Pb2+ was found to be ~1000-fold more potent than Ca2+ in driving the protein-membrane association. These differences can be attributed to the specifics of individual C2 domains, or the presence of two of them in synaptotagmin I, or both.
Based on the relative binding affinities of Pb2+ and Ca2+, it is likely that site (1) of C2α gets populated at low concentrations of Pb2+. The formation of the mixed Pb2+(1)/Ca2+(2)-bound species was suggested as a plausible explanation of why high concentrations of Ca2+ reverse the inhibitory behavior of Pb2+ in the full-length PKCα.10 To determine if the formation of the C2α·Pb·Ca species is feasible, we added Ca2+ ions to the preformed C2α·Pb complex. The analysis of the NMR data of Figure 5 revealed that the presence of Pb2+ at site (1) reduces the affinity of Ca2+ to site (2) ~50-fold. We conclude that given the 13 mM affinity of Ca2+ to site (2) in the presence of pre-bound Pb2+, the C2α·Pb·Ca complex is unlikely to be the most dominant species at physiological concentrations of Ca2+.
Pb2+ can successfully compete with Ca2+ for the C2α metal-binding sites in the presence of PtdSer-containing membranes. We demonstrated this by titrating Pb2+ into the pre-formed ternary complex of C2α, Ca2+, and PtdSer-containing LUVs (inset of Figure 6A). The displacement of Ca2+ by Pb2+ is evident from the change in FRET intensity, which reaches a plateau at ~[Pb2+]/[Ca2+] ratio of 0.5. This behavior is consistent with Pb2+ displacing Ca2+ from site (1) in the membrane-free form, and then re-routing the membrane-binding process through the Pb2+-driven path.
The activation sequence of PKCα requires an inter-domain rearrangement, which is initiated by the divalent metal ion binding. This rearrangement occurs concomitantly with the two membrane-binding events that involve both C2 and C1 domains of PKCα. Our data show that the affinity of C2α to PtdSer-containing membranes is comparable in the presence of Ca2+ and Pb2+. It appears that the partial activation of full-length PKCα at picomolar-to-nanomolar concentrations of Pb2+ can be due to the domain rearrangement triggered by Pb2+ binding to a single high-affinity site on the C2 domain.
Effect of Pb2+ on the structure of C2α
To evaluate the effect of Pb2+ binding on the structure of C2α, we used NMR chemical shift perturbation analysis combined with site-specific resonance assignments of the backbone 1H-15N groups. In addition, we determined the crystal structures of apo and Pb2+-bound C2α.
Chemical shift perturbation analysis enabled us to identify the high- (67 nM) and low-affinity (129 μM) Pb2+-binding sites. Binding of Pb2+ to the high-affinity site (1) results in the perturbation of the CBL1 and CBL3 regions (Figures 3A and B). In the crystal structure of the C2α·Pb2 complex, the coordination geometry of Pb(1) is holodirected, with uniform distribution of the nine oxygen-donating ligands in the coordination sphere. Binding of Pb2+ to the low-affinity site (2) affects only the CBL3 region. In contrast to Pb(1), Pb(2) adopts a hemidirected coordination geometry, in which eight ligands occupy only one coordination hemisphere. In this geometry, the stereochemically active 6s2 pair of Pb2+ that has a partial p-character occupies the “empty” hemisphere.
