

Abstract
There is growing recognition of an interaction between cerebrovascular disease and Alzheimer’s disease, but the mechanisms of this interaction remain poorly understood. While macroscopic stroke can clearly produce cognitive deficits and accelerate Alzheimer’s disease, the prevalence and implications of microvascular disease in Alzheimer’s disease pathogenesis has been harder to define. At present, white matter (WM) lesions, primarily defined as hyperintensities seen on T2-weighted magnetic resonance imaging (MRI), provide the best biomarker of cerebrovascular disease at the microvascular level. However, T2 hyperintensities in WM can also be caused by other mechanisms such as inflammation. Arterial spin labeled (ASL) perfusion MRI provides a noninvasive approach for quantifying cerebral blood flow (CBF). We explored CBF measurements with ASL in AD patients, mild cognitive impairment patients, and an age-matched control group to determine if CBF in gray matter or WM could be correlated with WM lesions, or to stratify patients by microvascular disease severity. In a retrospective sample, we were able to obtain credible measures of WM CBF using ASL MRI and observed trends suggesting that WM CBF may provide a useful biomarker of microvascular disease. Future prospective studies in larger cohorts with optimized ASL MRI protocols will be needed to validate these observations.
Keywords: Alzheimer’s disease, arterial spin labeling, cerebral blood flow, magnetic resonance imaging, perfusion, vascular cognitive impairment, white matter lesions
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disorder associated with the accumulation of amyloid-β peptide (Aβ) and hyperphosphorylated microtubule-associated protein tau [1]. Vascular cognitive impairment (VCI) is characterized by clinical evidence of stroke, or subclinical vascular brain injury based on neuroradiological features with impairment in at least one cognitive domain [2]. In practice for clinicians and neuropathologists alike, most dementias are mixed and it is often impossible to discern the relative contributions of each type of pathology [3].
AD and VCI are thought to be together responsible for the majority of dementia. While AD and VCI have traditionally been considered as distinct entities, there is growing evidence of an overlap between these categories such that mixed dementia may account for up to 50% of cases of dementia [4–6]. Currently, elucidating whether the cognitive deterioration is solely a consequence of vascular factors or underlying AD is difficult [7]. Several studies have found that in patients with combined AD and cerebrovascular disease, less AD pathology is needed to express a given degree of dementia severity [8, 9], and autopsy series have verified the high incidence of combined AD and vascular disease in patients with dementia [9, 10]. Accordingly, it appears that an interaction exists between AD and cerebrovascular pathologies [9], and these mixed pathologies are also common in mild cognitive impairment (MCI) [11, 12]. This overlap of pathologies has motivated an increase in research into the impact of vascular risk factors in dementia [3, 13].
Many of the traditional risk factors for stroke such as hypertension, hyperlipidemia, and apolipoprotein E (APOE) genotype are also risk markers for AD and VCI [14–16]. A convergence of pathogenic mechanisms in vascular and neurodegenerative processes causing impairment of cognition [17, 18] has also been postulated. Both the risk [8, 9] and pace [19] of dementia is increased in patients with acute and chronic stroke, especially subcortical infarcts in the basal ganglia, thalamus, and deep white matter (WM) [9]. Stroke has been found to double the risk of dementia in those above age 65 [20]. The cerebral vasculature, including both large and small vessels, is also thought to be affected by both vascular and neurodegenerative pathologies [5]. Large intracranial vessels exhibit atherosclerotic plaques in both VCI and AD [21, 22]. In addition, increased Aβ deposition [23] and tau phosphorylation have also been observed in experimental ischemia [24]. These interactions provide a mechanistic basis for a vascular hypothesis for AD pathogenesis [25–29].
Despite the well-recognized reciprocal interactions between cerebrovascular disease and dementia, clinical trials of vascular prophylaxis strategies have thus far failed to slow the progression of dementia due to AD, at least once it is established [5]. Furthermore, it remains difficult to effectively stratify patients with dementia according to the presence of AD, VCI, and particularly the extent of mixed dementia. Clinical features differentiating AD from VCI apply mainly to the most extreme cases. While significant progress has been made in the development of biomarkers that measure neurodegenerative change in AD such as structural magnetic resonance imaging (MRI), fluorodeoxyglucose positron emission tomography (FDG-PET), and, over the past decade, molecular markers of AD neuropathology [30–32], one of the limitations in advancing current understanding of the interactions between VCI and AD is the lack of reliable methods for quantifying VCI pathophysiology in vivo. Although computed tomography (CT) and particularly MRI scanning are highly effective in identifying macroscopic strokes, microvascular dysfunction has been particularly difficult to assess [33]. The presence of white matter lesions (WML) on CT and MRI, thought to be the result of hyaline fibrosis of small vessels, currently provides the most commonly accepted measure of microvacular ischemia, but the clinical relevance of WML remains ambiguous, as they are not specific to ischemic neuropathology and can also reflect inflammatory processes or gliosis [6]. WML are also found in healthy elderly patients [34], and are therefore not specific to patients with VCI, MCI, or AD. Dysfunction of the blood brain barrier (BBB) is an early finding in WML associated with VCI and AD [35]. BBB deficits can produce vascular inflammation and demyelination which might also contribute to the neural dysfunction that underlies cognitive impairment [36]. A potentially more specific marker of vascular amyloid is the presence of multifocal hemorrhage or microhemorrhage on susceptibility weighted MRI [37–39].
One potential means for more specifically characterizing vascular dysfunction in dementia is to quantify tissue-specific perfusion, or quantitative cerebral blood flow (qCBF). Arterial spin labeled (ASL) perfusion MRI provides a means for noninvasive quantification of CBF as part of a multimodal MRI examination. ASL MRI is a less invasive, less costly, and more widely available approach for qCBF imaging than prior radionuclide methods such as Xe-133 clearance or O-15 PET. In ASL MRI, ‘labeling’ or ‘tagging’ is achieved by manipulating the magnetic properties of in-flowing arterial blood proximal to the stationary tissues of interest [40]. This magnetically labeled arterial blood behaves as an endogenous contrast agent that produces transient signal changes as water, oxygen, and nutrients permeate the tissue at the capillary bed. The perfusion contrast in ASL is generated by subtracting images collected with arterial blood labeling from control images collected in the absence of labeling, and modeling this difference to derive CBF in classical units of ml/g/min [41] along with assumed or measured values for additional parameter such as the T1 relaxation rates of blood and brain and the blood:brain partition coefficient for water. Quantitative perfusion maps can be calculated from these difference images since the signal intensity is directly proportional to the movement of water molecules into the tissue, and the resulting CBF maps are in close agreement with PET measures [42, 43].
