

Abstract
Gold nanoparticles (AuNPs) allow the tuning of pharmacokinetic and pharmacodynamic properties by active or passive targeting of drugs for cancer and other diseases. We have functionalized gold nanoparticles by tethering specific ligands, agonists and antagonists, of adenosine receptors (ARs) to the gold surface as models for cell surface interactions with G protein-coupled receptors (GPCRs). The AuNP conjugates with chain-extended AR ligands alone (PEGylated nucleosides and nonnucleosides, anchored to the Au via thioctic acid) were found to be insoluble in water due to hydrophobic entities in the ligand. Therefore, we added a second, biologically inactive pendant moiety to increase the water solubility, consisting of a PEGylated chain terminating in a carboxylic or phosphate group. The purity and stability of the immobilized biologically active ligand were examined by ultrafiltration and HPLC. Pharmacological receptor binding studies on these GPCR ligand-derivatized AuNPs (2–5 nm in diameter), performed using membranes of mammalian cells stably expressing human A1, A2A, and A3ARs, showed that the desired selectivity was retained with Ki values (nanomolar) of A3AR agonist 21b and A2AAR antagonists 24 and 26a of 14 (A3), 34 (A2A), and 69 (A2A), respectively. The corresponding monomers displayed Ki values of 37, 61, and 1,420 nM, respectively. In conclusion, we have synthesized stable, water-soluble AuNP derivatives of tethered A3 and A2AAR ligands that retain the biological properties of their monomeric ligands and are intended for therapeutic and imaging applications. This is the first prototypical application to gold carriers of small molecule (nonpeptide) GPCR ligands, which are under investigation for treatment of cancer and inflammatory diseases.
Electronic supplementary material
The online version of this article (doi:10.1007/s11302-012-9338-z) contains supplementary material, which is available to authorized users.
Keywords: G protein-coupled receptor, Nanoparticle, Nucleoside, Adenosine, Radioligand binding
Introduction
Gold nanoparticles (AuNPs) are being developed as cancer therapeutic and diagnostic agents [1]. The ability to control the size and shape of these particles allows the tuning of their pharmacokinetic and pharmacodynamic properties in passive targeting, and the option of tethering specific ligands for cell surface receptors to the functionalized gold surface facilitates active targeting [2–4]. The advantages of the application of AuNPs for therapeutics and diagnostics are numerous and potentially applicable to many diseases. First, AuNPs can circulate systemically in the blood stream due to their nanometer size, and AuNPs are considered excellent candidates as drug delivery systems due to their biocompatibility. Secondly, they can easily be coated with multiple copies of biological ligands such as DNA, small interfering RNA, and drugs through several mechanisms (e.g., bioconjugation, noncovalent interactions, and direct surface bonding) [5]. In addition, immobilization of small molecules on a gold surface can be applied to imaging, plasmon resonance-based labeling, and optical and electrochemical sensing in cancer therapy and several other disease states (viz., Alzheimer’s disease, AIDS, hepatitis, tuberculosis, arthritis, and diabetes) [6, 7]. The cargo of the AuNPs may also be cytotoxic agents, for example in cancer chemotherapy, or other moieties for altering the fate of the particle in vivo or altering its ability to cross cell membranes [8]. A particularly clever application of AuNP conjugates is in thermoablation of tumor tissue by Au nanoshells, which are concentrated in the diseased tissue followed by irradiation to elevate the temperature [9]. AuNPs were used to interrogate the mechanistic pathways involved in killing cancer cells [10]. Also, because the shape of the particle determines optical properties, it is possible to apply such particles to in vivo imaging, for example, based on absorption in the near infrared (NIR) range [9]. Design parameters are being explored to obtain biocompatible AuNPs to serve as carriers of small molecules or biomacromolecules.
G protein-coupled receptors (GPCRs), which modulate intracellular pathways to alter cell survival, proliferation, and other functions, have recently become a focus in relation to the treatment of cancer [11]. GPCRs are involved in a wide range of diseases and serve as the basis of at least one third of the pharmaceuticals on the market. There are four known subtypes of adenosine receptors (ARs), which belong to the superfamily of GPCRs, referred to as A1, A2A, A2B, and A3—each of which has a unique pharmacological profile, tissue distribution, and combination of effects [12, 13]. An agonist of the A3AR is currently in clinical trials for hepatocellular carcinoma [13, 14]. Fishman et al. reported that growth of melanoma cells can be inhibited by activation of A3AR via deregulation of the NFκB and Wnt pathways [14]. They demonstrated that the small molecule nucleoside agonists IB-MECA 1 and Cl-IB-MECA 2 (Fig. 1), both invented in our laboratory, inhibit the growth of B16-F10 melanoma cells via activation of A3AR both in vitro and in vivo, and among these, 2 has already entered phase II clinical trials for hepatocellular carcinoma [14, 43]. Additionally, A2AAR agonists have anti-inflammatory properties and are used in cardiac imaging [13, 25]. A2AAR antagonists are potentially useful in the treatment of Parkinson’s disease, progressive neurodegeneration, and cancer [13]; cerebroprotection is observed either as a result of A2AAR antagonism or gene deletion [13, 15].
Fig. 1.

High affinity and chemically functionalized ligands for various ARs of interest. Agonists IB-MECA (1), Cl-IB-MECA (2), MRS 3558 (3) and 4 of the A3AR, selective agonist 5 and antagonist 6 of the A2AAR, and nonselective AR antagonist 7
A recurrent problem in the development of agonist and antagonist ligands for GPCRs is the widespread occurrence of a given receptor leading to side effects at undesired sites of action. Thus, there is a need for enhancing the selectivity of GPCR ligands for specific tissues, organs, or signaling pathways [16]. Devices and drugs that can be delivered with high accuracy to maximize efficacy and minimize side effects are needed. Thus, new methods for designing GPCR modulators that function as “smart drugs” are required. One possibility could be GPCR ligand-functionalized AuNPs, where proper adjustment of their pharmacokinetic properties may reduce off-target effects.
In this study, we explored the feasibility of using functionalized AuNPs as carriers for small GPCR ligands. Unlike the abovementioned applications of AuNPs in cancer therapy, their use in targeting cell surface GPCRs would not require dissociation of the therapeutic moiety from the carrier. In fact, the linkage of the GPCR ligand could be designed such that activity is retained in the intact tethered particle, which would bind to the surface of cells expressing the receptor for that ligand. Internalization of the particle would be a separate process that may or may not add to the efficacy of the gold nanoconjugate.
Materials and methods
Chemical synthesis
All reagents and solvents (regular and anhydrous) were of analytical grade and obtained from Sigma-Aldrich (St. Louis, MO, USA), unless noted, and used without further purification. Reactions were conducted under an atmosphere of nitrogen whenever anhydrous solvents were used. All reactions to prepare small molecules were monitored by thin layer chromatography using silica gel-coated plates with a fluorescence indicator which were visualized: (a) under UV light, (b) by dipping in a mixture of anisaldehyde (2.5 mL)/conc. H2SO4 (5 mL)/methanol (425 mL), or (c) by dipping the plate in a solution of ninhydrin (0.3 g in 100 mL EtOH, containing AcOH, 1.3 mL) followed by heating. Silica gel column chromatography was performed with silica gel (SiO2, 200–400 mesh, 60 Å, Sorbent Technologies, Atlanta, GA, USA) using moderate air pressure. Evaporation of solvents was carried out under reduced pressure at a temperature below 50 °C. After column chromatography, appropriate fractions were pooled, evaporated, and dried at high vacuum for at least 12 h to give the desired products in high purity. ESI-high resolution mass spectroscopic measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters) using external calibration with polyalanine. Observed mass accuracies are those expected on the basis of known performance of the instrument as well as the trends in masses of standard compounds observed at intervals during the series of measurements. Reported masses are observed masses uncorrected for this time-dependent drift in mass accuracy.
The synthetic intermediates and AR ligands (including AuNP conjugates) were characterized by 1H NMR. The spectra were recorded on a Bruker 400 MHz spectrometer. Chemical shifts are reported in parts per million (ppm) relative to tetramethylsilane or using deuterated solvent as the internal standard (δH: CDCl3 7.26 ppm). All J values are reported in Hertz (Hz). Electronic absorption spectra were recorded using an HP 8453 diode array spectrophotometer (Agilent Technologies, Santa Clara, CA, USA). The spectra were collected using quartz cuvettes (Spectrocell, Inc., Oreland, PA, USA) with 0.5 cm optical path length. The prepared AuNPs were characterized structurally by examination under a FEI Tecnai12 transmission electron microscope operating at a beam energy of 120 keV. Images were acquired by using a 2 k × 2 k cooled CCD camera from Gatan (Warrendale, PA, USA). Samples for transmission electron microscopy (TEM) were prepared by spreading a drop of the AuNP dispersion onto an ultrathin carbon support film on a fine mesh Cu grid (400 mesh) and letting it dry. The nanoparticles were imaged with a 1.8-nm pixel size. Dynamic light scattering measurements were carried out in a DynaPro NanoStar (Wyatt Technology, Santa Barbara, CA, USA). We purchased dialysis membranes (Spectra/Pore Membrane, MW cutoff 3,500, flat width 18 mm) from Spectrum Labs (Rancho Dominguez, CA, USA). The Amicon Ultra-15 centrifugal filter unit (30 K MW cutoff) was purchased from Millipore Ltd. (Billerica, MA, USA).