The differences between the Ca2+- and Pb2+-bound C2α structures are localized to the metal-coordination sites. In addition to the different coordination geometries, the positions of Ca(2) and Pb(2) in the C2α-metal complexes do not coincide. As a result, Pb(2) does not form a coordination bond with the carbonyl oxygen of Met186 like Ca(2) does, but instead acquires two additional bonds, one with the second carboxylate oxygen of Asp254 and the other with the water molecule. The sidechain of Asp254 may promote the hemidirected coordination of Pb(2) by serving as a bidentate ligand. Inspection of the metal-binding site in the apo C2α structure reveals that the sidechains of aspartate residues are not pre-organized for the metal binding (see Figure 4B). Thus, the metal coordination event involves a rotameric transition of the coordinating sidechains. Binding of the divalent metal ions to the CBL regions minimizes the electrostatic repulsion between the negatively charged aspartate sidechains, and results in a significant stabilization of the protein structure – the melting temperatures of the apo and Ca2+-bound C2α are 50 and 80 °C, respectively.43
The crystal structures of the apo, Ca2+-bound, and Pb2+-bound C2α are essentially superimposable, indicating that metal binding does not significantly influence the average conformation of the protein backbone. The same conclusion was drawn for the only other pair of apo and Ca2+-bound C2 structures, the C2A domain of synaptotagmin I.53 Given the negligible changes in the backbone conformation, the large differences in chemical shifts between the apo and metal-bound forms reflect the changes in the electrostatic environment and dynamics of the loops. In fact, the CBL1 and CBL3 regions in the crystal structure of the apo form have elevated temperature factors. We also detected the presence of microsecond-to-millisecond timescale motions in the CBL1 and CBL3 regions of apo C2α, as manifested in the elevated 15N transverse relaxation rate constants. These motions are attenuated upon addition of Ca2+ (K. A. M. and T. I. I., unpublished data).
Hemidirected coordination geometry of Pb(2) and its role in the C2α-membrane association
In view of our findings that the coordination sphere of Pb(2) in C2α is hemidirected, we wanted to assess the prevalence of this coordination geometry among protein-bound oxygen-coordinated Pb2+ ions. Recently, Kirberger and Yang conducted a thorough analysis of the Protein Data Bank (http://www.rcsb.org/) to compare the structural properties of Ca2+ and Pb2+ binding sites in proteins.46 The analysis provided invaluable information on the Pb-ligand bond lengths and coordination preferences. The holodirected and hemidirected Pb2+ coordination geometries were not explicitly addressed in that analysis.
Our search of PDB for Pb2+-biomacromolecule complexes produced a total of 36 structures. Of those 36, we retained 23 by eliminating the nucleic acid and duplicate structures, as well as those where oxygen was not present in the coordination sphere of Pb2+ sites. The PDB identifiers for the selected 23 structures are given in Section 7 of the Supporting Information. We further refined our criteria by requiring that (i) all ligands are oxygens and (ii) Pb2+ replaces the metal cofactor (i.e. some other divalent metal ion) rather then being non-specifically adsorbed on the protein surface. The 14 Pb2+ sites that met those criteria and their corresponding PDB identifiers are listed in Table S2. The Pb-O distances ranged from 2.0 Å to 3.5 Å, as evident from the histogram presented in Figure S6, with the mean of 2.63±0.43 Å. For the analysis of the coordination geometry (vide infra) we used the 3.5 Å cutoff, as prescribed by Kirberger and Yang.46
We found that only 5 stuctures from the original group of 23: 2G0A (mouse pyrimidine 5’-nucleotidase type 1, P5N-1),54 2XAL (inositol 1,3,4,5,6-pentakisphosphate 2-kinase),55 1E9N (human apurinic/apyrimidinic endonuclease),56 1QR7 (phenylalanine-regulated 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase, DAHPS),57 and 2V01 (vertebrate calmodulin),29 contained hemidirected Pb2+ that replaced the metal cofactor and had at least one oxygen in the coordination sphere. The coordination numbers, ligand identities, and natural metal cofactor for the unique hemidirected Pb2+ sites are summarized in Table 2. For two out of five proteins listed in Table 2, DAHPS (1QR7) and P5N-1 (2G0A), the inhibitory effect of Pb2+ on enzyme activity was linked to the coordination geometry of Pb2+. In both enzymes, Pb2+ binding to the active site compromises its geometry and proper positioning of the substrate(s).
Table 2.