A recent review by Alsop et al. summarized several ASL studies that have been carried out in AD patients [44], and have yielded patterns of hypoperfusion that are consistent with patterns of hypometabolism previously identified in FDG- PET studies. ASL has also been used to differentiate AD and MCI from healthy controls [45, 46]. However, most ASL studies in AD and MCI to date have focused on reductions in gray matter (GM) perfusion indicative of neural dysfunction, rather than on hypoperfusion as a measure of primary vascular dysfunction. While a few studies have attempted to associate qCBF changes with WML [47], these have not been considered in the setting of dementia.
A challenge in applying ASL to the investigation of microvascular dysfunction in dementia is the relatively poor sensitivity of ASL in WM, which has lower CBF and longer arterial transit times than GM [48]. While it was difficult to obtain any credible WM CBF measurements in early ASL studies of human brain, several methodological advances including higher field strength [49, 50] and more efficient labeling strategies such as pulsed continuous ASL [51] have greatly improved the sensitivity of ASL, and it is now possible to obtain qCBF measures in WM regions of interest (ROI) that are reproducible [52] and in agreement with PET values [43]. Van Osch et al. [53] argue that the combination of the labeling duration and use of a sufficient post-label delay in pulsed continuous ASL (pCASL) allows for adequate transit time of the spin label to most WM regions. This approach overcomes some of the limitations of early ASL techniques, such as pulsed ASL, that were not sensitive to long transit times associated with WM [48]. High-resolution anatomical segmentation can also reduce partial volume effects by excluding voxels that contain a mix of GM, WM, and cerebrospinal fluid (CSF) to provide a more specific measure of WM CBF [53].
Here we aimed to begin to explore the utility of microvascular perfusion measurements using previously acquired ASL perfusion MRI from control, MCI, and AD patients to specifically determine whether qCBF data from GM and particularly deep WM ROI could be reliably obtained and used to further stratify patients according to the likelihood of vascular dysfunction. We carried out an analysis combining existing MRI datasets that included both ASL MRI obtained at 3 T using pCASL and fluid attenuated inversion recovery (FLAIR) images.
MATERIALS AND METHODS
Subjects
Data from 120 subjects were included, comprising 30 probable AD, 26 MCI, and 64 age-matched healthy control subjects. Available data from 2 AD patients, 1 MCI patient, and 11 controls were discarded due to image artifacts on ASL MRI or non-physiological CBF values within segmented regions. Diagnosis of probable AD was made according to the criteria of National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association [54]. MCI was diagnosed according to the criteria proposed by Petersen [55] and Winblad [56]. Controls were recruited either from the healthy Control cohort of the Penn Alzheimer’s Disease Center, or by advertisement. The institutional review board of University of Pennsylvania approved the study protocols and informed written consent was obtained from all subjects or their legally designated representatives prior to participation in the study.
MR imaging protocol
MRI scans were acquired on a 3T whole-body Siemens TIM Trio scanner (Erlangen, Germany) with an 8-channel receive-only head coil and body coil transmission. High-resolution whole brain anatomic images were collected using 3-dimensional magnetization-prepared rapid gradient echo (MPRAGE) with the following parameters: TR/TE/TI = 1620/3.87/950 ms, 160 axial slices, and 1 mm isotropic resolution. FLAIR images were acquired with the following parameters: TR/TE/TI = 9190/103/2200 ms, FOV=18×24cm2, acquisition matrix = 320×144, slice thickness = 3 mm, slice gap = 1 mm, number of slices = 40. Resting CBF measurements were acquired using pCASL [51] with gradient-echo echo-planar imaging (EPI). TR/TE = 4000/17 ms, slice thickness = 6 mm, slice gap = 1.2 mm, voxel resolution = 3.5× 3.5 × 6 mm3, and the labeling duration is 1.5 s. Due to differences in acquisition protocols, the post-labeling delay time was 1.2 s for 29 of 64 controls and MCI patients and 1.5 s for the remainder of the controls and the AD patients. The labeling plane was positioned 90 mm below the center of the imaging slab composed of 18 axial slices. Fifty-nine pairs of interleaved control and tag images were acquired for 13 AD patients, 44 normal subjects, and all MCI patients; 45 pairs were obtained for other 17 AD patients and 20 normal subjects. The data from 44 normal subjects, 21 AD, and all MCI patients who had FLAIR images were used to analyze correlation between CBF and WMLs. Twenty four AD patients and 20 normal subjects were rescanned approximately 1 week to 1 month after the initial scan to assess the reproducibility of ASL MRI for measuring WM CBF (the data from these 20 normal subjects were not used for other analyses because their age were not matched with patients groups).
Image processing
ASL data preprocessing and CBF calculation were performed by fMRI Grocer 2.09, which is a homebrewed SPM toolbox (http://www.fil.ion.ucl.ac.uk/spm/ext/#Grocer).
Each subject’s MPRAGE dataset was segmented into probabilistic GM, WM, and CSF maps. Probabilistic GM and WM maps were converted to binary masks with a threshold >0.8, and then were subsampled to the resolution of the raw ASL images.
Raw ASL images were first coregistered to the MPRAGE images and motion-corrected using a 6-parameter rigid body spatial transformation. After motion correction, the ASL images were multiplied by the GM and WM masks; global cortical GM images and global WM images were then generated. Pairwise subtraction images were generated after smoothing by an 8 mm full-width half maximum (FWHM) kernel. Averaged difference images were converted to ml/100g/min GM and WM CBF maps using a single-compartment model [57].