Synthesis of monomeric AR ligands, linkers, and solubilizing moieties
Compound 12
(R)-Thioctic acid (0.19 g, 0.95 mmol, TA, also known as α-lipoic acid), N3-(PEG)10-NH2 (Aldrich, 0.50 g, 0.95 mmol), 4-(N,N-dimethylamino)-pyridine (0.03 g, 0.28 mmol), and CH2Cl2 (20 mL) were added in a 100-mL round bottom flask, which was cooled to 0 °C. N,N′-dicyclohexylcarbodiimide (DCC, 0.19 g, 0.95 mmol) was slowly added under N2 with stirring. The above mixture was stirred at 0 °C for 2 h, then warmed to room temperature, and further stirred overnight. The reaction mixture was filtered through Celite and the filter rinsed with ethyl acetate. The filtrate was evaporated, and the residue was chromatographed on silica gel with CHCl3 and then 10:1 CH2Cl2/MeOH as eluent to give 0.66 mmol (0.47 mg) of homogeneous TA-(PEG)10-N312 (yield 70 %). 1H NMR (400 MHz, CDCl3): δ 6.17 (br s, 1H), 3.55–3.65 (m, PEG H), 3.51 (m, 3H), 3.40 (m, 2H), 3.34 (m, 2H), 3.05–3.15 (m, 2H), 2.42 (m, 1H), 2.14 (t, 2H, J = 7.6), 1.87 (m, 1H), 1.62 (m, 4H), 1.42 (m, 2H). m/z (M + ESI MS) found: 715.3640; calc for C30H58N4O11S2: 715.3622.
Compound 13
To a mixture of TA-PEG-N3 (12) (11.4 mg, 0.016 mmol) and alkyne 4 (6 mg, 0.011 mmol, A3AR agonist) [17] in a 0.50 mL of (1:1) mixture of tBuOH and water was added tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl] amine (TBTA, 1 mg, 0.001 mmol) and freshly prepared 1 M aqueous sodium ascorbate solution (11.7 μL, 0.011 mmol) followed by 7.5 % aqueous copper sulfate pentahydrate solution (19.4 μL, 0.005 mmol). The reaction mixture was stirred overnight at room temperature, the solvent was evaporated, and the residue was purified on a flash silica gel chromatography column (CH2Cl2/MeOH = 30:1) to give A3AR agonist monomer 13 for Au complexation (7.5 mg, 55 %). 1H NMR (400 MHz, MeOD): δ 8.08 (s, 1H), 7.83 (s, 1H), 7.41 (s, 1H), 7.25–7.31 (m, 2H), 5.01 (d, 1H, J = 6.7 Hz,), 4.52 (dd, 2H, J1 = 5.08 Hz, J2 = 4.76 Hz), 3.98 (d, 1H, J = 5.9 Hz), 3.85 (t, 2H, J1 = 4.92 Hz), 3.5–3.75 (m, PEG H), 3.3–3.45 (m, 2H), 3.05–3.15 (m, 2H), 2.85 (s, 3H), 2.78 (t, 2H, J1 = 7.44 Hz), 2.51 (dd, 2H, J1 = 7.08 Hz, J2 = 6.7 Hz), 2.43 (m, 1H), 2.20 (dd, 2H, J1 = 7.24 Hz, J2 = 7.36 Hz), 2.06 (m, 1H), 1.86 (m, 4H), 1.6–1.8 (m, 8H), 1.3–1.5 (m, 2H). 13C NMR (400 MHz, MeOD): δ 174.6, 173.1, 133.8, 129.7, 126.6, 125.7, 87.7, 86.0, 86.8, 76.0, 71.9, 70.2, 69.8, 69.7, 69.0, 56.2, 50.3, 50.0, 39.9, 38.7, 35.4, 34.3, 28.4, 28.3, 27.3, 27.1, 25.3, 18.2, 13.4. m/z (M + ESI MS) found: 1,247.5565; calc for C53H88N12O16S2Cl: 1,247.5571.
Compound 15
The synthesis followed a published procedure [18]. TA-(PEG)10-N3 (12) (0.409 g, 0.572 mmol) and triphenylphosphine (0.211 g, 2 equiv. 0.817 mmol) were stirred with 6.3 mL of tetrahydrofuran (THF) at room temperature under a N2 atmosphere for 2 h. Water (0.318 mL, 18 mmol) was added to the mixture, and stirring of the reaction mixture under N2 was continued for 72 h. The solvent was evaporated, and the crude product was purified by silica gel column chromatography using first 10:1 dichloromethane (DCM)/MeOH and then 100:20:1 DCM/MeOH/Et3N as eluent. Compound 15 (0.343 g, 60 %, TA-(PEG)10-NH2) was obtained as a yellowish liquid. 1H NMR (300 MHz, CDCl3): δ 6.48 (brs, 1H), 3.61 (m, PEG H), 3.51 (m, 2H), 3.40 (m, 1H), 3.11 (m, 2H), 2.94 (t, 2H, J = 5.2), 2.76 (brs, 2H), 2.41 (m, 1H), 2.16 (t, 2H, J = 7.6), 1.85 (m, 1H), 1.63 (m, 4H), 1.42 (m, 2H). m/z (M + ESI MS) found: 689.9310; calc: 689.9318.
Compound 16
TA-(PEG)10-NH2 (12) (97.0 mg, 0.141 mmol), succinic anhydride (240 mg, 0.239 mmol), and dry pyridine 1.55 mL were added to round bottom flask under N2. The mixture was stirred at room temperature for 17 h. The solvent was evaporated and the crude product was purified by column chromatography as the eluent (10:1 CHCl3/MeOH) to give yellowish homogeneous compound 16 (57 mg, 52 %, TA-(PEG)10-NH-succinoyl, a carboxylate-solubilizing moiety for Au complexation). 1H NMR (400 MHz, CDCl3): δ 7.12 (brs, 1H), 6.45 (brs, 1H), 3.61 (m, PEG H), 3.52 (m, 7H), 3.40 (m, 5H), 3.11 (m, 2H), 2.63 (dd, 2H, J1 = 6.2, J2 = 6.8), 2.45 (dd, 2H, J1 = 6.4, J2 = 5.76), 2.43 (brs, 2H), 2.17 (t, 3H, J = 7.2), 1.88 (m, 3H), 1.63 (m, 8H), 1.42 (m, 4H). m/z (M + ESI MS) found: 787.3654; calc for C34H63N2O14S2: 787.3721.
Compound 14
The N-heptynoyl derivative of XAC (7, xanthine amine congener, 8-[4-[[[[(2-aminoethyl)amino]carbonyl]methyl)oxy]phenyl]-1,3-dipropylxanthine) was synthesized according to a published procedure [19]. The alkyne derivative (7) (3.65 mg, 5.10 μmol) and TA-PEG-N3 (12) (2 mg, 3.01 μmol) were dissolved in 0.30 mL of a (3:1) mixture of DMF and water in a round bottom flask, and TBTA (1.01 mg, 1.02 μmol) was added. Freshly prepared sodium ascorbate (1 M, 3.72 μL, 3.70 μmol) followed by 7.5 % aqueous copper sulfate pentahydrate solution (6.16 μL, 1.86 μmol) was added to the above mixture. The reaction mixture was stirred overnight at room temperature, the solvent was evaporated, and the residue was purified on a flash silica gel chromatography column (CHCl3/MeOH = 6:1) to give xanthine AR antagonist monomer 14 for Au complexation (3.02 mg, 2.40 μmol). 1H NMR (400 MHz, MeOD): δ 7.98 (d, 2H, J = 8.8 Hz), 7.72 (s, 1H), 7.08 (d, 2H, J = 8.8), 4.55 (s, 2H), 4.47 (t, 3H, J = 5.0), 4.09 (t, 3H, J = 7.2 Hz), 3.90 (t, 3H, J = 7.5), 3.86 (t, 3H, J = 5.1 Hz), 3.7–3.5 (m), 3.3–3.0 (m), 2.62 (m, 2H), 2.43 (m, 1H), 2.18 (m, 4H), 1.86–1.79 (m, 3H), 1.67–1.57 (m, 12H), 1.42 (m, 2H), 1.27 (m, 3H), 0.90 (dt, 6H, J1 = 14.4 Hz, J2 = 7.4 Hz). m/z (M + ESI MS) found: 1,273.6241; calc for C63H94N8O14S2Na: 1,273.6229.
