

Abstract
Li-Fraumeni syndrome (LFS) is a rare, inherited syndrome associated with increased risk of various early-onset tumors. Since the introduction of classic LFS criteria, various criteria have been proposed to include patients with incomplete LFS features, which make up Li-Fraumeni-like syndromes (LFL). Germline missense mutations of TP53 are the primary cause of LFS and LFL. Mutations mostly reside in the DNA-binding domain of the gene and have a dominant-negative effect (DNE) over alternate wild-type alleles. Germline TP53 mutation c.566C>T results in the missense mutation GCC (Ala) to GTC (Val) at codon 189 (A189V) and has been reported in a case of multiple primary colon tumors. Herein we report a second case of the same mutation in a breast cancer patient, who has familial history of late-onset malignancies. Due to the relatively late onset of malignancies, neither case fulfils previously defined criteria for the syndrome. Mutational analysis for breast tissue in this patient showed a loss of heterozygosity. These clinical features may suggest a relatively weak DNE of A189V compared to other TP53 mutations, and in silico predictions and in vitro findings of the function of A189V mutant protein are conflicting. Considering the increased risk of malignancies and the therapeutic implications for patients who have a TP53 mutation, care must be taken when treating those who are suspected of possessing cancer-prone traits due to TP53 mutation, especially when there is a family history of late-onset cancer with low penetrance.
Keywords: Li-Fraumeni syndrome, LFS, Li-Fraumeni-like syndrome, LFL, TP53, Breast cancer, Late onset, Penetrance
INTRODUCTION
Li-Fraumeni syndrome (LFS, OMIM #151623) is a rare, inherited syndrome with high penetrance, especially in women. It is associated with increased risk of various early-onset tumors. Since the introduction of the syndrome in 1969 by Li and Fraumeni [1], various criteria have been developed to include those with incomplete LFS features, referred to as Li-Fraumeni-like syndromes (LFL) [2-6].
Tumor suppressor gene TP53 (chromosome 17p13.1; OMIM #191170) encodes the transcription factor p53, which plays a fundamental role in the tumor suppressor signaling pathway. The gene is activated in response to various types of cellular stresses, and p53 stops replication of stressed cells by binding to specific DNA sequences and transactivating genes [7]. Germline mutations of TP53 are the primary cause of syndromes that include tendency to cancer, demonstrated in 70-80% of individuals fulfilling classic LFS criteria and 20-40% of those fulfilling LFL criteria [8].
Germline TP53 mutation c.566C>T causes a change from GCC (Ala) to GTC (Val) at codon 189 (A189V). This mutation was reported in a case of concurrent multiple primary colon tumors [9]. Although the patient was suspected to be prone to cancer, because of unusually late onset and absence of family history of cancer, none of the clinical criteria for the syndrome was met. We report a case of a germline A189V mutation and discuss TP53 mutations with low penetrance.
CASE REPORT
A 47-yr-old woman presented to a local hospital with a painless lump in the right breast that had been present for 1 month. She had no history of other breast complaints or surgeries. Her menarche occurred at age 14 yr and she gave birth to 3 children. There was no family history of breast or ovarian cancer, but 2 of her family members died from late-onset cancers (Fig. 1). On physical examination, a poorly defined, firm mass was noted in the medial upper portion of her right breast, measuring approximately 3 cm, and no axillary or cervical lymphadenopathy was detected. Mammography demonstrated a lesion highly suspicious of cancer, and histological examination of the core needle biopsy of the lesion revealed invasive ductal carcinoma (IDC) of the breast. The patient was then transferred to our hospital for further evaluation and management of the cancer. Results for complete cell count, blood chemistry, and tumor markers (carcinoembryonic antigen; 0.8 ng/mL, cancer antigen 15-3; 5.3 U/mL) were all within reference intervals. Screening for metastatic disease by computed tomography and positron emission tomography showed no evidence of adjacent lymphatic, pulmonary, bone, or hepatic metastases.
Fig. 1.