To minimize the contamination of WM CBF measurements with GM, WM ROIs were established with the following methods: the probabilistic WM map was smoothed with a 4 mm FWHM kernel, binarized with a threshold >0.8, and then eroded with a threshold 2, and finally subsampled to the resolution of the CBF images. The final WM ROI mainly consisted of periventricular WM regions, and was used to extract CBF value for WM from individual global WM CBF map. The binarized (threshold>0.8) and subsampled probabilistic GM map was used as ROI to extract CBF for GM from individual global GM CBF map. Representative CBF maps from ASL MRI and GM and WM masks are shown in Fig. 1. CBF was also calculated for a composite ROI developed by Landau et al. [58] consisting of regions in bilateral angular gyri, bilateral posterior cingulate, and left middle/inferior temporal gyrus previously shown to be sensitive to AD and MCI in FDG-PET studies. The composite ROI was spatially normalized to individual subject space using the normalization parameters calculated in the segmentation, and then subsampled to the resolution of the CBF map. The GM mask was also applied since the composite ROI consists of GM regions.
Fig. 1.

Sample axial FLAIR, MPRAGE, gray matter (GM) CBF map, and white matter (WM) CBF maps in a (A) control, (B) MCI patient, and (C) AD patient. The red regions represent the overlap between the GM and WM masks used to extract CBF within these segments and the GM and WM segmentations derived from anatomical data.
FLAIR images of each subject were assessed by an experienced rater (QZ), blind to the diagnosis, using the White Matter Lesions Semiquantitive Rating Scale [59]. The scale provides a semiquantitive measurement of the amount, size and location of WMLs. In the present study, a score of deep WM hyperintensities (dWMH) was computed from the sum of WMLs in frontal, parietal, temporal, and occipital lobes. A score of periventricular WM hyperintensities (PVH) was the sum of bands, frontal and occipital caps.
Statistical analysis
All data were analyzed using Statistical Package for Social Sciences for Windows (release 16.0; SPSS, Chicago, IL, USA). Group differences were analyzed using analysis of variance (ANOVA) together with least significant difference post hoc comparisons, to evaluate possible group differences in CBF value for composite ROI, GM, and WM, and clinical variables. A chi-squared (χ2) test was used to assess differences in gender and the number of APOE ε4 carriers. Group differences in dWMH and PVH were examined using a Kruskal-Wallis test and post hoc Mann-Whitney U test. The correlation between age and scores of WMLs were examined using Spearman rank correlation analysis. Partial correlation analysis with gender correction was applied to examine the relationship between CBF value and dWMH and PVH. Reproducibility of the ASL was assessed using Intraclass Correlation Coefficient (ICC) test and within-subject coefficient of variation (wsCV) [60]. The independent sample student’s t-test was used to detect the difference of time signal noise ratio (tSNR, defined as mean CBF divided by standard deviation of CBF) of WM CBF in subjects with test-retest data. All statistical tests were regarded as significant at p<0.05.
RESULTS
Demographics and clinical information of subjects are summarized in Table 1. There were no significant differences in age and gender between groups. Years of education in AD group was lower than in the MCI and control groups (p<0.05). The number of APOE ε4 carriers was significantly higher in AD group than MCI and control group (p<0.05). Group differences in Mini Mental State Examination, Animal Fluency, Boston Naming Test, Digit Span Backward, and 10-item Delayed Word-List Recall from the Consortium to Establish a Registry for AD battery were significant (p<0.001); post hoc tests revealed that patients with AD had lower scores than MCI patients and control group (both p<0.001), and MCI patients had lower scores than control group (p<0.05). There were also significant group differences in Digit Span Forward (p<0.05); post hoc tests show that the control group had higher scores than AD and MCI patients (both p<0.05), but there was no significant difference between AD and MCI groups (p=0.438).
Table 1.
Demographic and neuropsychological summary of subjects.
| Control (n=44) | MCI (n=26) | AD (n=30) | p | |
|---|---|---|---|---|
| Age (years) | 69.8±8.5 | 71.6±6.7 | 72.9±7.4 | 0.228 a |
| Gender (male/female) | 15/29 | 12/14 | 13/17 | 0.552 b |
| Education (years) | 16.5±2.9 | 17.0±2.5 | 15.0±3.0 | 0.019 a |
| MMSE | 29.4±1.1 | 27.3±1.6 | 19.8±5.9 | 0.000 a |
| APOE ε4 Carrier (%) | 35 | 59 | 69 | 0.051 b |
| Animal Fluency | 23.5±5.3 | 16.1±4.9 | 9.1±4.2 | 0.000 a |
| Boston Naming Test | 29.0±1.3 | 27.1±3.3 | 19.4±6.6 | 0.000 a |
| Digit Span Forward | 9.0±1.9 | 8.0±1.9 | 7.4±2.3 | 0.014 a |
| Digit Span Backward | 7.5±2.8 | 6.0±1.3 | 4.3±1.6 | 0.000 a |
| Delayed Word-List Recall | 8.8±1.4 | 3.1±2.0 | 0.9±1.4 | 0.000 a |
MMSE, Mini-Mental State Examination score.
p value from ANOVA test.
p value from chi-squared (χ2) test
The mean CBF values for composite ROI, GM, and WM in control, MCI, and AD groups are presented in Table 2. There was a significant group difference in mean CBF value for composite ROI between groups ((F(2, 97)=12.076, p=0.000). Post hoc tests revealed significant differences between control and MCI (p=0.020), control and AD (p=0.000), and MCI and AD groups (p=0.036). The difference in mean CBF value for GM between groups was also significant (F(2, 97)=4.443, p=0.014). Post hoc tests revealed that MCI patients had lower mean CBF value for GM than controls (p=0.005), but differences between MCI and AD groups (p=0.260) or between controls and AD groups (p=0.083) within the entire GM segment were not significant. Although there were no significant differences in the mean CBF value for WM between groups (F(2, 97)=2.816, p=0.065), group effects approached significance and the MCI group had lowest CBF value.
Table 2.
Cerebral blood flow (mean±sd) for composite ROI, gray matter, and white matter in Alzheimer’s disease, mild cognitive impairment, and control groups.
| Control (n=44) | MCI (n=26) | AD (n=30) | p | |
|---|---|---|---|---|
| Composite ROI | 49.8±12.4 | 43.7±7.9 | 37.8±8.9 | 0.000 |
| Gray matter | 39.4±9.2 | 33.5±7.0 | 36.0±7.5 | 0.014 |
| White matter | 19.2±5.7 | 16.4±3.8 | 19.2±5.5 | 0.065 |
CBF in ml/100g/min
p values are from ANOVA test
Excellent reproducibility for WM CBF was observed for the 20 normal subjects (ICC= 0.736, p=0.000 and wsCV±95% confidence interval (CI) was14.98%±1.39%) and reasonable reproducibility was also observed for 24 AD patients (ICC=0.529, p=0.004 and wsCV±95% CI was 20.37%±2.39%). The tSNR values for WM CBF was significantly higher in normal subjects than in AD patients were (2.7±0.6 versus 2.1±0.5, t=−5.332, p=0.000).