Compound 17
The PEGylated amine 15 (34 mg, 50 μmol) and 5 (CGS21680) (12.1 mg, 20.1 μmol) as a hydrochloride salt were dissolved in 1 mL DMF. Then, diisopropylethylamine (DIEA, 9 μL, 50.3 μmol) was added to the reaction mixture and stirring continued for 10 min. Then, benzotriazol-1-yloxy-tripyrrolidino-phosphonium hexafluorophosphate (PyBOP, 11.2 mg, 20.6 μmol) was added to the solution, and the mixture was stirred for 18 h at room temperature. The solvent was removed under vacuum, and the crude product was dissolved in a minimum volume (200 μL) of methanol. An excess volume (5–7 mL) of dry diethyl ether was added, and the mixture was left overnight at 4 °C, leading to the precipitation of the product. The ether supernatant was then removed using a Pasteur pipette, and the remaining solid was dried under vacuum to obtain the pure product 17, AR agonist monomer for Au complexation (6.01 mg, 5.0 μmol), in 25 % yield as a gummy, yellowish solid. 1H NMR (300 MHz, DMSO): δ 8.07 (br s, 2H), 7.85, 7.78 (d (each), J = 6.8 Hz, 4H), 5.95 (d, J = 6.4 Hz, 1H), 4. 89 (m, 1H), 4. 45 (s, 1H), 4.41 (s, 1H), 3. 68 (m, peg H), 3.51 (m, 2H), 3.45 (m, 1H), 3.32 (m, 6H), 2.93 (m, 6H), 2.51 (m, 2H), 2.24 (t, J = 5.4 Hz, 2H), 2.15 (m, 2H), 1.91 (m, 1H), 1.65 (m, 4H), 1.43 (m, 2H), 0.98 (t, J = 6.8, 3H). 13C NMR (400 MHz, MeOD): δ 170.6, 156.0, 137.7, 137.5, 128.6, 128.1, 113.3, 88.5, 84.1, 73.3, 69.8, 69.2, 56.1, 45.4, 42.9, 39.9, 39.1, 38.9, 37.9, 37.5, 34.3, 33.7, 31.1, 28.4, 25.3, 23.8, 13.3. m/z (M + ESI MS) found: 1,170.5773; calc for C53H88N9O16S2: 1,170.5790.
Compound 18
The PEGylated acid derivative 16 (12.4 mg, 15.1 μmol) and compound 6 (5.01 mg, 10.0 μmol) were dissolved in 0.9 mL DMF. DIEA (8.3 μL, 26.0 μmol) was added to the reaction mixture, and it was stirred for 10 min. Then, PyBOP (5.46 mg, 10.2 μmol) was added to the solution, and the mixture was stirred for 18 h at room temperature. The solvent was removed under vacuum, and the crude product was purified using a silica column (solvent gradient MeOH/CH2Cl2 0:100 to 20:80 in 45 min). The product 18, A2AAR antagonist monomer for Au complexation, was isolated as a white solid (9.0 mg, 7.0 μmol) in 70 % yield. 1H NMR (400 MHz, MeOD): δ 8.0 (s, 1H), 7.83 (s, 1H), 7.41 (d, 1H, J = 3.28 Hz), 7.0 (d, 2H, J = 8.44 Hz), 6.81 (d, 2H, J = 8.28 Hz), 6.63 (m, 1H), 4.38 (s, 2H), 4.33 (t, 2H, J = 6.7 Hz), 3.65–3.52 (PEG H), 2.57 (t, 3H, J = 7.42 Hz), 2.41 (m, 8H), 2.18 (m, 5H), 1.83 (m, 2H), 1.61 (m, 5H), 1.40 (m, 3H). m/z (M-ES MS) found: 1,244.5645; calc for C57H86N11O6S2: 1,244.5695.
Compound 20
3-Butyne 1-monophosphate 19 (20 mg, 0.059 mmol) [20] and PEGylated azide 12 (55 mg, 78 μmol) were dissolved in a 3:1 mixture of DMF and water (3.9 mL) in a round bottom flask, and TBTA (2.0 mg, 2.0 μmol) was added. Freshly prepared aqueous sodium ascorbate (1 M, 59 μL, 59 μmol) and 7.5 % aqueous copper sulfate pentahydrate solution (150 μL, 44 μmol) were added sequentially to the above mixture. The reaction mixture was stirred overnight at room temperature, and the solvent was evaporated. The residue was purified by semipreparative HPLC as described above to obtain homogeneous compound 20, phosphate-solubilizing monomer for Au complexation, using as mobile phase a gradient of triethylammonium acetate/CH3CN 100:0 to 80:20 in 20 min. 1H NMR (400 MHz, D2O): δ 8.01 (s, 1H), 4.62 (t, 1H,), 4.10 (m, 2H), 3.99 (t, 2H, J = 5.02 Hz), 3.71–3.62 (PEG H), 3.40 (t, 3H, J = 5.2 Hz,), 3.25 (m, 1H), 3.0 (m, 2H), 2.51 (m, 1H), 2.28 (t, 2H, J = 7.2 Hz), 2.00 (m, 1H), 1.76 (m, 1H), 2.62 (m, 4H), 1.43 (m, 3H). m/z (M-ES MS) found: 863.3543; calc for C34H64N4O15S2P: 863.3547.
Synthesis and characterization of AuNP conjugates
AuNP conjugate 21a
Synthesis
When the same procedure as for 21b (except redissolving the crude product in a water/methanol mixture (75:25)) was carried out using a 20:1 ratio of hydrogen tetrachloroaurate(III) tetrahydrate to each monomer 13 and 16, a larger AuNP conjugate, 21a, was obtained.
Purification process
The deep red-colored solution of the crude complex was filtered using a Millipore Amicon Ultra-15 centrifugal filter (30 K MW cutoff) and the solid remaining on the filter redissolved for five cycles. Each time, the residue was dissolved in 15 mL of water/methanol mixture (70:30) and refiltered. After the fifth filtration cycle, the residue was dissolved in water and the solution filtered for four additional cycles with a 50 K MW cutoff filter. Then, the residue retained on the filter was dissolved in 10 mL water and extracted five times with CHCl3 (15 mL each time). The solvent was removed from the last CHCl3 extract, and the organic phase residue was examined by mass spectroscopy to confirm that no small MW contaminants remained at detectable levels. After the final extraction step, the complex in the aqueous phase was lyophilized to obtain 21a, AuNP conjugate (larger diameter) containing an AR agonist and a carboxylate-solubilizing moiety, as a brown/black-colored residue.
AuNP conjugate 21b
Synthesis
Compounds 13 (2.51 mg, 2.12 μmol) and 16 (1.57 mg, 2.13 μmol) and DMF (3 mL) were added to a 25-mL round bottom flask. Hydrogen tetrachloroaurate(III) tetrahydrate (4.62 mg, 3 equiv., 12.2 μmol) was dissolved in 0.380 mL of water and added to the above flask, and the mixture was stirred for 30 min at room temperature. Sodium tetrahydroborate (1.37 mg, 36.1 μmol) in 2.25 mL water was added to the mixture dropwise. The pale yellowish color of the reaction mixture turned red immediately and was stirred for 4 h at room temperature. The solvent was evaporated at ~40 °C, and the residue containing 21b, AuNP conjugate (smaller diameter) containing an AR agonist, and a carboxylate-solubilizing moiety, was redissolved in 15 mL of water.
Purification process
The deep red-colored solution of the crude complex was filtered using a Millipore Amicon Ultra-15 centrifugal filter (30 K MW cutoff) and the solid remaining on the filter redissolved for five cycles. Each time, the residue was dissolved in 15 mL of water and refiltered. After the fifth filtration cycle, the residue was dissolved in water, and the solution was lyophilized. The lyophilized solid was dissolved in 1 mL of water and treated with 6 mL of THF, and the colloidal solution was centrifuged at 4,000 × g for 8 min to precipitate the AuNPs. This cycle of dissolving the residue in water followed by the addition of THF and finally centrifugation was repeated three times. The final residue was dissolved in water (2 mL) and transferred to a pre-weighed glass vial and lyophilized to obtain 21b, as a brown/black-colored residue. Subsequent bioassays of supernatant fractions from this procedure demonstrated that they were free of small MW contaminants binding to ARs.
General procedure for the synthesis of AuNP conjugate 22
Synthesis
Compounds 14 (2.10 mg, 1.06 μmol) and 16 (1.25 mg, 1.02 μmol) and DMF (2.4 mL) were added with stirring to a 25-mL round bottom flask. Hydrogen tetrachloroaurate(III) tetrahydrate (3.74 mg, 3 equiv., 9.04 μmol) was dissolved in 0.304 mL of water, and the solution was added to the above flask while stirring continuously for 30 min at room temperature. A solution of sodium tetrahydroborate (1.08 mg, 28.6 μmol) in 1.8 mL water was added to the mixture dropwise. The pale yellowish color of the reaction mixture turned red immediately and was stirred for 4 h at room temperature. The solvent was evaporated, and the residue containing 22, a xanthine AR antagonist AuNP conjugate containing a carboxylate-solubilizing moiety, was redissolved in 15 mL of a water/methanol mixture (75:25).