Unique Pb2+ sites with hemidirected coordination geometry.
| PDB ID | Pb SNa | CNb | Coordination sphere atoms | Displaced cofactor |
|---|---|---|---|---|
| 1E9N | 4339 | 7 | 7O | Mg(II) |
| 1E9N | 4340 | 5 | 3O and 2N | Mg(II) |
| 1QR7 | 10296 | 5 | 3O, 1N and 1S | Mg(II) |
| 2G0A | 4655 | 7 | 7O | Mg(II) |
| 2XAL | 6758 | 6 | 4O | Mn(II) |
| 2XAL | 6759 | 5 | 6O | Mn(II) |
| 2V01 | 1117 | 7 | 7O | Ca(II) |
| 2V01 | 1118 | 7 | 7O | Ca(II) |
| 2V01 | 1120 | 6 | 6O | Ca(II) |
|
| ||||
| TOTAL number of unique all-oxygen Pb2+ hemidirected sites: 7 | ||||
SN is the serial number of Pb in the PDB file.
CN is the coordination number of Pb2+ ion.
7 out of 9 Pb2+ sites listed in Table 2 have an all-oxygen coordination sphere. Comparing the results of our PDB analysis presented in Tables 2 and S2, we conclude that half, 7 out of 14 total all-oxygen coordination sites that Pb2+ occupies in lieu of the metal cofactor, have hemidirected coordination geometry. These findings are in general agreement with the conclusions of the Cambridge Structural Database (CSD) analysis conducted by Shimoni-Livny et al.58 It was found that for the divalent Pb2+ complexed with small molecules, hemidirected and holodirected geometries are preferred when the coordination number is <5 and 9-10, respectively. For the coordination numbers between 6 and 8, Pb2+ can adopt either coordination geometry. The nature of Pb2+ ligand is also a factor, with hard ligands such as oxygen and nitrogen favoring the hemidirected geometry. The structure of the C2α-Pb2+ complex that was determined in our study provides a remarkable example of how both holodirected and hemidirected Pb2+ ions, both oxygen-coordinated with similar coordination numbers, can coexist within one macromolecular structure. A recent ab initio study of Pb2+ substitution in the C2B domain of synaptotagmin I59 predicts that (i) the displacement of Ca2+ with Pb2+ would result in the rearrangement of the coordination sphere with Pb2+ adopting a hemidirected coordination; and (ii) a single site replacement of Ca2+ with Pb2+ would be energetically favorable, implying higher binding affinities of Pb2+ to C2B. It appears that for the calcium-binding C2 domains in general, one would expect a combination of holodirected and hemidirected Pb2+ geometries, depending on the identity and properties of the oxygen-donating amino acid sidechains.
It has been hypothesized that divalent metal ions play three roles in the C2 domains of conventional PKC isoenzymes: (i) modulation of the electrostatic potential to facilitate C2 insertion into lipid bilayers, (ii) serving as a “bridge” between the protein and PtdSer head-group, and (iii) modulation of the intra- and inter-domain interactions in PKC.14 The hypothesis that Ca2+ is involved in forming a protein-membrane “bridge” by coordinating PtdSer oxygens was formulated based on the crystal structures of C2α in complex with Ca2+ and short-chain PtdSer molecules.41,49,50 As shown in Figure 4, in the crystal structures the phosphoryl oxygens of the lipid provide an axial ligand to the Ca(1) site, while Ca(2) site coordinates water41 or phosphate group of the crystallization buffer.49 Mutations of Ca(1)- and Ca(2)-coordinating aspartate residues to asparagines in the C2α domain resulted in diminished binding of the full-length PKCα to lipid vesicles, with site (1) having a more pronounced effect.60 Thus, if we assume that both metal sites are filled prior to membrane-binding and both of them form coordination bonds with oxygens of PtdSer headgroups, then Pb(2) is likely to undergo hemidirected to holodirected conversion. The activation energy for such a conversion was estimated to be 8-12 kcal/mol for the tetracoordinated Pb2+,58 but would be expected to be much lower for coordination numbers of 6-8 that could be accommodated by either geometry.