There were no significant differences between groups in dWMH scores (χ2=0.646, p=0.724), but differences in PVH scores between groups were significant (χ2=7.391, p=0.025). Mann Whitney U-testing revealed that PVH scores were higher in AD group than that in control (p=0.008) or MCI groups (p=0.044), but there was no significant difference between control and MCI groups (p=0.680). Details of the dWMH and PVH analyses are presented in Table 3.
Table 3.
Periventricular white matter hyperintensities (PVH) and deep white matter hyperintensities (dWMH) in 3 groups.
| Control (n=44) | MCI (n=26) | AD (n=21) | |
|---|---|---|---|
| PVH | |||
| Prevalence | 35 (77.8%) | 21 (80.8%) | 18 (85.7%) |
| Median | 1 | 1.5 | 3 |
| dWMH | |||
| Prevalence | 42 (93.3%) | 23 (88.5%) | 18 (85.7%) |
| Median | 6.5 | 7.5 | 7 |
Significant correlations were observed between age and dWMH and PVH scores in control (dWMH: r=0.493, p=0.001; PVH: r=0.531, p=0.000) and MCI groups (dWMH: r=0.397, p =0.044; PVH: r=0.410, p=0.038), but not in the AD group (dWMH: r=0.345, p=0.125; PVH: r=0.367, p=0.102). Partial correlation analysis showed no significant correlations between mean CBF values for GM and WM and WML scores in all three groups. However, as illustrated in Fig. 2, a negative trend approaching significance in control group (GM and PVH: r=−0.274, p=0.075; WM and PVH: r=−0.244, p=0.115) and a weaker trend in MCI group (GM and PVH: r=−0.286, p=0.166; WM and PVH: r=−0.191, p=0.361) were observed, but not in the AD group (GM and PVH: r= 0.018, p=0.939; WM and PVH: r=0.091, p=0.703).
Fig. 2.
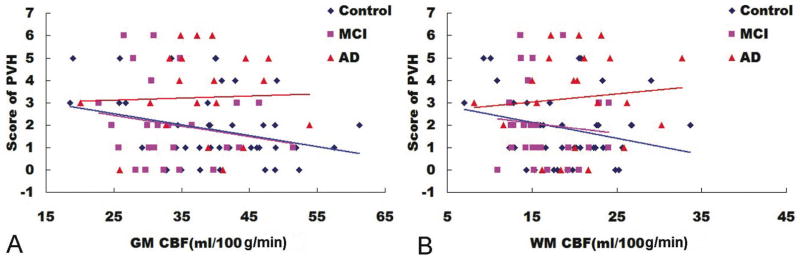
The relationship between scores of periventricular white matter hyperintensities (PVH) and CBF values for (A) gray matter (GM) and (B) white matter (WM). A negative trend was found between scores of PVH and CBF values in control (GM, p=0.075; WM, p=0.115) and MCI (GM, p=0.166; WM, p=0.361) groups.
DISCUSSION
In present study, we carried out a retrospective analysis of ASL perfusion MRI data from patients with AD, MCI, and age-matched controls and then compared these data to structural MRI measures of WMH. We found that CBF within a composite ROI sensitive to regional brain dysfunction in AD and MCI [58] also showed significant differences by diagnostic group when applied to ASL MRI data. This result supports the notion that CBF measurement with ASL MRI is sensitive to AD neuropathology, though the mechanism for focal CBF reductions within the composite ROI is not likely to be primarily vascular. Instead, it most likely reflects a reduction in neural activity within these brain regions since similar patterns are also seen for FDG-PET hypometabolism. We also found much less significant group effects in GM CBF, which are consistent with prior reports of ASL MRI in these populations [45, 61, 62]. The observed CBF differences across GM are also likely dominated by changes in regional neural activity.
The main aim of present study was to explore the potential of ASL perfusion MRI as a measure of microvascular perfusion deficits in AD neuropathology. Microvascular effects might be expected to be more widespread in the brain, and may be most notable within deep WM, which is the location where structural changes associated with WM disease are typically observed. To that end, one of the main goals of this study was to assess whether WM CBF could be reliably measured with ASL MRI using currently available 3T pCASL data. To try to obtain reliable CBF values for WM, the ROI for WM was restricted to periventricular WM by using a high threshold (probability >80%) for segmentation and then eroding the resulting ROI to further limit it to subcortical WM, and minimize the potential for contamination from GM. The qCBF values obtained from WM in 20 normal subjects showed excellent reproducibility and were in agreement with published values [43, 47]. No significant differences in WM CBF were observed between controls, MCI patients, and AD patients. There was a trend toward lower CBF in MCI patients than in either AD patients or controls, but additional data will be required to determine whether this trend becomes significant.
While Van Gelderen et al. [48] have argued that ASL MRI at 3T is not capable of obtaining reliable measures of WM CBF, our data suggest that WM CBF measurements from ASL MRI at 3T are reliable. The improved reliability we observed can be explained by the methodical use of pCASL, high-resolution segmentation, and ROI analysis. Van Gelderen et al. [48] make the assumption that the transit delays expected in ASL are equivalent to the delay in contrast bolus arrival from a Gd-based contrast agent injected into the antecubital vein. A difference in bolus arrival time between GM and WM was estimated to be ~0.65 seconds [48], which is compensated for in that our effective post-label delays were 2.7 seconds for the MCI cohort and 3.0 seconds for the AD cohort in our study. The T1 relaxation time of arterial blood at 3T is on the order of 1600 ms [63], which suggests that the ASL is still viable at these longer effective post-labeling delays. Also of note, contrast bolus arrival times are inherently affected by bolus dispersion and arrival time heterogeneity [64] that are not truly representative of an ASL label bolus arriving at a voxel. Perhaps a more representative measure of ASL transit delay times could be measured either by generating a low-resolution transit delay map [65], or Hadamard encoded ASL methods [66, 67]. Another key criticism of WM CBF measurements discussed by van Gelderen et al. [48] is the potential for measurement bias due to partial volume effects in voxels that include a mix of tissues. By employing MP-RAGE segmentation with a high threshold and by further eroding the mask, we were able to exclude voxels with mixed GM and WM from our analysis. Furthermore, by using a large ROI analysis instead of evaluating the CBF on a voxel-by-voxel basis, we were able to spatially signal average CBF values from voxels with a similar arterial transit delay time [65]. Although van Osch et al. [53] recently estimated that 75 measurements were required to determine CBF in ~70% of the WM voxels and that 150 measurements were required to obtain significant ASL effects in deep WM, by averaging ASL effect from approximately 1000 voxels we are able to obtain meaningful measurements with a fewer number of averages.