Purification process
The deep red-colored complex was filtered using Millipore Amicon Ultra-15 centrifugal filter (30 K MW cutoff) five times in succession. Each time, the residue was dissolved in 15 mL of water/methanol mixture (70:30). After the fifth time, the residue was dissolve in water and filtered four times with a 50 K MW cutoff filter. The residue was dissolved in 10 mL water and extracted five times with CHCl3 (15 mL). The solvent was removed from the final CHCl3 extract, which was subjected to mass spectroscopy and a competitive binding assay at the A2AAR to confirm that no starting material was left in the complex. After the final extraction step, the complex was lyophilized to obtain 22 as a brown/black-colored residue.
General procedure for the synthesis of AuNP conjugates 23 and 24
Synthesis
A mixture of compound 16 (1.80 mg, 2.01 μmol) and either 17 (2.36 mg, 2.02 μmol) or 18 (2.51 mg, 2.06 μmol) in DMF (23 mL) was added to a 100-mL round bottom flask. Hydrogen tetrachloroaurate(III) tetrahydrate (30.7 mg, 20 equiv., 81.0 μmol) dissolved in 2.46 mL of water was added to the above flask, and the mixture was stirred 30–45 min at room temperature. Sodium tetrahydroborate (9.12 mg, 242 μmol) in 14.7 mL water was added to the mixture slowly (using a dropping funnel over 30 min). The pale yellowish color of the reaction mixture turned deep red immediately, and the mixture was stirred for 4 h at room temperature. The solvent was evaporated, and the residue was dialyzed against water for 24 h, changing the water hourly. This residue contained either 23, a nucleoside AR agonist AuNP conjugate containing a carboxylate-solubilizing moiety, or 24, an A2AAR antagonist AuNP conjugate containing a carboxylate-solubilizing moiety.
Purification process
The deep red-colored complex was filtered using Millipore Amicon Ultra-15 centrifugal filter (30 K MW cutoff) five times in succession. Each time, the residue was dissolved in 15 mL of water/methanol mixture (70:30). After the fifth time, the residue was dissolved in water and filtered four times with a 50 K MW cutoff filter. The residue was then dissolved in 10 mL of water and extracted with CHCl3 (20 mL each, five times) until no starting material remained in the organic phase (each organic fraction was analyzed using mass spectroscopy), and aqueous phase was lyophilized to obtain 23 and 24.
AuNP conjugate 26a
Synthesis
Compounds 14 (2.51 mg, 2.01 μmol) and 20 (1.80 mg, 2.02 μmol) and DMF (23 mL) were added to a 100-mL round bottom flask. Hydrogen tetrachloroaurate(III) tetrahydrate (30.7 mg, 20 equiv., 80.2 μmol) dissolved in 2.46 mL of water, and the solution added to the above flask. The pale yellow mixture was stirred for 30–45 min at room temperature. A solution of sodium tetrahydroborate (9.02 mg, 0.24 mmol) in 14.7 mL water was added slowly to the mixture (using a dropping funnel over 30 min), and the color changed from pale yellow to deep red indicating the formation of the AuNP conjugate. The solvent was evaporated, and the residue was dialyzed against water for 24 h, changing the water hourly, to obtain 26a, a xanthine AR antagonist AuNP conjugate containing a phosphate-solubilizing moiety (larger diameter).
AuNP conjugate 26b
Synthesis
Compounds 14 (2.51 mg, 2.01 μmol) and 20 (1.80 mg, 2.02 μmol) and DMF (6 mL) were added to a 100-mL round bottom flask. Hydrogen tetrachloroaurate(III) tetrahydrate (9.22 mg, 6 equiv. 24.2 μmol) was dissolved in 0.76 mL of water, the solution was added to the above flask, and the mixture stirred for 30–45 min at room temperature. Sodium tetrahydroborate (2.74 mg, 72.0 μmol) in 4.5 mL water was added slowly (during 30 min) to the mixture, using a dropping funnel. The pale yellowish color of the reaction mixture turned deep red immediately, and the mixture was stirred for 4 h at room temperature. The solvent was evaporated, and the residue was dialyzed against water for 24 h, changing the water hourly, to obtain 26b, a xanthine AR antagonist AuNP conjugate containing a phosphate-solubilizing moiety (smaller diameter).
Purification process
Each residue was then dissolved in 10 mL of water and extracted with CHCl3 (20 mL each, five times), until no starting material remained in the organic phase (each organic fraction was analyzed successively using mass spectroscopy), and the aqueous phase was lyophilized to obtain compounds 26a and 26b.
Ultrapurification method
Solutions (4 μM) of the AuNP conjugates were prepared in 50 mM Tris–HCl (pH 7.5) and 10 mM MgCl2 buffer. Wash buffer solutions, whose concentrations of Tris–HCl, MgCl2, and DMSO matched those of the solutions of AuNPs, were also prepared for rinsing the centrifugal filters prior to ultrafiltration. Centrifugal filters (Millipore Ltd., Amicon Ultra-0.5, 10 K MW cutoff) were rinsed with 400 μL of wash buffer for 5 min at 14,000 × g, after which the filter units were placed into fresh microcentrifuge tubes.
Ultrafiltration of the colloidal solutions of AuNPs was then performed by adding 400 μL of each AuNP solution to a filter assembly and then spinning the filters for 15 min at 14,000 × g. The filtrate was transferred to a fresh microcentrifuge tube for analysis in a radioligand binding assay, and the solid material remaining on each filter was resuspended in 400 μL of buffer. The resuspended solution of AuNPs was collected by inverting and transferring the contents of the filter assembly to a fresh microcentrifuge tube and spinning the colloidal solution for 2 min at 1,000 × g to precipitate the larger particles that would interfere in the assay. Once collected, the remaining supernatant from each AuNP sample (approx. 20 μL) was diluted in a total of 400 μL of 50 mM Tris–HCl (pH 7.5) buffer containing 10 mM MgCl2. Finally, the fractions collected from the initial filter rinse and the ultrafiltration steps and the resuspended AuNPs were tested in a radioligand binding assay, as described below.
Calculation of the number of ligands per AuNP using NMR studies
NMR spectra of the AuNP conjugates (2–8 mg) were measured in D2O (300 μL). After that, to the same NMR tube, a known amount of DMF was added as an internal standard and the NMR peak intensities of the DMF formyl proton at 8.04 ppm and PEG methylene groups at 3.62 ppm were compared. The ratio was used to calculate the number of PEG-containing groups on average attached to each AuNP (see Supporting Information).
Assessment by analytical HPLC of the stability of the monomeric arms containing AR ligands (14, 18) in the presence of either CHO or human embryonic kidney 293 cell membranes
Assay solutions containing a monomeric arm (diluted from a 5-mM DMSO stock solution of either 14 or 18, 10 μM final concentration) and either Chinese hamster ovary (CHO) or human embryonic kidney 293 (HEK293) cell membrane suspensions (100 μg/mL) were prepared in 50 mM Tris–HCl (pH 7.5) and 10 mM MgCl2 buffer. The solutions in glass culture tubes (20 × 75 mm) were incubated at 25 °C for 1 h in a shaking water bath. Then, the solutions were transferred to 1.5 mL microcentrifuge tubes and centrifuged for 15 min at 14,000 × g to separate the cell membrane fragments from the solution. The supernatants of the solutions were then analyzed for degradation of the monomeric arms using an analytical Hewlett-Packard 1100 HPLC equipped with a Zorbax Eclipse 5-μm XDB-C18 analytical column (250 × 4.6 mm; Agilent Technologies Inc, Palo Alto, CA, USA). Mobile phase: linear gradient solvent system: 5 mM TBAP-CH3CN from 80:20 to 40:60 in 6.5 min; the flow rate was 1 mL/min system. Peaks (retention time was 3.22 min for 14 and 7.62 min for 18) were detected by UV absorption with a diode array detector at 254, 275, and 280 nm, and AUC was calculated based on the peak at 254 nm.
Competitive radioligand binding assays at ARs
Competition radioligand binding experiments were conducted to determine the binding affinities of conjugated AuNPs (see Supporting Information). A range of concentrations of either AuNPs or monomeric arms between 0.1 nM and 10 μM was tested in competing for binding to human (h) A3 or A2AARs on cell membranes derived from A3AR-expressing CHO cells or A2AAR-expressing HEK293 cells. The radioligands used were [125I]I-AB-MECA (N6-(4-amino-3-iodobenzyl)-5′-N-methylcarboxamidoadenosine) and [3H]CGS21680 (2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine) for A3AR and A2AAR binding, respectively, by methods described [17]. Selected compounds were tested at the hA1AR using A1AR-expressing CHO cells and [3H]PIA (N6-R-phenylisopropyladenosine) as the radioligand [17]. For some of the larger AuNP derivatives, an increase in radioactivity bound to the glass fiber filter was observed with increasing concentrations of AuNPs, which precluded determination of a binding curve.
