

I. Introduction
1. Background
Heparin, first isolated in 1917, was found to be highly effective as an anticoagulant, and within two decades, it was being used clinically.1–4 Besides their anticoagulation activities, heparin and the related heparan sulfate (HS) play important roles in a wide range of biological functions such as cell differentiation, viral infection, and cancer metastasis.5–12
Heparin is a member of the glycosaminoglycan (GAG) family, which ranges from the unsulfated polymer hyaluronan to chondroitin and dermatan sulfates, and to the most complex examples, heparin and HS.13 Heparin and HS share the basic disaccharide components, composed of D-glucosamine (GlcN) α-(1→4)-linked to a uronic acid (Scheme 1A). The GlcN component has a high degree of variability, as its O-6 and O-3 positions can be free or sulfated, and the amino group can be sulfated, acylated, or unmodified. The uronic acid can be either D-glucuronic acid (D-GlcA) or its C-5 epimer, L-iduronic acid (L-IdoA), both of which can be sulfated at the O-2 position.
Scheme 1.
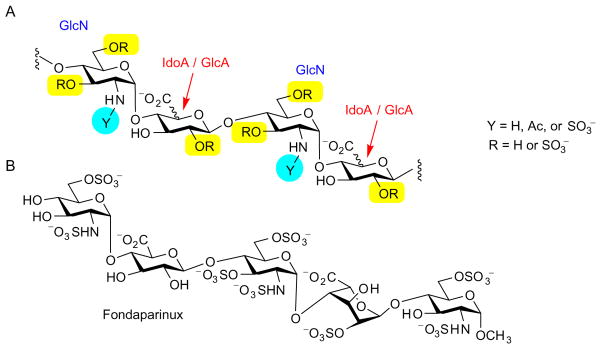
(A) Structures of heparin/HS; (B) structure of fondaparinux (Arixtra®). (Note idose and iduronic acid are arbitrarily presented in the 1C4 conformation following common usage in the field. This does not necessarily represent the conformations in solution of the various heparin derivatives depicted throughout the article.)
Heparin and HS are differentiated by their tissue location and their detailed structures. Heparin has a higher degree of sulfation, with around 2.7 sulfate groups per disaccharide unit, and contains about 90% of its uronic acid as L-IdoA. Heparin is selectively synthesized in mast cells, whereas HS is omnipresent on cell surfaces and in the extracellular matrix as part of the proteoglycan complex.14 More prevalent and heterogeneous, HS has on average one sulfate group per disaccharide, but it includes areas of high sulfation and swaths of unsulfated disaccharides.15 The backbone sequence of HS is also more varied, in that the uronic acid residue is around 40% L-IdoA, with the major entity being D-GlcA.5,16
Although the naturally occurring heparin/HS is an exceedingly heterogeneous mixture, its interactions with biological receptors can be highly specific, as is evident from its binding to antithrombin III (ATIII).17 Thorough structural analysis has demonstrated that the oligosaccharide sequence in heparin responsible for ATIII binding is a rare pentasaccharide fragment that is sulfated at O-3 in the middle GlcN component.3,17–20 Removal of this O-3 sulfate group diminished its antithrombin affinity 10,000-fold.21,22 The understanding of this structure–activity relationship led to the development of the drug fondaparinux (trade name: Arixtra, Scheme 1B), a fully synthetic pentasaccharide approved by the US Food and Drug Administration for the treatment of deep-vein thrombosis.21
Despite the success in establishing the ATIII-binding site, the heterogeneities of heparin and HS from natural sources present a major challenge in obtaining sufficient quantities of pure materials for the determination of detailed structure–activity relationships. To overcome this limitation, a frequently employed strategy has involved chemical modification of natural heparin and HS. However, this approach can give a complex mixture of many partially modified products from incomplete reactions.23,24 Thus, the synthesis of pure and homogenous oligosaccharide sequences of the parent heparin and HS polysaccharides becomes crucial in facilitating biological studies. Commercially, Arixtra has been prepared by pure chemical synthesis, which is an impressive accomplishment considering there are over 50 synthetic steps. For Arixtra synthesis and other synthetic work prior to 2000, the reader should refer to several excellent reviews.21,25–29 This article focuses on the advancement of heparin and HS component synthesis since 2000.