The reproducibility of WM CBF observed in AD patients in our data was slightly lower than that in normal subjects. The tSNR was also significantly lower in AD patients than controls, suggesting increased physiological noise in AD ASL data. For future work, physiological noise can be reduced through the use of background suppressed ASL [68].
Additional methodological developments are also capable of further improving the qCBF measurements in both GM and WM. MRI scanners with ultra-high magnetic field strengths are now becoming more widely available. Although there are challenges to ultra-high field ASL, by moving to higher field strengths (e.g., 7 Tesla) ASL benefits from both an increased SNR owing to the increased spin polarity and the prolongation of the T1 of arterial blood [63]. This increased SNR and prolonged T1 will produce a greater signal difference between label and control images, allowing for more accurate qCBF measurements in weakly perfused tissue. Furthermore, the increased SNR could be converted into increased spatial resolution, allowing for better differentiation between GM and WM by reducing partial volume effects [48]. The longer arterial T1 at higher field strength also increases the time window during which the magnetic label is viable, allowing for better qCBF measurements in tissues with a long transit time, particularly deep WM [6, 69].
In this study, the prevalence of total WML (combined deep and periventricular WML) on structural MRI was more than 92% in all groups. WML were mainly distributed in the frontal and parietal lobes and were correlated with age in control and MCI groups. These results are consistent with previous reports [70, 71]. WML are almost universally observed on brain MRI in elderly subjects, and are attributed to age-related arteriovascular disease [71, 72]. The periventricular WM is primarily supplied by lenticulostriate and long medullary vessels, which converge toward the periventricular region [73]. This structural characteristic results in an arterial border zone that makes the periventricular areas prone to ischemia [69]. The correlation between age and WMH did not reach significance in the AD group, though the PVH score was significantly higher in AD group than in control and MCI groups. This may simply reflect a lack of power in our AD data, but alternatively could suggest that WML in AD are multifactorial in etiology. Supporting this notion are the group trends observed in the correlation between CBF and WMH. While there were inverse trends between CBF values and PVH scores in control and MCI groups, consistent with an ischemic etiology, this trend was not observed in the AD data (Fig. 2). Furthermore, while we found significant correlations between age and scores for dWMH and PVH in control and MCI groups, we did not observe these correlations in AD group. An inflammatory mechanism due to BBB damage [36], demyelination [35], or the accumulation of effects of Aβ accumulation in AD patients [36, 74, 75] may lead to greater heterogeneity in WML etiology in patients with advanced AD. Amyloid angiopathy or Wallerian degeneration secondary to cortical atrophy could also contribute to WMLs in AD patients [76].
This study was limited by its use of retrospective data that were not obtained for the purposes of assessing microvascular perfusion per se. Future work employing ASL MRI protocols specifically optimized for WM CBF in larger cohorts will be needed to confirm these preliminary findings. Another limitation of this work is the lack of assessment for microhemorrhage, which is thought to be an important structural correlate of vascular amyloid. Future studies should also include gradient-echo or susceptibility weighted imaging to specifically assess for the burden of microhemorrhage and its associations with microvascular perfusion.
CONCLUSION
A growing literature suggests an interaction between vascular disease and AD pathogenesis, but the precise nature of this interaction has yet to be established, in part because it has been difficult to assess the pathophysiology of small vessel ischemic disease. ASL MRI is capable of quantifying microvascular CBF, including CBF in deep WM regions affected by small vessel disease. The reliability of these measures can be increased by using ASL MRI protocols optimized for quantifying WM CBF, and this could be an important application of ASL MRI at ultra-high field strengths. Future studies correlating qCBF by ASL MRI with structural, molecular, and behavioral indices of AD should be useful in better characterizing the nature of the interaction between vascular dysfunction and dementia.
Acknowledgments
This study was supported by NIH grants NS058386, EB015893, EB000814.
Footnotes
Authors’ disclosures available online (http://www.j-alz.com/disclosures/view.php?id=1413).