Pedigree of the TP53 A189V family. Generations are in Roman numerals and individuals are in Arabic numbers (without parenthesis). Individuals with a clinical diagnosis of a cancer are presented with a solid symbol and a footnote regarding the specific cancer type (age of onset). Individuals without a history of cancer are presented with open symbols. The proband is marked by an arrow.
Abbreviation: IDC, invasive ductal carcinoma.
A pathologist reviewed slides from the other hospital and confirmed the diagnosis of IDC. The patient then underwent a partial mastectomy of the right breast with ipsilateral, axillary sentinel lymph node biopsies. The final pathology of the tumor was IDC, and the cancer was staged as pT2N0M0. The specimen showed comedo-type necrosis with calcification and had a maximum diameter of 2.2 cm. The Nottingham classification [10] was II (moderately differentiated), and the total point score was 7 (2-3-2). The tumor cells stained positive for p53, and results for Ki67 were positive in 5% of the tumor cells. The carcinoma displayed no nuclear reactivity for estrogen receptors, progesterone receptors, or HER-2. Biopsy findings for 7 sentinel lymph nodes were all negative. Although frozen sections, removed during the surgery, were negative for malignancy, final pathology revealed malignant infiltrations in the deep layers of the inferior and medial margins.
Given the extensive family history of cancer, samples of peripheral blood (PB) lymphocytes and tumor tissue were assessed for TP53 mutation. Briefly, DNA was extracted from breast cancer tissue and mononuclear cells from PB, and 5 to 9 TP53 exons and intron-exon boundaries were studied. Analysis of PB revealed a germline heterozygotic A189V mutation that was homozygous in the IDC specimen (Fig. 2). Later, her 3 offsprings underwent genetic screening, and 2 daughters (aged 20 and 16 yr) were found to carry the mutation (Fig. 1 and 2) despite absence of signs or symptoms of cancer. Patients (or their parents, if younger than 14 yr) gave informed consent for all molecular testing.
Fig. 2.

Nucleotide sequence analysis of the TP53 gene of family members. DNA-segments include the coding sequence 566 of the TP53 gene (marked by black arrows). The patient had a compound heterozygous missense mutation in exon 6 (c.566C>T; p.Ala189Val) and IDC tissue was homozygous for the mutation. The patient's 2 daughters (III-1 and III-2) were found to be heterozygous carriers for the mutation, whereas the patient's son (III-3) had a wild type TP53 gene.
Abbreviations: PB, peripheral blood; IDC, invasive ductal carcinoma.
After confirmation of the patient's TP53 mutation status, an interdisciplinary panel met to determine the utility of localized radiotherapy, based on the suspected radiosensitivity of individuals harboring a deleterious mutation in the TP53 gene [11, 12]. As partial mastectomy is generally followed by radiotherapy to increase the probability of tumor-free resection margins, risk of cancer relapse was determined to be greater than the risk of secondary carcinogenesis due to radiation. The patient has completed 4 cycles of adjuvant chemotherapy consisting of doxorubicin (60 mg, 32.8 mg/m2) and cyclophosphamide (500 mg, 273.2 mg/m2), and 6,400 cGy of adjuvant radiotherapy is planned. The patient is currently well, without any signs or symptoms of cancer recurrence.
DISCUSSION
Majority (75%) of TP53 mutations are missense substitutions [13]. Thus, understanding structure-function relationships is useful for comprehending the consequence of a TP53 mutation. The protein product p53 contains 393 amino acids constituting multiple functional domains. The core region (amino acids 96-292) serves the most crucial functions as a DNA-binding domain (DBD), which recognizes cognate sequences in the regulatory region of the target gene [14]. Mutation in the DBD would disrupt DNA binding ability of p53, and for this reason, mutations in the domain are assumed to be pathogenic. The International Agency for Research on Cancer (IARC) TP53 database (version R15, released in 2010, http://www-p53.iarc.fr/index.html) [15] indicates that most germline missense mutations (1,394, or 79.4%) are also located in the DBD. Codon 189 resides in the L2 loop motif of the DBD, which is necessary for the binding of p53 to the minor groove of the DNA. In silico assessment of gene function, using both SIFT and Align-GVGD, predicted A189V to be deleterious [16]. However, experimental data in yeast demonstrated that the A189V mutant p53 retained fair transactivity compared with wild-type in vitro [17], complicating the prediction of its function in vivo.