Assessment by radioligand binding of the stability of a representative AuNP conjugate (26a) in the presence of HEK293 cell membranes
A solution containing compound 26a, AR antagonist AuNP conjugate containing a phosphate-solubilizing group, from an H2O stock solution (1 μM, final volume 250 μM) and membranes of control HEK293 cells not expressing an AR (100 μg/mL) was prepared in 50 mM Tris–HCl (pH 7.5) and 10 mM MgCl2 buffer (binding buffer). In addition, a 250-μL solution containing only control HEK293 cell membranes and another one containing control HEK293 cell membranes in the presence of 10 μM nonselective agonist 5′-N-ethylcarboxamidoadenosine (NECA) were prepared. These solutions in glass culture tubes (20 × 75 mm) were incubated for 1 h at 25 °C in a shaking water bath. Then, the solutions were transferred to 1.5 mL microcentrifuge tubes and centrifuged for 20 min at 14,000 × g to pellet the cell membrane fragments from the solutions. The supernatants of the solutions were run through centrifugal filter units (Millipore Ltd., Amicon Ultra-0.5, 10 K MW cutoff) to collect filtrates (approximate volume of each filtrate 220 μL). Then, the material remaining on the centrifugal filter units (approximate volume 30 μL) was collected and resuspended in binding buffer to the starting volume (250 μL). Then, the filtrates and recovered materials were each tested for binding activity in a radioligand binding assay using cell membranes from HEK293 cells stably expressing the hA2AAR, as described above.
Results
Design and chemical synthesis
We have explored strategies for conjugating small molecular ligands for ARs to AuNPs with retention of biological activity. A3AR agonists 1 and 2 have shown similar hA3AR binding affinities (Ki ~ 1 nM) and have been used widely as pharmacological probes [13]. A ring-constrained North (N)-methanocarba derivative of 2 was recently reported to be a highly selective A3AR agonist, MRS3558 (3), having a Ki value in binding to the hA3AR of 0.3 nM [12]. Tosh et al. modified 3 at the adenine C2 position with a dialkyne to form 4 that retained hA3AR affinity (Ki = 23 nM) and was suitable for linking at the terminal alkyne with an azide through a 2 + 3 cycloaddition (click) reaction [17]. We also used a moderately potent and selective A2AAR agonist, CGS21680 (5), a C2 derivatized adenosine 5′-carboxamide, for immobilization on AuNPs through condensing its terminal carboxylic acid with an amine. Nevertheless, it is recognized that the A2AAR selectivity may be limited because the hA3AR affinity of this nucleoside is substantial. Selective A2AAR antagonists, pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine-5-amine (PTP) derivatives, were explored for chain functionalization to arrive at amine functionalized congener 6 to provide a means of covalent attachment by amide formation of sterically bulky groups without losing the ability to bind to the A2AAR with high affinity [21]. The structure–activity relationship of xanthine derivatives as AR antagonists has been exhaustively explored [22]. Heptynoyl-XAC (7), an analogue of the 8-phenyl derivative xanthine amine congener (XAC), is relatively potent but nonselective in binding to hARs [19]. This terminal alkyne can be reacted with azides by click chemistry with retention of AR affinity, as was shown previously with dendrimer conjugates [19].
In Fig. 2, we describe the synthesis of PEGylated derivatives of a selective A3AR agonist 13 and nonselective AR antagonist 14, for AuNP conjugation from a commercially available polyethylene glycol (PEG) azide 11 and TA 10 via amide coupling to obtain 12 [18, 23]. The key triazole compounds, 13 and 14, were obtained after a click reaction of azide 12 and an alkyne, e.g., 4 or 7 [17].
Fig. 2.

Functionalization of A3AR agonist 4 and AR antagonist 7 for coupling to AuNPs. Reagents and conditions: a DCC, DMAP, DCM, 0 °C, 2 h, rt, overnight; b (1 eq) sodium ascorbate, (0.5 eq) CuI, H2O/DMF (1:3), TBTA, rt, overnight
PEGylated derivatives of the selective A2AAR agonist 5 and antagonist 6, armed with TA for AuNP conjugation (compounds 17 and 18, respectively) were synthesized according to Fig. 3. The PEGylated azide 12 was reduced to its amine 15 using a Staudinger reaction, followed by coupling with succinic anhydride in pyridine to provide the carboxyl-terminated compound 16 [18]. The coupling of compounds 5 with 15 and 6 with 16 using PyBOP and DIEA gave the desired intermediates 17 and 18.
Fig. 3.

Functionalization of A2AAR agonist 5 and antagonist 6 for coupling to AuNPs. Reagents and conditions: a PPh3, THF, H2O; b succinic anhydride, pyridine; c PyBOP, DIEA, DMF, overnight
Initial preparation of AuNPs coated with our agonists/antagonists alone resulted in particles with low aqueous solubility. This prompted the use of solubilizing “doping” agents in the nanoparticle synthesis. An obvious choice was the carboxylic acid-terminated linker 16 (previously synthesized in Fig. 3). In addition, we prepared the monophosphoryl linker 20 for increasing aqueous solubility from azide 12 and alkynyl phosphate 19 using click chemistry (Fig. 4). Both molecules 16 and 20 alone did not show any AR binding activity.
Fig. 4.
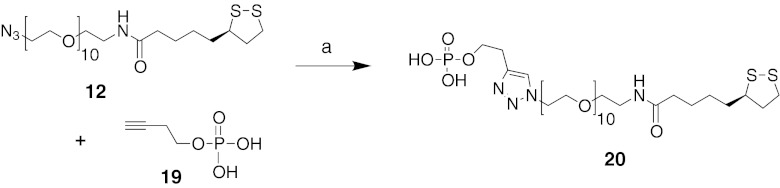
Synthesis of a water-solubilizing ligand. Reagents and conditions: a sodium ascorbate (1 eq), CuSO4, (0.5 eq), H2O/DMF (1:3), rt, overnight
The synthesis of AuNPs coated with AR ligands 13, 14, 17, and 18 along with solubilizing carboxylate chain 16 is shown in Fig. 5. Alternately, ligand 14 was also coated on AuNPs that were doped with solubilizing monophosphate 20 (Fig. 6). The diameters of the AuNPs were 1.8–4.5 nm, as shown by transmission electron microscopy (Fig. 7a–f). This size range was chosen because it was small enough for imaging purposes, and similarly sized AuNPs have shown slower kinetics of cellular uptake than larger particles [24]. The anchor point of the tethered GPCR ligands consisted of a TA moiety containing two thiol groups to achieve a relatively stable bond with the nanoparticle [37]. A segment of ten PEG units attached to the carboxylic group of the thioctic acid contributed to water solubility and was intended to allow the interaction of the immobilized GPCR ligand with the adaptable binding site of the GPCR. In a typical reaction, an equimolar amount of the AR ligand and either carboxylate 16 or monophosphate 20 and 3 M equivalents of tetrachloroauric(III) acid (HAuCl4⋅3H2O) in DMF were reduced with 6 M equivalents of NaBH4 [10]. Using this synthetic approach, we expected (a) to achieve the ability to functionalize the AuNPs with multiple ligands, (b) to increase the solubility of the AuNPs by attaching a water-solubilizing ligand, and (c) to favorably alter the AuNP surface with anionic ligands containing COOH groups to lower the cellular uptake of the particles.
Fig. 5.

Coupling of AR agonists 13 and 17 and antagonists 14 and 18 to AuNPs. For the A3AR agonist, both large diameter 21a (4–5 nm) and smaller diameter 21b (~2 nm) conjugates were prepared. Reagents and conditions: a HAuCl4, NaBH4, DMF
Fig. 6.

Coupling of AR antagonist 14 to AuNPs. Reagents and conditions: a HAuCl4, NaBH4, DMF
Fig. 7.

Characterization of AuNP conjugates: TEM images (average AuNP diameter) of a 21b. Scale bar = 10 nm (2.00 ± 0.07 nm). b 22. Scale bar = 20 nm (2.56 ± 0.09 nm). c 23. Scale bar = 20 nm (3.0 ± 0.12 nm). d 24. Scale bar = 20 nm (4.24 ± 0.10 nm). e 26a. Scale bar = 20 nm (4.51 ± 0.07 nm). f 26b. Scale bar = 10 nm (1.89 ± 0.12 nm). g UV–visible absorption spectra for AuNP conjugates in H2O, and h DLS profiles of 24, 26a
The GPCR ligand-conjugated AuNPs were characterized using UV–visible absorbance spectroscopy, TEM, and NMR. Figure 7g shows the UV–visible absorption spectra collected from a set of AuNP samples with different tethered ligands; some representative TEM images collected from these samples are also shown. The absorption spectra show that at low Au-to-ligand (smaller size NPs) molar ratios, the characteristic shape-dependent surface plasmon absorption band (SPB) located at 520 nm is weak (22 (2.5 nm diameter) and 23 (3 nm)). However, the well-defined and narrow SPB peaks at higher Au/ligand molar ratios (relatively larger NPs (24 (4.2 nm) and 26a (4.5 nm))) indicated a narrow size distribution of these AuNPs. DLS studies confirmed that 24 and 26a have a larger diameter and a narrow size distribution (Fig. 7h).
The stoichiometry of substitution was determined using an NMR method [42] and compared to the calculated substitution based on reactants. The AuNPs were examined for the presence of noncovalently attached small molecules that could complicate the biological characterization by leaching from the particles. These noncovalently attached small molecules were removed via filtration and/or extraction techniques. For several conjugates, because of complications in the binding assay, it was necessary to prepare two sizes of AuNP conjugates, i.e., both large diameter (21a and 26a, 4–5 nm) and smaller diameter (21b and 26b, ~2 nm).