References
- 1.Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 2.Roman GC, Tatemichi TK, Erkinjuntti T, Cummings JL, Masdeu JC, Garcia JH, Amaducci L, Orgogozo JM, Brun A, Hofman A, et al. Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology. 1993;43:250–260. doi: 10.1212/wnl.43.2.250. [DOI] [PubMed] [Google Scholar]
- 3.Kling MA, Trojanowski JQ, Wolk DA, Lee V-Y, Arnold SE. Vascular disease and dementias: paradigm shifts to drive research in new directions. Alzheimer’s &dementia. 2012 doi: 10.1016/j.jalz.2012.02.007. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sahathevan R, Brodtmann A, Donnan GA. Dementia, stroke, and vascular risk factors; a review. Int J Stroke. 2012;7:61–73. doi: 10.1111/j.1747-4949.2011.00731.x. [DOI] [PubMed] [Google Scholar]
- 5.Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, Petersen RC, Schneider JA, Tzourio C, Arnett DK, Bennett DA, Chui HC, Higashida RT, Lindquist R, Nilsson PM, Roman GC, Sellke FW, Seshadri S. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke. 2011;42:2672–2713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iadecola C. The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol. 2010;120:287–296. doi: 10.1007/s00401-010-0718-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chui HC, Zarow C, Mack WJ, Ellis WG, Zheng L, Jagust WJ, Mungas D, Reed BR, Kramer JH, Decarli CC, Weiner MW, Vinters HV. Cognitive impact of subcortical vascular and Alzheimer’s disease pathology. Ann Neurol. 2006;60:677–687. doi: 10.1002/ana.21009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petrovitch H, Ross GW, Steinhorn SC, Abbott RD, Markesbery W, Davis D, Nelson J, Hardman J, Masaki K, Vogt MR, Launer L, White LR. AD lesions and infarcts in demented and non-demented Japanese-American men. Ann Neurol. 2005;57:98–103. doi: 10.1002/ana.20318. [DOI] [PubMed] [Google Scholar]
- 9.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA. 1997;277:813–817. [PubMed] [Google Scholar]
- 10.Schneider JA, Wilson RS, Bienias JL, Evans DA, Bennett DA. Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology. 2004;62:1148–1155. doi: 10.1212/01.wnl.0000118211.78503.f5. [DOI] [PubMed] [Google Scholar]
- 11.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol. 2009;66:200–208. doi: 10.1002/ana.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petersen RC, Parisi JE, Dickson DW, Johnson KA, Knopman DS, Boeve BF, Jicha GA, Ivnik RJ, Smith GE, Tangalos EG, Braak H, Kokmen E. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol. 2006;63:665–672. doi: 10.1001/archneur.63.5.665. [DOI] [PubMed] [Google Scholar]
- 13.Roman G. Vascular dementia: a historical background. Int Psychogeriatr. 2003;15(Suppl 1):11–13. doi: 10.1017/S1041610203008901. [DOI] [PubMed] [Google Scholar]
- 14.Launer LJ, Ross GW, Petrovitch H, Masaki K, Foley D, White LR, Havlik RJ. Midlife blood pressure and dementia: the Honolulu-Asia aging study. Neurobiol Aging. 2000;21:49–55. doi: 10.1016/s0197-4580(00)00096-8. [DOI] [PubMed] [Google Scholar]
- 15.Kivipelto M, Helkala EL, Laakso MP, Hanninen T, Hallikainen M, Alhainen K, Iivonen S, Mannermaa A, Tuomilehto J, Nissinen A, Soininen H. Apolipoprotein E epsilon4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life Alzheimer disease. Ann Intern Med. 2002;137:149–155. doi: 10.7326/0003-4819-137-3-200208060-00006. [DOI] [PubMed] [Google Scholar]
- 16.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103:5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iadecola C, Gorelick PB. Converging pathogenic mechanisms in vascular and neurodegenerative dementia. Stroke. 2003;34:335–337. doi: 10.1161/01.str.0000054050.51530.76. [DOI] [PubMed] [Google Scholar]
- 18.Iadecola C, Davisson RL. Hypertension and cerebrovascular dysfunction. Cell Metab. 2008;7:476–484. doi: 10.1016/j.cmet.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Helzner EP, Luchsinger JA, Scarmeas N, Cosentino S, Brickman AM, Glymour MM, Stern Y. Contribution of vascular risk factors to the progression in Alzheimer disease. Arch Neurol. 2009;66:343–348. doi: 10.1001/archneur.66.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Savva GM, Stephan BC. Epidemiological studies of the effect of stroke on incident dementia: a systematic review. Stroke. 2010;41:e41–46. doi: 10.1161/STROKEAHA.109.559880. [DOI] [PubMed] [Google Scholar]
- 21.Beach TG, Wilson JR, Sue LI, Newell A, Poston M, Cisneros R, Pandya Y, Esh C, Connor DJ, Sabbagh M, Walker DG, Roher AE. Circle of Willis atherosclerosis: association with Alzheimer’s disease, neuritic plaques and neurofibrillary tangles. Acta Neuropathol. 2007;113:13–21. doi: 10.1007/s00401-006-0136-y. [DOI] [PubMed] [Google Scholar]
- 22.Honig LS, Kukull W, Mayeux R. Atherosclerosis and AD: analysis of data from the US National Alzheimer’s Coordinating Center. Neurology. 2005;64:494–500. doi: 10.1212/01.WNL.0000150886.50187.30. [DOI] [PubMed] [Google Scholar]
- 23.Garcia-Alloza M, Gregory J, Kuchibhotla KV, Fine S, Wei Y, Ayata C, Frosch MP, Greenberg SM, Bacskai BJ. Cerebrovascular lesions induce transient beta-amyloid deposition. Brain. 2011;134:3697–3707. doi: 10.1093/brain/awr300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wen Y, Yang SH, Liu R, Perez EJ, Brun-Zinkernagel AM, Koulen P, Simpkins JW. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim Biophys Acta. 2007;1772:473–483. doi: 10.1016/j.bbadis.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 25.de la Torre JC. Pathophysiology of neuronal energy crisis in Alzheimer’s disease. Neurodegener Dis. 2008;5:126–132. doi: 10.1159/000113681. [DOI] [PubMed] [Google Scholar]
- 26.de la Torre JC, Mussivand T. Can disturbed brain microcirculation cause Alzheimer’s disease? Neurol Res. 1993;15:146–153. doi: 10.1080/01616412.1993.11740127. [DOI] [PubMed] [Google Scholar]
- 27.de la Torre JC. Critically attained threshold of cerebral hypoperfusion: the CATCH hypothesis of Alzheimer’s pathogenesis. Neurobiol Aging. 2000;21:331–342. doi: 10.1016/s0197-4580(00)00111-1. [DOI] [PubMed] [Google Scholar]