Genetic study of a well-designed control group is crucial to distinguish whether a missense nucleotide substitution is a polymorphism or a potentially deleterious mutation. Control subjects who are already past the mean age of onset are needed for genetic studies of late-onset cancers [18]. Currently, there are no studies of TP53 in elderly Korean individuals who are confirmed to be cancer free and without any known family history of cancer. Yet, molecular tests for TP53 of 693 sporadic Korean breast cancer patients without family history of cancer (median age of 49 yr, range 24-91 yr) found no case of A189V (unpublished data).
As p53 naturally binds DNA as a homotetramer, presence of a mutant protein interferes with a wild-type protein by forming hetero-oligomers, which might reduce transcriptional activity (dominant negative effect; DNE). Over 80% of mutated p53 are predicted to feature such an effect [13], in which case carcinogenesis will begin early in life. In contrast, among mutations lacking a DNE, a single allelic mutation would not be sufficient to cause oncogenesis; alteration of another modifier gene would be necessary. Determination of loss of heterozygosity (LOH) status within tumors has been reported to be important in carriers of a heterozygous TP53 mutation [19]. However, only a few p53 mutants have been studied with respect to their DNE, and A189V has been studied in none. Given the lack of experimental data, clinical observations of time of disease onset and accompanying genetic alterations would be helpful in assessing the DNE of the mutant.
The penetrance of TP53-mediated cancer is generally high, especially in women, because of breast cancer [8]. Patients with typical LFS develop sarcomas at a young age, including breast cancers and brain tumors at less than 44 yr [20]. According to the IARC TP53 database, 13 somatic and 1 germline A189V mutations have been identified. The previously reported case of a germline A189V mutation occurred in a patient with concurrent colon carcinomas at a late age without a familial history of cancer [9]. Somatic APC and/or KRAS mutations were found in all tumors, and, of note, the advanced carcinoma had an additional somatic mutation in the alternate TP53 allele (Table 1). In our case, the proband's breast cancer developed at a relatively late age compared with other breast cancers in carriers having a TP53 mutation [20]. The patient's father and half-brother both died from cancer that developed at a late age. However, the significance of this is unknown, given the lack of information about their genotypes. Genetic information about the patient's healthy siblings was not available. The patient's breast IDC tissue also demonstrated LOH (Fig. 2), possibly because of loss of the alternate allele. Segregational analysis was not possible because of incomplete genetic studies of family members; LOH in cancer tissues of the patient is the available evidence of pathogenicity. Also, clinical features of both cases of A189V mutation suggest that the DNE of A189V may be relatively weak compared with other, typical germline TP53 mutations.
Table 1.
Germline TP53 mutation c.566C>T, p.Ala189Val from the literature review and the present case
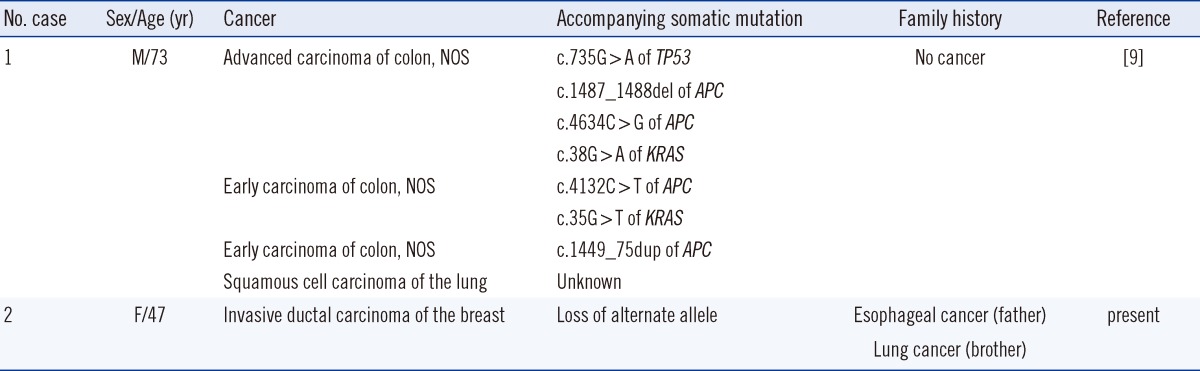
Abbreviations: M, male; F, female; NOS, not otherwise specified.