Association of Pb2+ with PtdSer-containing membranes
Two types of membrane-binding behavior of C2α in the presence of Pb2+ are illustrated in Figures 6A and 7A. Protein-to-membrane FRET experiments of Figure 6A were conducted under “dilute” conditions – 0.5 μM C2α and 150 μM total lipid – and showed full C2α membrane binding at 100 μM Pb2+. Ultracentrifugation experiments of Figure 7A were conducted with a 10-fold proportional increase in the concentration of all components – 5 μM C2α and 1.5 mM total lipid. Under those conditions, we observed a lag period at low Pb2+ concentrations, and partial dissociation of C2α from the PtdSer-containing membranes at high Pb2+ concentrations. This result is not technique-dependent. Both features were reproduced in the FRET experiment of Figure 7B, in which the concentration of all components matched exactly the conditions of the ultracentrifugation experiments. The pronounced dependence of the membrane association of C2α on the total concentration of all reactants in the system implies the presence of competing binding processes that involve Pb2+ ions, PtdSer-containing membranes, and C2α.
The partial dissociation of C2α from membranes at Pb2+ concentrations exceeding 0.5 mM could in principle be caused by the effect of Pb2+ on the LUV properties. The majority of research efforts to understand the interactions of divalent metal ions with PtdSer-containing membranes have so far focused on Ca2+. It is well-documented that Ca2+ promotes the fusion of both pure PtdSer vesicles61 and multilamellar structures,62 and that Ca2+-PtdSer interactions can be weakened substantially by incorporating neutral lipids into the system.63 In addition to fusion, Ca2+ induces lateral segregation of lipid components in the membrane, as monitored by the auto-quenching of fluorescent lipids.64 Similar to Ca2+, Pb2+ promotes lateral segregation in PtdSer vesicles.65 Although these findings indicate that Pb2+ could potentially change the physical properties of the PtdSer-containing LUVs in our experiments, the extent of segregation decreases significantly with decreasing the molar fraction of PtdSer to <~ 0.3 and metal-to-lipid ratio to <~1-2.64,65 Because our experiments were conducted with a 33 % molar fraction of PtdSer and Pb2+-to-lipid ratios less than 0.7, Pb2+-induced segregation of lipid components is not likely to be a major factor that influences the affinity of C2α to the membranes.
A comparison of the cryoEM images of 100 nm LUVs in the presence of Ca2+ and Pb2+ (Figure S4) revealed that 1 mM Pb2+ but not Ca2+ promotes the aggregation of vesicles. We hypothesized that the vesicle aggregation occurs because Pb2+ interacts with the PtdSer component of the LUVs. The ICP analysis of the membrane-bound and free Pb2+ concentrations confirmed that Pb2+ associates with LUVs, with an apparent Kd of 119±12 μM. We conclude that in the low mM range, Pb2+ can successfully compete with C2α for the PtdSer binding sites on the membrane. The 10-fold reduction in the concentration of all components in the FRET experiments of Figure 6A results in the smaller fractional population of membrane-bound Pb2+. As a result, there is sufficient free Pb2+ to enable full membrane binding of C2α.
Implications for Pb2+-dependent activity of PKCα
Given the complexity of the binding equilibria that involve Pb2+, PtdSer-containing lipid membranes, and C2α, how do we extrapolate our findings to explain the modulation of PKCα activity by Pb2+? Pb2+ has both activating and inhibitory effects on full-length PKCα.10 In vitro studies of Pb2+-dependent activity of recombinant PKCα showed that the enzyme is activated in the range of Pb2+ concentrations from 0.1 to 10 nM, reaching 40% of the maximum Ca2+-dependent activity. The stimulatory effect of Pb2+ was attributed to its interactions with the high-affinity metal-binding site residing on the C2 domain. The inhibitory effect of Pb2+ at concentrations exceeding 10 nM was attributed to two factors: binding of Pb2+ to the second site on the C2 domain, and the interaction of Pb2+ with the catalytic domain at micromolar concentrations of metal ion. The work was carried out with 0.13–0.25 nM PKCα in the presence of 100% PtdSer.10
The blood level of 10 μg/dL translates into 0.48 μM concentration of Pb2+. The concentration of bioavailable Pb2+ is much lower and is on the order of picomolar to nanomolar.47 Based on the Pb2+ affinities to C2α determined in this work, we expect the high-affinity metal-binding site (1) of C2α to be at least partially occupied in vivo. We did not detect any substantial inhibitory effect of Pb(2) on the interactions of C2α with membranes – Pb2+ is almost as effective as Ca2+ in driving the membrane association process (Figure 6A). We conclude that the C2α-mediated activation of PKCα by Pb2+ is likely to occur through a change in the inter-domain orientation in the enzyme, rather than through a direct modulation of metal-dependent membrane binding.