- 28.de la Torre JC. The vascular hypothesis of Alzheimer’s disease: bench to bedside and beyond. Neurodegener Dis. 2010;7:116–121. doi: 10.1159/000285520. [DOI] [PubMed] [Google Scholar]
- 29.de la Torre JC. A turning point for Alzheimer’s disease? Biofactors. 2012;38:78–83. doi: 10.1002/biof.200. [DOI] [PubMed] [Google Scholar]
- 30.Hampel H, Wilcock G, Andrieu S, Aisen P, Blennow K, Broich K, Carrillo M, Fox NC, Frisoni GB, Isaac M, Lovestone S, Nordberg A, Prvulovic D, Sampaio C, Scheltens P, Weiner M, Winblad B, Coley N, Vellas B. Biomarkers for Alzheimer’s disease therapeutic trials. Prog Neurobiol. 2011;95:579–593. doi: 10.1016/j.pneurobio.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 31.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 32.Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gouw AA, Seewann A, van der Flier WM, Barkhof F, Rozemuller AM, Scheltens P, Geurts JJ. Heterogeneity of small vessel disease: a systematic review of MRI and histopathology correlations. J Neurol Neurosurg Psychiatry. 2011;82:126–135. doi: 10.1136/jnnp.2009.204685. [DOI] [PubMed] [Google Scholar]
- 34.Kim JH, Hwang KJ, Lee YH, Rhee HY, Park KC. Regional white matter hyperintensities in normal aging, single domain amnestic mild cognitive impairment, and mild Alzheimer’s disease. J Clin Neurosci. 2011;18:1101–1106. doi: 10.1016/j.jocn.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 35.Farrall AJ, Wardlaw JM. Blood-brain barrier: ageing and microvascular disease--systematic review and meta-analysis. Neurobiol Aging. 2009;30:337–352. doi: 10.1016/j.neurobiolaging.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 36.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 37.Sperling RA, Jack CR, Jr, Black SE, Frosch MP, Greenberg SM, Hyman BT, Scheltens P, Carrillo MC, Thies W, Bednar MM, Black RS, Brashear HR, Grundman M, Siemers ER, Feldman HH, Schindler RJ. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7:367–385. doi: 10.1016/j.jalz.2011.05.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schrag M, McAuley G, Pomakian J, Jiffry A, Tung S, Mueller C, Vinters HV, Haacke EM, Holshouser B, Kido D, Kirsch WM. Correlation of hypointensities in susceptibility-weighted images to tissue histology in dementia patients with cerebral amyloid angiopathy: a postmortem MRI study. Acta Neuropathol. 2010;119:291–302. doi: 10.1007/s00401-009-0615-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nandigam RN, Viswanathan A, Delgado P, Skehan ME, Smith EE, Rosand J, Greenberg SM, Dickerson BC. MR imaging detection of cerebral microbleeds: effect of susceptibility-weighted imaging, section thickness, and field strength. AJNR Am J Neuroradiol. 2009;30:338–343. doi: 10.3174/ajnr.A1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams DS, Detre JA, Leigh JS, Koretsky AP. Magnetic resonance imaging of perfusion using spin inversion of arterial water. Proc Natl Acad Sci U S A. 1992;89:212–216. doi: 10.1073/pnas.89.1.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Detre JA, Leigh JS, Williams DS, Koretsky AP. Perfusion imaging. Magn Reson Med. 1992;23:37–45. doi: 10.1002/mrm.1910230106. [DOI] [PubMed] [Google Scholar]
- 42.Ye FQ, Berman KF, Ellmore T, Esposito G, van Horn JD, Yang Y, Duyn J, Smith AM, Frank JA, Weinberger DR, McLaughlin AC. H(2)(15)O PET validation of steady-state arterial spin tagging cerebral blood flow measurements in humans. Magn Reson Med. 2000;44:450–456. doi: 10.1002/1522-2594(200009)44:3<450::aid-mrm16>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 43.Xu G, Rowley HA, Wu G, Alsop DC, Shankaranarayanan A, Dowling M, Christian BT, Oakes TR, Johnson SC. Reliability and precision of pseudo-continuous arterial spin labeling perfusion MRI on 3.0 T and comparison with 15O-water PET in elderly subjects at risk for Alzheimer’s disease. NMR Biomed. 2010;23:286–293. doi: 10.1002/nbm.1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alsop DC, Dai W, Grossman M, Detre JA. Arterial spin labeling blood flow MRI: its role in the early characterization of Alzheimer’s disease. J Alzheimers Dis. 2010;20:871–880. doi: 10.3233/JAD-2010-091699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson NA, Jahng GH, Weiner MW, Miller BL, Chui HC, Jagust WJ, Gorno-Tempini ML, Schuff N. Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: initial experience. Radiology. 2005;234:851–859. doi: 10.1148/radiol.2343040197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dai W, Lopez OL, Carmichael OT, Becker JT, Kuller LH, Gach HM. Mild cognitive impairment and alzheimer disease: patterns of altered cerebral blood flow at MR imaging. Radiology. 2009;250:856–866. doi: 10.1148/radiol.2503080751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Firbank MJ, He J, Blamire AM, Singh B, Danson P, Kalaria RN, O’Brien JT. Cerebral blood flow by arterial spin labeling in poststroke dementia. Neurology. 2011;76:1478–1484. doi: 10.1212/WNL.0b013e318217e76a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Gelderen P, de Zwart JA, Duyn JH. Pittfalls of MRI measurement of white matter perfusion based on arterial spin labeling. Magn Reson Med. 2008;59:788–795. doi: 10.1002/mrm.21515. [DOI] [PubMed] [Google Scholar]
- 49.Wang J, Alsop DC, Li L, Listerud J, Gonzalez-At JB, Schnall MD, Detre JA. Comparison of quantitative perfusion imaging using arterial spin labeling at 1.5 and 4.0 Tesla. Magn Reson Med. 2002;48:242–254. doi: 10.1002/mrm.10211. [DOI] [PubMed] [Google Scholar]
- 50.Wang J, Zhang Y, Wolf RL, Roc AC, Alsop DC, Detre JA. Amplitude-modulated continuous arterial spin-labeling 3.0-T perfusion MR imaging with a single coil: feasibility study. Radiology. 2005;235:218–228. doi: 10.1148/radiol.2351031663. [DOI] [PubMed] [Google Scholar]