2. Challenges in Synthesis of Oligosaccharides of Heparin and HS
The synthesis of heparin/HS oligosaccharides presents a major challenge. Multiple factors must be considered for a successful synthetic design. These include (a) synthetic access to L-iduronic acid and L-idose; (b) the choice of uronic acid or the corresponding pyranoside as building blocks; (c) formation of the 1,2-cis linkage from the GlcN donor; (d) suitable protecting-group strategy to install sulfate groups at desired locations; and (e) methods used for elongation of the backbone sequence.
a. Preparation of L-Iduronic Acid and L-Idose
L-Iduronic acid (L-IdoA) and the corresponding idopyranosides are not available from natural sources in large quantities and must be synthesized. There has been much research in order to access L-IdoA and its derivatives efficiently. Many approaches start from the commercially available 1,2:5,6-di-O-isopropylidene-α-D-glucofuranose (1), followed by the inversion of the configuration at C-5 through formation of an L-ido epoxide as in 3 (Scheme 2A).30–32 Other routes employing compound 1 involve oxidation of the 5-hydroxyl group to aldehyde 4 through a three-step process, followed by stereo-selective addition of a cyano group (Scheme 2B) or elimination of the primary hydroxyl group with subsequent hydroboration to invert the stereochemistry at C-5 (compound 6 in Scheme 2C).33–36
Scheme 2.
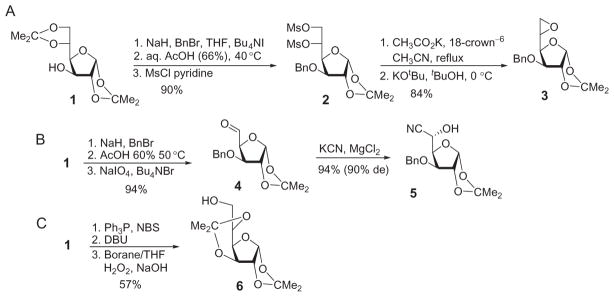
Various routes for inverting D-glucose to L-idose derivatives.
Alternative routes to L-IdoA have been reported.32–45 As an example, Seeberger and coworkers have spearheaded research in the de novo synthesis of L-IdoA. Early work from their laboratory started from L-arabinose, but the low selectivity in the Mukaiyama aldol reaction with aldehyde 7 resulted in a low overall yield (6%) (Scheme 3A).39 Starting from D-xylose and switching the aldol reaction to a more-selective cyanation furnished the L-IdoA building block 11 in 24% overall yield (Scheme 3B).38 However, despite the many routes developed toward the preparation of L-IdoA or L-idose,32–45 the synthesis of heparin/HS oligosaccharides remains difficult as long as 8–12 synthetic steps are required for the preparation of a single monosaccharide building block.
Scheme 3.
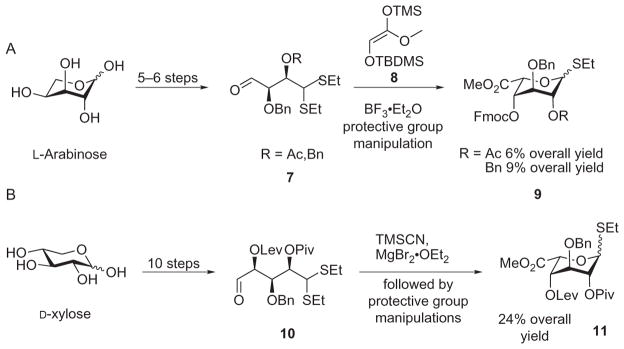
Recent routes to monosaccharide precursors of L-IdoA.