The association of Pb2+ with PtdSer-containing membranes can in part be responsible for the inbitory effect of Pb2+. At low concentrations of Pb2+, we observed a lag period in the C2α membrane-binding curve of Figure 7A. The lag is likely caused by the sequestration of Pb2+ by the PtdSer-containing membranes (inset of Figure S5). High concentration of PtdSer facilitates the formation of the Pb2+-PtdSer complex resulting in a more pronounced lag period as shown in Figure 7C. Based on these considerations, it is feasible that Pb2+ can interfere with the membrane binding of C2α (and hence PKC) even at low concentrations. This process would be facilitated by high local concentration of acidic lipids and phosphoinositides.
CONCLUSIONS
We investigated the effect of Pb2+ on the structure and membrane-binding properties of C2α, a Ca2+-dependent membrane-binding domain of PKCα. We found that Pb2+ binds C2α with higher affinity than Ca2+, and can displace Ca2+ from the C2α in the presence of PtdSer-containing membranes. A comparison of the apo and Pb2+-bound C2α crystal structures revealed that the rotation of the metal-coordinating sidechains is required for the metal binding to occur. The two Pb2+ ions adopt different coordination geometries – holodirected and hemidirected. We demonstrated that Pb2+ could associate with PtdSer-containing membranes and thereby compete with C2α for the PtdSer binding sites even at low concentrations. This process can contribute to the inbitory effect of Pb2+. The activation of PKCα by low concentrations of Pb2+ likely occurs through a change in the inter-domain orientation, rather than through direct modulation of metal-dependent membrane binding.
Supplementary Material
Acknowledgments
The authors thank Dr. Robert Taylor (Trace Element Research Laboratory, College of Veterinary Medicine, Texas A&M University) for conducting the ICP analysis; Samir Moussa (Department of Biochemistry and Biophysics, Texas A&M University) and Dr. Zhiping Luo (Microscopy and Imaging Center, Texas A&M University) for the cryoelectron microscopy measurements; and Mukundan Ragavan for collecting the NMR data of Figure 5. This work was supported by the startup funds from Texas A&M University and Ralph E. Powe junior faculty enhancement award from Oak Ridge Associated Universities (T.I.I.); NIH GM68849 (W.C.); and NIH Training Grant 2-T32GM008523-13 (K.A.M.).