Pharmacological characterization
AR ligand-bearing AuNPs were characterized pharmacologically for their binding affinities and functional effects in mammalian cells expressing three subtypes of hARs. Radioligand binding assays were carried out using standard 3H and 125I-labeled nucleosides (Table 1) [17, 21]. The membrane preparations were prepared from CHO cells (A1 and A3) or HEK293 cells (A2A) stably expressing a hAR subtype. The solubilizing moieties alone 16, 20 or as an AuNP conjugate 25, 27 did not bind appreciably to ARs. Before testing the AuNP conjugates, we ascertained that low MW components of significant biological activity (i.e., that passed through ultrafiltration with 10–30 kDa cutoff) were not present. Also, the stability of the monomeric ligands with attached tether (14, 18) in the presence of HEK293 cell membranes was demonstrated (Supporting Information).
Table 1.
AR binding affinity of AuNP conjugates and intermediates
| Compound | A2AAR, Ki (nM) or % inhibition, 1 μM | A3AR, Ki (nM) or % inhibition, 1 μM | Average no. of ligands per AuNP |
|---|---|---|---|
| AR ligand monomers | |||
| 13 | ND | 37.4 ± 9.0 | N/A |
| 14 | 1,420 ± 570 | 3,040 ± 1,050 | N/A |
| 17 | 44.0 ± 3.0 | 245 ± 18 | N/A |
| 18 | 61.0 ± 3.0 | 4,060 ± 830 | N/A |
| Au conjugates of AR ligands | |||
| 21a | ND | NDa | 600 |
| 21b | ND | 14.8 ± 1.8 | 42 |
| 22 | 21 % | 262 ± 29 | 36 |
| 23 | 28 % | 240 ± 39 | 36 |
| 24 | 34.9 ± 3.5 | ND | 140 |
| 26a | 69.0 ± 9.0 | NDa | 770 |
| 26b | 370 ± 40 | 750 ± 120 | 20 |
| Solubilizing moieties and Au conjugates | |||
| 16 | 0 % | 0 % | N/A |
| 20 | 0 % | 0 % | N/A |
| 25 | 30 % | 0 % | N/A |
| 27 | 28 % | NDa | N/A |
Using standard radioligands (A1: [3H]N6-R-phenylisopropyladenosine, A2A: [3H]2-[p-(2-carboxyethyl)-phenylethylamino]-5′-N-ethylcarboxamido-adenosine, or A3: [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyl-uronamide) [17, 25]. Values shown as mean ± SEM were for three determinations done in duplicate. The concentration of the AuNP was used in the calculation, not the concentration of individual pharmacophoric units. Three compounds were tested at A1 (Ki, nM): 24, 211; 26a, 211; 18, 35 % inhibition at 10 μM
ND not determined, N/A not applicable
aTotal binding in the A3AR assay increased with increasing concentration of ligand for 21a and 26a (larger diameter particles) likely because of trapping of the AuNPs in the filter, but this phenomenon did not occur with 21b and 26b (smaller diameter particles)
The A3AR affinity of 2-alkynyl adenosine conjugate 21b of smaller diameter (Ki 14.8 nM) was increased by twofold in comparison to the monomeric agonist ligand 13. A Ki value for the corresponding large diameter particle 21a at the A3AR could not be obtained because of an unexpected increase of radioligand bound, rather than inhibition. The A2AAR affinity of monomeric PTP antagonist derivative 18 was also well preserved in the corresponding AuNP conjugate 24 (Ki 34.9 nM). Binding of 24 at the A1AR was also shown. However, the moderate A2AAR affinity (Ki 1.42 μM) of XAC monomeric antagonist derivative 14 was not maintained in the AuNP conjugate 22 containing carboxylate water-solubilizing groups, but the A3AR affinity of 22 (Ki 262 nM) was 12-fold greater than the monomeric precursor 14. The XAC conjugate containing phosphate water-solubilizing groups 26a (large diameter) did bind potently to both A1 (Ki 211 nM) and A2AARs (Ki 69 nM). A Ki value for this large diameter particle 26a at the A3AR could not be obtained because of increased radioligand bound. The smaller diameter AuNP conjugate 26b was shown to bind to both A2A and A3ARs with submicromolar Ki values. Thus, the phosphate water-solubilizing moiety was advantageous for retaining receptor recognition. The A2AAR agonist conjugate 23 was inactive in binding to that receptor. Similarly linked derivatives of 17 on quantum dots also failed to bind appreciably to the A2AAR [23], suggesting that there might be differences in the tolerance of the various AR subtypes for solid particle attachment. However, 23 bound to the A3AR with a Ki of 240 nM, suggesting a difference between the A2AAR and A3AR structures in the steric restrictions for binding conjugates.
Functional data were determined in an assay consisting of hA3AR-induced inhibition of the production of cAMP in membranes of CHO cells expressing the hA3AR [38]. The C2 alkynyl AuNP conjugate 21b was a full agonist at the A3AR, showing a relative efficacy of 100 % (relative to inhibition by 10 μM NECA). A full concentration response curve in hA3AR-mediated inhibition of adenylate cyclase provided an EC50 value of 13.6 nM (Fig. 8), i.e., identical to its binding affinity.
Fig. 8.

Functional agonism by 21b in an assay of adenylate cyclase in CHO cells expressing the hA3AR (EC50 value 13.6 nM). The activity of 10 μM 5′-N-ethyluronamidoadenosine represented 100 % efficacy. The experiment was repeated three times, and the average is shown
Discussion
GPCRs are a superfamily of cell surface signaling receptors having important roles in many physiological functions and disease states, including cancer and cardiovascular disease [12]. Many of the currently approved drugs that target GPCRs achieve excellent therapeutic benefits. Therefore, the development of new GPCR-based drugs is a well-validated and ongoing process in the development of therapies for many diseases including cancer [11]. There is increasing interest in the therapeutic potential of selective AR agonists/antagonists for treating a wide range of diseases including cancer.
The ARs are overexpressed in various tumor cells, and their activation by certain selective agonist ligands has been shown to inhibit tumor growth via a range of signaling pathways. The A3AR can be found in different types of tumor cells such as K562 human leukemia, Jurkat lymphoma, U937 human monocytic/macrophagic cells, Nb2 rat lymphoma, A375 human melanoma, PGT-beta mouse pineal gland tumor cells, human glioblastoma, and human prostatic cells [26–29]. The A3AR expression level is high in human melanoma, colon, breast, small cell lung, and pancreatic carcinoma as compared to normal tissue, suggesting upregulation of the A3AR in cancer growth. The A3AR agonists were found to inhibit growth of various tumors in vivo. These findings of A3AR overexpression in different tumor cell types suggest that this receptor may be a specific target in novel cancer treatment.
The A2AAR is expressed on many lymphoid cells, including neutrophils, monocytes, macrophages, T cells, and natural killer cells [13]. Activation of this receptor by adenosine or adenosine analogues (A2AAR agonist) results in a wide range of anti-inflammatory and immunosuppressive responses [12, 13]. In vitro studies showed A2AAR agonists to protect against excessive inflammation and tissue injury in the liver, heart, kidney, and vasculature. The A2AAR is overexpressed in the striatum and olfactory tubercle of the brain. The A2AAR is coupled to stimulation of adenylyl cyclase, and its antagonism mediates many of the CNS stimulatory effects of caffeine in humans [13]. Other potential therapeutic applications of selective A2AAR agonists are as anti-aggregatory, anti-psychotic, and anti-Huntington’s disease agents.
Nanoparticles such as AuNPs could offer many advantages in the development of therapeutics due to their multivalent binding abilities and biocompatibility. Toxicity of AuNPs depends on their size, shape, surface functionality, surface charge, and hydrodynamic radius [39, 40]. The low in vivo toxicity of AuNPs, especially of relatively small diameter, has been described [39]. Although many in vitro toxicity studies of various AuNPs have been done, more in vivo studies are needed [40]. We envision the use of these AuNP conjugates for both diagnostic and therapeutic purposes. The presence of the attached Au particle could affect the action of the drug molecules by altering pharmacokinetic or biodistribution properties. The nanometer size of these particles may either help to avoid penetration of the tethered drug to certain tissues in the body or it may enhance the exposure of larger particles to certain tissues, such as tumors by the enhanced permeability and retention (EPR) effect [1]. Targeting extracellular GPCRs that are known to be overexpressed in tumor cells, such as ARs, with ligand-conjugated NPs may have an added advantage for targeted therapy over the EPR effect alone. Many studies have shown that attaching a targeted ligand to NPs enhances their biodistribution within solid tumors [30, 31]. Thus, tethering an AR ligand that may have its own anticancer activity, such as an A3AR agonist or A2AAR antagonist [32], could serve both targeting and therapeutic purposes. The methodology introduced in this study for AR agonists and antagonists could potentially be applied to other GPCR ligands.