- 51.Dai W, Garcia D, de Bazelaire C, Alsop DC. Continuous flow-driven inversion for arterial spin labeling using pulsed radio frequency and gradient fields. Magn Reson Med. 2008;60:1488–1497. doi: 10.1002/mrm.21790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Y, Wang DJ, Detre JA. Test-retest reliability of arterial spin labeling with common labeling strategies. J Magn Reson Imaging. 2011;33:940–949. doi: 10.1002/jmri.22345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Osch MJ, Teeuwisse WM, van Walderveen MA, Hendrikse J, Kies DA, van Buchem MA. Can arterial spin labeling detect white matter perfusion signal? Magn Reson Med. 2009;62:165–173. doi: 10.1002/mrm.22002. [DOI] [PubMed] [Google Scholar]
- 54.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 55.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183–194. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]
- 56.Winblad B, Palmer K, Kivipelto M, Jelic V, Fratiglioni L, Wahlund LO, Nordberg A, Backman L, Albert M, Almkvist O, Arai H, Basun H, Blennow K, de Leon M, DeCarli C, Erkinjuntti T, Giacobini E, Graff C, Hardy J, Jack C, Jorm A, Ritchie K, van Duijn C, Visser P, Petersen RC. Mild cognitive impairment--beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J Intern Med. 2004;256:240–246. doi: 10.1111/j.1365-2796.2004.01380.x. [DOI] [PubMed] [Google Scholar]
- 57.Wang J, Alsop DC, Song HK, Maldjian JA, Tang K, Salvucci AE, Detre JA. Arterial transit time imaging with flow encoding arterial spin tagging (FEAST) Magn Reson Med. 2003;50:599–607. doi: 10.1002/mrm.10559. [DOI] [PubMed] [Google Scholar]
- 58.Landau SM, Harvey D, Madison CM, Koeppe RA, Reiman EM, Foster NL, Weiner MW, Jagust WJ. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging. 2011;32:1207–1218. doi: 10.1016/j.neurobiolaging.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scheltens P, Barkhof F, Leys D, Pruvo JP, Nauta JJ, Vermersch P, Steinling M, Valk J. A semiquantative rating scale for the assessment of signal hyperintensities on magnetic resonance imaging. J Neurol Sci. 1993;114:7–12. doi: 10.1016/0022-510x(93)90041-v. [DOI] [PubMed] [Google Scholar]
- 60.Bland JM, Altman DG. Measurement error proportional to the mean. BMJ. 1996;313:106. doi: 10.1136/bmj.313.7049.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alsop DC, Detre JA, Grossman M. Assessment of cerebral blood flow in Alzheimer’s disease by spin-labeled magnetic resonance imaging. Ann Neurol. 2000;47:93–100. [PubMed] [Google Scholar]
- 62.Yoshiura T, Hiwatashi A, Yamashita K, Ohyagi Y, Monji A, Takayama Y, Nagao E, Kamano H, Noguchi T, Honda H. Simultaneous measurement of arterial transit time, arterial blood volume, and cerebral blood flow using arterial spin-labeling in patients with Alzheimer disease. AJNR Am J Neuroradiol. 2009;30:1388–1393. doi: 10.3174/ajnr.A1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dobre MC, Ugurbil K, Marjanska M. Determination of blood longitudinal relaxation time (T1) at high magnetic field strengths. Magnetic resonance imaging. 2007;25:733–735. doi: 10.1016/j.mri.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 64.Ko L, Salluzzi M, Frayne R, Smith M. Reexamining the quantification of perfusion MRI data in the presence of bolus dispersion. Journal of magnetic resonance imaging: JMRI. 2007;25:639–643. doi: 10.1002/jmri.20781. [DOI] [PubMed] [Google Scholar]
- 65.Dai W, Robson PM, Shankaranarayanan A, Alsop DC. Reduced resolution transit delay prescan for quantitative continuous arterial spin labeling perfusion imaging. Magn Reson Med. 2012;67:1252–1265. doi: 10.1002/mrm.23103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wells JA, Lythgoe MF, Gadian DG, Ordidge RJ, Thomas DL. In vivo Hadamard encoded continuous arterial spin labeling (H-CASL) Magn Reson Med. 2010;63:1111–1118. doi: 10.1002/mrm.22266. [DOI] [PubMed] [Google Scholar]
- 67.Dai W, Shankaranarayanan A, Alsop DC. Volumetric measurement of perfusion and arterial transit delay using hadamard encoded continuous arterial spin labeling. Magn Reson Med. 2012 doi: 10.1002/mrm.24335. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu WC, Edlow BL, Elliot MA, Wang J, Detre JA. Physiological modulations in arterial spin labeling perfusion magnetic resonance imaging. IEEE Trans Med Imaging. 2009;28:703–709. doi: 10.1109/TMI.2008.2012020. [DOI] [PubMed] [Google Scholar]
- 69.Moody DM, Bell MA, Challa VR. Features of the cerebral vascular pattern that predict vulnerability to perfusion or oxygenation deficiency: an anatomic study. AJNR Am J Neuroradiol. 1990;11:431–439. [PMC free article] [PubMed] [Google Scholar]
- 70.Artero S, Tiemeier H, Prins ND, Sabatier R, Breteler MM, Ritchie K. Neuroanatomical localisation and clinical correlates of white matter lesions in the elderly. J Neurol Neurosurg Psychiatry. 2004;75:1304–1308. doi: 10.1136/jnnp.2003.023713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Targosz-Gajniak M, Siuda J, Ochudlo S, Opala G. Cerebral white matter lesions in patients with dementia - from MCI to severe Alzheimer’s disease. J Neurol Sci. 2009;283:79–82. doi: 10.1016/j.jns.2009.02.314. [DOI] [PubMed] [Google Scholar]
- 72.Longstreth WT, Jr, Manolio TA, Arnold A, Burke GL, Bryan N, Jungreis CA, Enright PL, O’Leary D, Fried L. Clinical correlates of white matter findings on cranial magnetic resonance imaging of 3301 elderly people. The Cardiovascular Health Study. Stroke. 1996;27:1274–1282. doi: 10.1161/01.str.27.8.1274. [DOI] [PubMed] [Google Scholar]
- 73.Moody DM, Brown WR, Challa VR, Ghazi-Birry HS, Reboussin DM. Cerebral microvascular alterations in aging, leukoaraiosis, and Alzheimer’s disease. Ann N Y Acad Sci. 1997;826:103–116. doi: 10.1111/j.1749-6632.1997.tb48464.x. [DOI] [PubMed] [Google Scholar]
- 74.Kalaria RN. The blood-brain barrier and cerebral microcirculation in Alzheimer disease. Cerebrovasc Brain Metab Rev. 1992;4:226–260. [PubMed] [Google Scholar]
- 75.Gurol ME, Irizarry MC, Smith EE, Raju S, Diaz-Arrastia R, Bottiglieri T, Rosand J, Growdon JH, Greenberg SM. Plasma beta-amyloid and white matter lesions in AD, MCI, and cerebral amyloid angiopathy. Neurology. 2006;66:23–29. doi: 10.1212/01.wnl.0000191403.95453.6a. [DOI] [PubMed] [Google Scholar]
- 76.Debette S, Markus HS. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta-analysis. BMJ. 2010;341:c3666. doi: 10.1136/bmj.c3666. [DOI] [PMC free article] [PubMed] [Google Scholar]