Footnotes
Supporting Information Available Over-expression and purification protocol for C2α; description of the X-ray diffraction data collection and structure determination for apo and Pb2+-bound C2α; tabulated statistics of the crystallographic analysis; estimation of the KdCa; estimation of the binding affinity of Ca2+ to the preformed C2α·Pb complex; cryoelectron microscopy images of LUV suspensions; ICP measurements of membrane-bound Pb2+; and coordination preferences of protein-bound oxygen-coordinated Pb2+. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Center for Disease Control and Prevention. 2011 http://www.cdc.gov/nceh/lead/
- 2.Canfield RL, Henderson CR, Jr, Cory-Slechta DA, Cox C, Jusko TA, Lanphear BP. N Engl J Med. 2003;348:1517. doi: 10.1056/NEJMoa022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiodo LM, Jacobson SW, Jacobson JL. Neurotoxicol Teratol. 2004;26:359. doi: 10.1016/j.ntt.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 4.Abadin H, Ashizawa A, Stevens Y-W, Llados F, Diamond G, Sage G, Citra M, Quinones A, Bosch SJ, Swarts SG. U S Department of Health and Human Services, Agency for Toxic Substances and Disease Registry. 2007 [PubMed] [Google Scholar]
- 5.Godwin HA. Curr Opin Chem Biol. 2001;5:223. doi: 10.1016/s1367-5931(00)00194-0. [DOI] [PubMed] [Google Scholar]
- 6.Suszkiw JB. Neurotoxicology. 2004;25:599. doi: 10.1016/j.neuro.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 7.Rosse C, Linch M, Kermorgant S, Cameron AJ, Boeckeler K, Parker PJ. Nat Rev Mol Cell Biol. 2010;11:103. doi: 10.1038/nrm2847. [DOI] [PubMed] [Google Scholar]
- 8.Newton AC. Chem Rev. 2001;101:2353. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- 9.Steinberg SF. Physiol Rev. 2008;88:1341. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun XY, Tian XT, Tomsig JL, Suszkiw JB. Toxicol Appl Pharmacol. 1999;156:40. doi: 10.1006/taap.1999.8622. [DOI] [PubMed] [Google Scholar]
- 11.Markovac J, Goldstein GW. Nature. 1988;334:71. doi: 10.1038/334071a0. [DOI] [PubMed] [Google Scholar]
- 12.Tomsig JL, Suszkiw JB. J Neurochem. 1995;64:2667. doi: 10.1046/j.1471-4159.1995.64062667.x. [DOI] [PubMed] [Google Scholar]
- 13.Long GJ, Rosen JF, Schanne FA. J Biol Chem. 1994;269:834. [PubMed] [Google Scholar]
- 14.Cho W, Stahelin RV. Biochim Biophys Acta-Molecular and Cell Biology of Lipids. 2006;1761:838. doi: 10.1016/j.bbalip.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 15.Kohout SC, Corbalan-Garcia S, Torrecillas A, Gomez-Fernandez JC, Falke JJ. Biochemistry. 2002;41:11411. doi: 10.1021/bi026041k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stahelin RV, Rafter JD, Das S, Cho W. J Biol Chem. 2003;278:12452. doi: 10.1074/jbc.M212864200. [DOI] [PubMed] [Google Scholar]
- 17.Murray D, Honig B. Mol Cell. 2002;9:145. doi: 10.1016/s1097-2765(01)00426-9. [DOI] [PubMed] [Google Scholar]
- 18.Kohout SC, Corbalan-Garcia S, Gomez-Fernandez JC, Falke JJ. Biochemistry. 2003;42:1254. doi: 10.1021/bi026596f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lai CL, Landgraf KE, Voth GA, Falke JJ. J Mol Biol. 2010;402:301. doi: 10.1016/j.jmb.2010.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guerrero-Valero M, Marin-Vicente C, Gomez-Fernandez JC, Corbalan-Garcia S. J Mol Biol. 2007;371:608. doi: 10.1016/j.jmb.2007.05.086. [DOI] [PubMed] [Google Scholar]