Moreover, AuNPs have been extensively developed for selectively inducing hyperthermia for localized ablation of cancerous tumors. Thus, AuNPs are delivered to a tumor either by active or passive targeting, and the tumor then is exposed to the radiation such as NIR laser light, radiowaves, or an alternating magnetic field [33–36]. Another very important property of AuNPs is that they can be used as imaging agents for cancer detection. The versatile optical properties of AuNPs have enabled optical imaging of cells with a wide variety of contrast mechanisms.
In the present study, we have demonstrated the feasibility of coupling functionalized small GPCR ligands to AuNPs with retention of high affinity binding to the receptor. As a first example of this approach, we selected agonist and antagonist ligands for A2A and A3ARs. We functionalized AuNPs by tethering specific ligands, agonists and antagonists, of ARs to the gold surface as models for cell surface interactions with GPCRs. The AuNP conjugates with chain-extended AR ligands alone (PEGylated nucleosides and nonnucleosides, anchored to the Au via thioctic acid) were found insoluble in water due to hydrophobic entities in the ligand. Therefore, we added a second, biologically inactive pendant moiety, to increase the water solubility, consisting of a PEGylated chain terminating in a carboxylic or phosphate group. Pharmacological receptor binding studies on these GPCR ligand-derivatized AuNPs (2–5 nm in diameter) performed using membranes of mammalian cells stably expressing hA1, A2A, and A3ARs, showed that the desired selectivity was retained with Ki values (nanomolar) of A3AR agonist 21b and A2AAR antagonists 24 and 26a of 14 (A3), 34 (A2A), and 69 (A2A), respectively. The corresponding monomers displayed Ki values of 37, 61, and 1,420 nM, respectively. Thus, the A2AAR affinity of the antagonist conjugate 26a was 21-fold greater than the xanthine monomer 14. The monomer 18 of antagonist conjugate 24 was 67-fold selective for the A2AAR in comparison to the A3AR, and its AuNP conjugate had 2-fold greater affinity than the monomer. Also, the nucleoside conjugate 21b potently activated the A3AR.
Several parallel synthetic approaches were used to prepare new PEGylated derivatives via conjugating the above AR ligands to TA-PEG-N312, TA-PEG-NH215, or TA-PEG-NHCO(CH2)2COOH 16 with click or amide coupling chemistry. TA groups and PEG chains were known to increase the water solubility, but AuNP conjugates of a PEGylated adenosine derivative were found insoluble in water due to aromatic and aliphatic entities in the ligand. Addition of a second, negatively charged ligand (TA-PEG-NHCO(CH2)2COOH 16 or monophosphate tether 20) together with the PEGylated AR ligand increased the nanoparticle water solubility and allowed biological evaluation. TEM images revealed that the gold core had an average diameter of ∼2 nm for complex 21b (Fig. 7a). The diameter of the AuNPs could be reliably controlled by the initial molar ratio of Au to the combination of AR ligand monomer and solubilizing moiety, if present, during the preparation. This ratio varied from 3 (e.g., smaller particle 21b) to 20 (e.g., larger particle 21a) [41].
Purification of multivalent AuNP conjugates was very challenging because one is limited mainly to dialysis, filtration, or centrifugation. Additionally, the effect from untethered/trapped small molecules on the AuNP conjugates can affect the biological results. We found that purification of AuNP conjugates via dialysis and/or filtration did not eliminate all small MW contamination. However, extraction of the AuNPs with CHCl3 or THF (for 21b) was sufficient to remove such contaminants prior to biological evaluation.
In conclusion, we have introduced a strategy for applying the potential of nanotechnology for targeted drug delivery for GPCRs. These encouraging initial results—that GPCR affinity can be maintained in AuNP conjugates with appropriate functionalization—need to be followed by in vivo pharmacokinetic studies. By combining nanomedicine approaches based on AuNPs targeting specific GPCRs that are associated with or are known to be dysregulated in disease states, it may be possible to enhance the functionality of such conjugates over the parent small molecule GPCR ligands. Although we have demonstrated the feasibility of coupling small AR agonists and antagonists to AuNPs, in some cases with retention of high affinity and/or selectivity, this strategy could become a general approach for a wide range of GPCR ligands. The feasibility of performing such imaging studies using AuNPs functionalized with ligands for ARs and other GPCRs can be carried out initially in in vitro studies. Such conjugates may eventually prove useful in the diagnosis or treatment of cancer and other diseases.
Electronic supplementary material
Supporting information with details of the AuNP characterization and biological and stability assays is available online at http://. (DOCX 11420 kb)
Acknowledgments
We thank Dr. Noel Whittaker (NIDDK) for MS measurements. This research was supported by the Intramural Research Programs of NIDDK and NCI, National Institutes of Health.
Abbreviations
- AR
Adenosine receptor
- DLS
Dynamic light scattering
- DMSO
Dimethylsulfoxide
- EDC
N-ethyl-N′-dimethylamino-propylcarbodiimide
- cAMP
Adenosine 3′,5′-cyclic phosphate
- CHO
Chinese hamster ovary
- Cl-IB-MECA
2-Chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamido-adenosine
- DCC
N,N′-dicyclohexylcarbodiimide
- DMAP
4-Dimethylaminopyridine
- DCM
Dichloromethane
- DIEA
Diisopropylethylamine
- DMF
Dimethylformamide
- DMEM
Dulbecco’s modified Eagle’s medium
- AuNP
Gold nanoparticle
- GPCR
G protein-coupled receptor
- HEK
Human embryonic kidney
- CGS21680
2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamido-adenosine
- MS
Mass spectrometry
- NECA
5′-N-ethylcarboxamidoadenosine
- PTP
Pyrazolo[4,3-e][1, 2, 4]triazolo[1,5-c]pyrimidine-5-amine
- PyBOP
Benzotriazol-1-yloxy-tripyrrolidino-phosphonium hexafluorophosphate
- SPB
Surface plasmon absorption band
- TEM
Transmission electron microscopy
- THF
Tetrahydrofuran
- I-AB-MECA
N6-(4-amino-3-iodobenzyl)-adenosine-5′-N-methyluronamide
- PEG
Polyethylene glycol
- PIA
N6-(2-phenylisopropyl)adenosine
- Tris
2-amino-2-hydroxymethyl-propane-1,3-diol
References
- 1.Arvizo R, Bhattacharya R, Mukherjee P. Gold nanoparticles: opportunities in nanomedicine. Expert Opin Drug Deliv. 2010;7:753–763. doi: 10.1517/17425241003777010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chitrani DB. Intracellular uptake, transport, and processing of gold nanostructures. Mol Membr Biol. 2010;27:299–311. doi: 10.3109/09687688.2010.507787. [DOI] [PubMed] [Google Scholar]
- 3.Huang X, Peng X, Wang Y, Shin DM, El-Sayed MA, Nie S. A reexamination of active and passive tumor targeting by using rod-shaped gold nanocrystals and covalently conjugated peptide ligands. ACS Nano. 2010;4:5887–5896. doi: 10.1021/nn102055s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khlebtsov N, Dykman L. Biodistribution and toxicity of engineered gold nanoparticles: a review of in vitro and in vivo studies. Chem Soc Rev. 2011;40:1647–1671. doi: 10.1039/c0cs00018c. [DOI] [PubMed] [Google Scholar]
- 5.Han G, Ghosh P, Rotello VM. Functionalized gold nanoparticles for drug delivery. Nanomedicine (London) 2007;2:113–123. doi: 10.2217/17435889.2.1.113. [DOI] [PubMed] [Google Scholar]
- 6.Cai W, Gao T, Hong H, Sun J. Applications of gold nanoparticles in cancer. Nanotechnol Sci Appl. 2008;1:17–32. doi: 10.2147/NSA.S3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boisselier E, Astruc D. Gold nanoparticles in nanomedicine: preparations, imaging, diagnostics, therapies and toxicity. Chem Soc Rev. 2009;38:1759–1782. doi: 10.1039/b806051g. [DOI] [PubMed] [Google Scholar]
- 8.Patel PC, Giljohann DA, Daniel WL, Zheng D, Prigodich AE, Mirkin CA. Scavenger receptors mediate cellular uptake of polyvalent oligonucleotide-functionalized gold nanoparticles. Bioconjug Chem. 2010;21:2250–2256. doi: 10.1021/bc1002423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bardhan R, Chen W, Perez-Torres C, Bartels M, Huschka RM, Zhao LL, Morosan E, Pautler RG, Joshi A, Halas NJ. Nanoshells with targeted simultaneous enhancement of magnetic and optical imaging and photothermal therapeutic response. Adv Funct Mater. 2009;19:3901–3909. doi: 10.1002/adfm.200901235. [DOI] [Google Scholar]