Li and Fraumeni first described LFS in 1969 based on retrospective analysis of families with soft tissue sarcomas during infancy or childhood [1]. The spectrum of cancers typical of TP53 mutations has been identified as breast cancer, sarcomas, brain tumors, adrenocortical carcinoma, and leukemia [21-23]. Studies in search of specific genotype-phenotype associations in subjects with TP53 mutation found a significant association between missense mutations in the L2 and L3 loops and brain tumors [20]. Both A189V cases exhibited varied spectrums of cancers without a predilection for a certain type of cancer, and no brain tumors were found. With respect to breast cancer and the syndrome, a recent study revealed a significantly higher prevalence of HER2-positive tumors in subjects with the mutation compared to those who did not have the mutation [24]. However, the breast IDC in our case tested negative for HER2.
Radiotherapy has been demonstrated to play a small role in secondary carcinogenesis, with increasing doses of radiation for breast cancer treatment [11], and an even higher risk of developing radiation-induced malignancies was noted in breast cancer patients with germline TP53 mutations [12]. Yet, there are no generally accepted contraindications for any type of radiation-based treatment or diagnostic modality. Physicians should assess the safety of radiotherapy with regard to possible benefits and risks. In our case, malignant cells were present in resection margins of the partial mastectomy specimen and the conclusion of interdisciplinary consultation was that local radiation therapy with adjuvant chemotherapy outweighed risks associated with the use of radiation.
The final issue we address is the clinical utility of diagnostic criteria for cancer-prone syndrome in carriers of TP53 germline mutations. Since the introduction of classic LFS criteria [1], supplementary LFL criteria have been suggested [2, 3]. Chompret et al. [4-6] proposed modified criteria that demonstrated maximum clinical utility in combination with classic LFS [23]. However, approximately 4% of families harboring TP53 mutations are missed with the current criteria [23]. Neither of the A189V cases fulfills any of the criteria, mainly because of the late onset of cancer and the lack of information about specific cancers in familial histories. As discussed here, certain TP53 mutations lack a DNE, delaying clinical manifestations and reducing disease penetrance. Carriers having such mutations may not be indicated for TP53 testing and miss the opportunity for cancer screening and optimal treatment. Thus, cancer-prone traits should be considered even in patients with late-onset cancers and low penetrance in their family, because TP53 mutation may be present despite absence of currently adopted LFS or LFL criteria.
Footnotes
No potential conflicts of interest relevant to this article were reported.
References
- 1.Li FP, Fraumeni JF., Jr Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med. 1969;71:747–752. doi: 10.7326/0003-4819-71-4-747. [DOI] [PubMed] [Google Scholar]
- 2.Birch JM, Hartley AL, Tricker KJ, Prosser J, Condie A, Kelsey AM, et al. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res. 1994;54:1298–1304. [PubMed] [Google Scholar]
- 3.Eeles RA. Germline mutations in the TP53 gene. Cancer Surv. 1995;25:101–124. [PubMed] [Google Scholar]
- 4.Chompret A, Brugières L, Ronsin M, Gardes M, Dessarps-Freichey F, Abel A, et al. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer. 2000;82:1932–1937. doi: 10.1054/bjoc.2000.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chompret A, Abel A, Stoppa-Lyonnet D, Brugières L, Pagés S, Feunteun J, et al. Sensitivity and predictive value of criteria for p53 germline mutation screening. J Med Genet. 2001;38:43–47. doi: 10.1136/jmg.38.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tinat J, Bougeard G, Baert-Desurmont S, Vasseur S, Martin C, Bouvignies E, et al. 2009 version of the Chompret criteria for Li Fraumeni syndrome. J Clin Oncol. 2009;27:e108–e109. doi: 10.1200/JCO.2009.22.7967. author reply e110. [DOI] [PubMed] [Google Scholar]