- 21.Manna D, Bhardwaj N, Vora MS, Stahelin RV, Lu H, Cho W. J Biol Chem. 2008;283:26047. doi: 10.1074/jbc.M802617200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Warren MJ, Cooper JB, Wood SP, Shoolingin-Jordan PM. Trends Biochem Sci. 1998;23:217. doi: 10.1016/s0968-0004(98)01219-5. [DOI] [PubMed] [Google Scholar]
- 23.Erskine PT, Duke EM, Tickle IJ, Senior NM, Warren MJ, Cooper JB. Acta Crystallogr D Biol Crystallogr. 2000;56:421. doi: 10.1107/s0907444900000597. [DOI] [PubMed] [Google Scholar]
- 24.Gourlaouen C, Parisel O. Angew Chem Int Ed Engl. 2007;46:553. doi: 10.1002/anie.200603037. [DOI] [PubMed] [Google Scholar]
- 25.Jaffe EK, Martins J, Li J, Kervinen J, Dunbrack RL., Jr J Biol Chem. 2001;276:1531. doi: 10.1074/jbc.M007663200. [DOI] [PubMed] [Google Scholar]
- 26.Payne JC, ter Horst MA, Godwin HA. J Am Chem Soc. 1999;121:6850. [Google Scholar]
- 27.Magyar JS, Weng TC, Stern CM, Dye DF, Rous BW, Payne JC, Bridgewater BM, Mijovilovich A, Parkin G, Zaleski JM, Penner-Hahn JE, Godwin HA. J Am Chem Soc. 2005;127:9495. doi: 10.1021/ja0424530. [DOI] [PubMed] [Google Scholar]
- 28.Neupane KP, Pecoraro VL. Angew Chem Int Ed Engl. 2010;49:8177. doi: 10.1002/anie.201004429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kursula P, Majava V. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;63:653. doi: 10.1107/S1744309107034525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butt TR, Edavettal SC, Hall JP, Mattern MR. Protein Expr Purif. 2005;43:1. doi: 10.1016/j.pep.2005.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muhandiram DR, Kay LE. J Magn Reson Ser B. 1994;103:203. [Google Scholar]
- 32.Montelione GT, Lyons BA, Emerson SD, Tashiro M. J Am Chem Soc. 1992;114:10974. [Google Scholar]
- 33.Yamazaki T, Lee W, Arrowsmith CH, Muhandiram DR, Kay LE. J Am Chem Soc. 1994;116:11655. [Google Scholar]
- 34.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 35.Goddard TD, Kneller DG. Sparky 3. University of California; San Francisco: [Google Scholar]
- 36.Wilcox CS. In: Frontiers in Supramolecular Organic Chemistry and Photochemistry. Schneider HJ, Dürr H, editors. Wiley-VCH; Weinheim: 1991. p. 123. [Google Scholar]
- 37.Stahelin RV, Cho W. Biochemistry. 2001;40:4672. doi: 10.1021/bi0020325. [DOI] [PubMed] [Google Scholar]
- 38.Giorgione J, Newton AC. Methods in Molecular Biology. In: Newton AC, editor. Protein Kinase C protocols. Vol. 233. 2003. p. 105. [DOI] [PubMed] [Google Scholar]
- 39.Barenholz Y, Amselem S. Liposome Technology. 2. Vol. 1 CRC Press; Boca Raton, FL: 1993. [Google Scholar]
- 40.Nalefski EA, Falke JJ. In: Methods in Molecular Biology. Vogel HJ, editor. Vol. 172. Humana Press Inc.; 2002. p. 295. [DOI] [PubMed] [Google Scholar]
- 41.Verdaguer N, Corbalan-Garcia S, Ochoa WF, Fita I, Gomez-Fernandez JC. EMBO J. 1999;18:6329. doi: 10.1093/emboj/18.22.6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shao X, Davletov BA, Sutton RB, Sudhof TC, Rizo J. Science. 1996;273:248. doi: 10.1126/science.273.5272.248. [DOI] [PubMed] [Google Scholar]
- 43.Torrecillas A, Laynez J, Menendez M, Corbalan-Garcia S, Gomez-Fernandez JC. Biochemistry. 2004;43:11727. doi: 10.1021/bi0489659. [DOI] [PubMed] [Google Scholar]