- 10.Li L, Zhang Q, Liu A, Li X, Zhou H, Liu Y, Yan B. Proteome interrogation using nanoprobes to identify targets of a cancer-killing molecule. J Am Chem Soc. 2011;133:6886–6889. doi: 10.1021/ja111137n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lappano R, Maggiolini M. G protein-coupled receptors: novel targets for drug discovery in cancer. Nat Rev Drug Discov. 2011;10:47–60. doi: 10.1038/nrd3320. [DOI] [PubMed] [Google Scholar]
- 12.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nat Rev Drug Discov. 2006;5:247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller C. Nomenclature and classification of adenosine receptors—an update. Pharmacol Rev. 2011;63:1–34. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Madi L, Bar-Yehuda S, Barer F, Ardon E, Ochaion A, Fishman PA. A3 adenosine receptor activation in melanoma cells. J Biol Chem. 2003;278:42121–42130. doi: 10.1074/jbc.M301243200. [DOI] [PubMed] [Google Scholar]
- 15.Yu L, Coelho JE, Zhang X, Fu Y, Tillman A, Karaoz U, Fredholm BB, Weng Z, Chen JF. Uncovering multiple molecular targets for caffeine using a drug target validation strategy combining A2A receptor knockout mice with microarray profiling. Physiol Genomics. 2009;37:199–210. doi: 10.1152/physiolgenomics.90353.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith NJ, Bennett KA, Milligan G. When simple agonism is not enough: emerging modalities of GPCR ligands. Mol Cell Endocrinol. 2011;331:241–247. doi: 10.1016/j.mce.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 17.Tosh DK, Chinn M, Yoo LS, Kang DW, Luecke H, Gao ZG, Jacobson KA. 2-Dialkynyl derivatives of (N)-methanocarba nucleosides: “clickable” A3 adenosine receptor-selective agonists. Bioorg Med Chem. 2010;18:508–517. doi: 10.1016/j.bmc.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Susumu K, Uyeda HT, Medintz IL, Pons T, Delehanty JB, Mattoussi H. Enhancing the stability and biological functionalities of quantum dots via compact multifunctional ligands. J Am Chem Soc. 2007;129:13987–13996. doi: 10.1021/ja0749744. [DOI] [PubMed] [Google Scholar]
- 19.Kecskés A, Tosh DK, Wei Q, Gao ZG, Jacobson KA. GPCR ligand dendrimer (GLiDe) conjugates: adenosine receptor interactions of a series of multivalent xanthine antagonists. Bioconjug Chem. 2011;22:1115–1127. doi: 10.1021/bc1005812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das A, Ko H, Burianek LE, Barrett MO, Harden TK, Jacobson KA. Human P2Y14 receptor agonists: truncation of the hexose moiety of uridine-5′-diphosphoglucose and its replacement with alkyl and aryl groups. J Med Chem. 2010;53:471–480. doi: 10.1021/jm901432g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar TS, Mishra S, Deflorian F, Yoo SL, Phan K, Kecskés M, Szabo A, Shinkre B, Gao ZG, Trenkle W, Jacobson KA. Molecular probes for the A2A adenosine receptor based on a pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine scaffold. Bioorg Med Chem Lett. 2011;21:2740–2745. doi: 10.1016/j.bmcl.2010.11.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baraldi PG, Tabrizi MA, Gessi S, Borea PA. Adenosine receptor antagonists: translating medicinal chemistry and pharmacology into clinical utility. Chem Rev. 2008;108:238–263. doi: 10.1021/cr0682195. [DOI] [PubMed] [Google Scholar]
- 23.Das A, Sanjayan GJ, Kecskés M, Yoo L, Gao ZG, Jacobson KA. Nucleoside conjugates of quantum dots for characterization of G protein-coupled receptors: strategies for immobilizing A2A adenosine receptor agonists. J Nanobiotechnol. 2010;8:11–40. doi: 10.1186/1477-3155-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verma A, Stellacci F. Effect of surface properties on nanoparticle–cell Interactions. Small. 2010;6:12–21. doi: 10.1002/smll.200901158. [DOI] [PubMed] [Google Scholar]
- 25.Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7:759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gessi S, Varani K, Merighi S, Cattabriga E, Iannotta V, Leung E, Baraldi PG, Borea PA. A3 adenosine receptors in human neutrophils and promyelocytic HL60 cells: a pharmacological and biochemical study. Mol Pharmacol. 2002;61:415–424. doi: 10.1124/mol.61.2.415. [DOI] [PubMed] [Google Scholar]
- 27.Gessi S, Merighi S, Varani K, Borea PA. Adenosine receptors in health and disease. Adv Pharmacol. 2011;61:41–75. doi: 10.1016/B978-0-12-385526-8.00002-3. [DOI] [PubMed] [Google Scholar]
- 28.Jajoo S, Mukherjea D, Watabe K. Adenosine A3 receptor suppresses prostate cancer metastasis by inhibiting NADPH oxidase activity. Neoplasia. 2009;11:1132–1145. doi: 10.1593/neo.09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fishman P, Bar-Yehuda S, Synowitz M, Powell JD, Klotz KN, Gessi S, Borea PA. Adenosine receptors and cancer. Handb Exp Pharmacol. 2009;193:399–441. doi: 10.1007/978-3-540-89615-9_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pirollo KF, Chang EH. Does a targeting ligand influence nanoparticle tumor localization or uptake? Trends Biotechnol. 2008;26:552–558. doi: 10.1016/j.tibtech.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 31.Hu-Lieskovan S, Heidel JD, Bartlett DW, Davis ME, Triche TJ. Sequence-specific knockdown of EWS-FLI1 by targeted, nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing's sarcoma. Cancer Res. 2005;65:8984–8992. doi: 10.1158/0008-5472.CAN-05-0565. [DOI] [PubMed] [Google Scholar]
- 32.Ohta A, Kjaergaard J, Sharma S, Mohsin M, Goel N, Madasu M, Fradkov E, Sitkovsky M. In vitro induction of T cells that are resistant to A2 adenosine receptor-mediated immunosuppression. Br J Pharmacol. 2009;156:297–306. doi: 10.1111/j.1476-5381.2008.00019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kennedy LC, Bickford LR, Lewinski NA, Coughlin AJ, Hu Y, Day ES, West JL, Drezek RA. A new era for cancer treatment: gold-nanoparticle-mediated thermal therapies. Small. 2011;7:169–183. doi: 10.1002/smll.201000134. [DOI] [PubMed] [Google Scholar]
- 34.Hirsch LR, Stafford RJ, Bankson JA, Sershen SR, Rivera B, Price RE, Hazle JD, Halas NJ, West JL. Nanoshell-mediated near-infrared thermal therapy of tumors under magnetic resonance guidance. Proc Natl Acad Sci U S A. 2003;100:13549–13554. doi: 10.1073/pnas.2232479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gannon CJ, Patra CR, Bhattacharya R, Mukherjee P, Curley SA. Intracellular gold nanoparticles enhance non-invasive radiofrequency thermal destruction of human gastrointestinal cancer cells. J Nanobiotechnol. 2008;6:2. doi: 10.1186/1477-3155-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johannsen M, Gneveckow U, Eckelt L, Feussner A, Waldofner N, Scholz R, Deger S, Wust P, Loening SA, Jordan A. Clinical hyperthermia of prostate cancer using magnetic nanoparticles: presentation of a new interstitial technique. Int J Hyperthermia. 2005;21:637–647. doi: 10.1080/02656730500158360. [DOI] [PubMed] [Google Scholar]
- 37.Henderson LC, Altimari JM, Dyson G, Servinis L, Niranjan B, Risbridger GP. A comparative assessment of α-lipoic acid N-phenylamides as non-steroidal androgen receptor antagonists both on and off gold nanoparticles. Bioorg Chem. 2011;40:1–5. doi: 10.1016/j.bioorg.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 38.Nordstedt C, Fredholm BB. A modification of a protein-binding method for rapid quantification of cAMP in cell-culture supernatants and body fluid. Anal Biochem. 1990;189:231–234. doi: 10.1016/0003-2697(90)90113-N. [DOI] [PubMed] [Google Scholar]
- 39.Longmire M, Choyke PL, Kobayshi H. Clearance properties of nano-sized particles and molecules as imaging agents: considerations and caveats. Nanomedicine. 2008;3:703–717. doi: 10.2217/17435889.3.5.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aillon KL, Xie Y, El-Gendy N, Berkland CJ, Forrest ML. Effects of nanomaterial physicochemical properties on in vivo toxicity. Adv Drug Deliv Rev. 2009;61:457–466. doi: 10.1016/j.addr.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oh E, Susumu K, Goswami R, Mattoussi H. One-phase synthesis of water-soluble gold nanoparticles with control over size and surface functionalities. Langmuir. 2010;26:7604–7613. doi: 10.1021/la904438s. [DOI] [PubMed] [Google Scholar]
- 42.Manea F, Bindoli C, Polizzi S, Lay L, Scrimin P. Expeditious synthesis of water-soluble, monolayer-protected gold nanoparticles of controlled size and monolayer composition. Langmuir. 2008;24:4120–4124. doi: 10.1021/la703558y. [DOI] [PubMed] [Google Scholar]
- 43.Cohen S., Stemmer SM, Zozulya G, Ochaion, Patoka R, Barer F, Bar-Yehuda S, Rath-Wolfson L, Jacobson KA, Fishman P. (2011) CF102 an A3 adenosine receptor agonist mediates anti-tumor and antiinflammatory effects in the liver. J Cell Physoil 226:2438–2447 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information with details of the AuNP characterization and biological and stability assays is available online at http://. (DOCX 11420 kb)