- 7.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 8.Nagy R, Sweet K, Eng C. Highly penetrant hereditary cancer syndromes. Oncogene. 2004;23:6445–6470. doi: 10.1038/sj.onc.1207714. [DOI] [PubMed] [Google Scholar]
- 9.Miyaki M, Iijima T, Ohue M, Kita Y, Hishima T, Kuroki T, et al. A novel case with germline p53 gene mutation having concurrent multiple primary colon tumours. Gut. 2003;52:304–306. doi: 10.1136/gut.52.2.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elston CW, Ellis IO. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology. 1991;19:403–410. doi: 10.1111/j.1365-2559.1991.tb00229.x. [DOI] [PubMed] [Google Scholar]
- 11.Rubino C, de Vathaire F, Shamsaldin A, Labbe M, Lê MG. Radiation dose, chemotherapy, hormonal treatment and risk of second cancer after breast cancer treatment. Br J Cancer. 2003;89:840–846. doi: 10.1038/sj.bjc.6601138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heymann S, Delaloge S, Rahal A, Caron O, Frebourg T, Barreau L, et al. Radio-induced malignancies after breast cancer postoperative radiotherapy in patients with Li-Fraumeni syndrome. Radiat Oncol. 2010;5:104. doi: 10.1186/1748-717X-5-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26:2157–2165. doi: 10.1038/sj.onc.1210302. [DOI] [PubMed] [Google Scholar]
- 14.Hainaut P, Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res. 2000;77:81–137. doi: 10.1016/s0065-230x(08)60785-x. [DOI] [PubMed] [Google Scholar]
- 15.Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 16.Mathe E, Olivier M, Kato S, Ishioka C, Vaisman I, Hainaut P. Predicting the transactivation activity of p53 missense mutants using a four-body potential score derived from Delaunay tessellations. Hum Mutat. 2006;27:163–172. doi: 10.1002/humu.20284. [DOI] [PubMed] [Google Scholar]
- 17.Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, et al. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci U S A. 2003;100:8424–8429. doi: 10.1073/pnas.1431692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collins JS, Schwartz CE. Detecting polymorphisms and mutations in candidate genes. Am J Hum Genet. 2002;71:1251–1252. doi: 10.1086/344344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dearth LR, Qian H, Wang T, Baroni TE, Zeng J, Chen SW, et al. Inactive full-length p53 mutants lacking dominant wild-type p53 inhibition highlight loss of heterozygosity as an important aspect of p53 status in human cancers. Carcinogenesis. 2007;28:289–298. doi: 10.1093/carcin/bgl132. [DOI] [PubMed] [Google Scholar]
- 20.Olivier M, Goldgar DE, Sodha N, Ohgaki H, Kleihues P, Hainaut P, et al. Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res. 2003;63:6643–6650. [PubMed] [Google Scholar]
- 21.Li FP, Fraumeni JF, Jr, Mulvihill JJ, Blattner WA, Dreyfus MG, Tucker MA, et al. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988;48:5358–5362. [PubMed] [Google Scholar]
- 22.Garber JE, Goldstein AM, Kantor AF, Dreyfus MG, Fraumeni JF, Jr, Li FP. Follow-up study of twenty-four families with Li-Fraumeni syndrome. Cancer Res. 1991;51:6094–6097. [PubMed] [Google Scholar]
- 23.Gonzalez KD, Noltner KA, Buzin CH, Gu D, Wen-Fong CY, Nguyen VQ, et al. Beyond Li Fraumeni Syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol. 2009;27:1250–1256. doi: 10.1200/JCO.2008.16.6959. [DOI] [PubMed] [Google Scholar]
- 24.Melhem-Bertrandt A, Bojadzieva J, Ready KJ, Obeid E, Liu DD, Gutierrez-Barrera AM, et al. Early onset HER2-positive breast cancer is associated with germline TP53 mutations. Cancer. 2012;118:908–913. doi: 10.1002/cncr.26377. [DOI] [PMC free article] [PubMed] [Google Scholar]