- 44.Stahelin RV, Wang J, Blatner NR, Rafter JD, Murray D, Cho W. J Biol Chem. 2005;280:36452. doi: 10.1074/jbc.M506224200. [DOI] [PubMed] [Google Scholar]
- 45.Conesa-Zamora P, Lopez-Andreo MJ, Gomez-Fernandez JC, Corbalan-Garcia S. Biochemistry. 2001;40:13898. doi: 10.1021/bi011303o. [DOI] [PubMed] [Google Scholar]
- 46.Kirberger M, Yang JJ. J Inorg Biochem. 2008;102:1901. doi: 10.1016/j.jinorgbio.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Claudio ES, Godwin HA, Magyar JS. Prog Inorg Chem. 2003;51:1. [Google Scholar]
- 48.Palmer AG, Kroenke CD, Loria JP. Methods Enzymol. 2001;339:204. doi: 10.1016/s0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- 49.Ochoa WF, Corbalan-Garcia S, Eritja R, Rodriguez-Alfaro JA, Gomez-Fernandez JC, Fita I, Verdaguer N. J Mol Biol. 2002;320:277. doi: 10.1016/S0022-2836(02)00464-3. [DOI] [PubMed] [Google Scholar]
- 50.Guerrero-Valero M, Ferrer-Orta C, Querol-Audi J, Marin-Vicente C, Fita I, Gomez-Fernandez JC, Verdaguer N, Corbalan-Garcia S. Proc Natl Acad Sci USA. 2009;106:6603. doi: 10.1073/pnas.0813099106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bouton CM, Frelin LP, Forde CE, Arnold Godwin H, Pevsner J. J Neurochem. 2001;76:1724. doi: 10.1046/j.1471-4159.2001.00168.x. [DOI] [PubMed] [Google Scholar]
- 52.Walters JD, Johnson JD. J Biol Chem. 1990;265:4223. [PubMed] [Google Scholar]
- 53.Shao X, Fernandez I, Sudhof TC, Rizo J. Biochemistry. 1998;37:16106. doi: 10.1021/bi981789h. [DOI] [PubMed] [Google Scholar]
- 54.Bitto E, Bingman CA, Wesenberg GE, McCoy JG, Phillips GN., Jr J Biol Chem. 2006;281:20521. doi: 10.1074/jbc.M602000200. [DOI] [PubMed] [Google Scholar]
- 55.Gonzalez B, Banos-Sanz JI, Villate M, Brearley CA, Sanz-Aparicio J. Proc Natl Acad Sci USA. 2010;107:9608. doi: 10.1073/pnas.0912979107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beernink PT, Segelke BW, Hadi MZ, Erzberger JP, Wilson DM, 3rd, Rupp B. J Mol Biol. 2001;307:1023. doi: 10.1006/jmbi.2001.4529. [DOI] [PubMed] [Google Scholar]
- 57.Shumilin IA, Kretsinger RH, Bauerle RH. Structure. 1999;7:865. doi: 10.1016/s0969-2126(99)80109-9. [DOI] [PubMed] [Google Scholar]
- 58.Shimoni-Livny L, Carrell HL, Wagner T, Kaufman Katz A, Afshar C, Mitchell LW, Volin M, Jaffe EK, Glusker JP. Acta Crystallogr D Biol Crystallogr. 1998;54:438. doi: 10.1107/s0907444997010925. [DOI] [PubMed] [Google Scholar]
- 59.van Severen MC, Piquemal JP, Parisel O. J Phys Chem B. 2010;114:4005. doi: 10.1021/jp910131r. [DOI] [PubMed] [Google Scholar]
- 60.Medkova M, Cho W. J Biol Chem. 1998;273:17544. doi: 10.1074/jbc.273.28.17544. [DOI] [PubMed] [Google Scholar]
- 61.Wilschut J, Papahadjopoulos D. Nature. 1979;281:690. doi: 10.1038/281690a0. [DOI] [PubMed] [Google Scholar]
- 62.Feigenson GW. Biochemistry. 1986;25:5819. doi: 10.1021/bi00367a071. [DOI] [PubMed] [Google Scholar]
- 63.Coorssen JR, Rand RP. Biophys J. 1995;68:1009. doi: 10.1016/S0006-3495(95)80276-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hoekstra D. Biochemistry. 1982;21:1055. doi: 10.1021/bi00534a036. [DOI] [PubMed] [Google Scholar]
- 65.Adonaylo VN, Oteiza PI. Toxicology. 1999;132:19. doi: 10.1016/s0300-483x(98)00134-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.