b. The Choice of Uronic Acid Versus Pyranoside as Building Blocks
As glycosyl donors based on uronic acids can potentially be epimerized during their preparation, and they are typically less reactive than the corresponding glycopyranosides, the latter are commonly used as surrogate glycosyl donors. However, this approach requires adjustment of the oxidation state on the oligosaccharide after its assembly. As the size of the oligosaccharide increases, high-yielding oxidation can become very difficult.46,47 Early syntheses relied on the Jones oxidation or the use of similar chromium reagents, which are toxic and frequently give low yields of the desired products (Scheme 4A).31,48–51 This problem was subsequently overcome by using the mild TEMPO-mediated oxidations, which are typically effected with a co-oxidant such as NaOCl52–54 or iodobenzene diacetate (BAIB; Scheme 4B),55,56 and this can be followed by Pinnick oxidation to achieve high yields (Scheme 4C).46,47,57,58 Alternatively, glycopyranosides could be used to prepare disaccharide intermediates as precursors for longer oligosaccharides by taking advantage of the high anomeric reactivity of the pyranoside donors. Adjustment of the oxidation state can then be performed on the disaccharide through oxidation at C-6 of the nonreducing end to the uronic acid, thus avoiding a late-stage oxidation of the more-valuable larger oligosaccharides (Scheme 4D).59,60
Scheme 4.

Conversion of glycopyranosides into uronic acids in synthesis of heparin/HS oligosaccharides.
The monosaccharide glucuronic and iduronic acids, suitably derivatized, can be used directly as donors. Sinaÿ’s synthesis of the ATIII-binding pentasaccharide used uronic acid-based glycosyl bromide donors, which gave glycosylation yields typically around 50%.30,31 The availability of newer glycosylation methods and an understanding of the effects of protecting groups on anomeric reactivities have potentially circumvented this issue.61,62 Bonnaffé and coworkers synthesized the disaccharide building block 22 in 75% yield by using the bromide donor 20 (Scheme 5A). The yield was increased to 91% employing the trichloroacetimidate donor 23 (Scheme 5B).41,63 The resultant disaccharide was then used in a highly convergent manner to afford a dodecasaccharide derivative that was used for the synthesis of an HS proteoglycan analogue (see Scheme 18).64
Scheme 5.
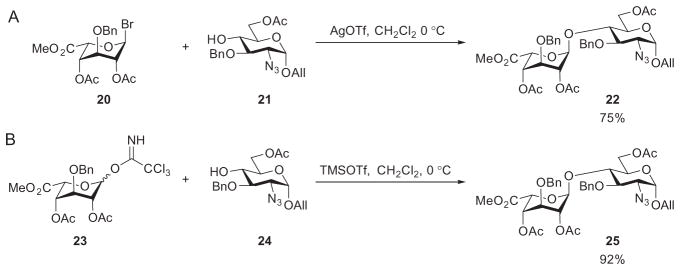
Comparison of glycosyl bromide and trichloroacetimidate donors in glycosylation.
Scheme 18.

Active–latent synthesis of dodecamer 109.
c. Stereochemical Control in Glycosylation
Stereochemical control is a crucial issue in the synthesis of heparin/HS components. The 1,2-trans linkage from the uronic acid to glucosamine is usually achieved through use of a participating group at the 2-position of the uronic acid. However, formation of the 1,2-cis linkage from the glucosamine donor can be difficult to control. The azido group, as a nonparticipating functionality, is widely employed as a precursor for the nitrogen atom at C-2 of glucosamine.65 Such 2-azido glucosamine precursors can lead to the thermodynamically more-stable α glycosides.29 This route generally provides high stereoselectivities in reactions with L-idosyl acceptors. However, for D-glucuronic acid-based acceptors, anomeric mixtures often result from the glycosylation, and this requires fine tuning of protecting groups to achieve high stereoselectivities.58,66 For example, substituting the 4-benzyl ether in donor 26 by a 4-t-butyldimethylsilyl ether (donor 29) led to formation of the α-linked disaccharide 28b exclusively (Scheme 6A and B).58 Bulky protecting groups at O-6 of the glucosamine component have also been explored to decrease the proportion of β anomer formed.55 In addition to the protecting groups, the conformation of the acceptor can play an important role in determining the stereochemical outcome of the glycosylation. While glycosylation of pentenyl glycoside 31 with trichloroacetimidate 30 gave the disaccharide derivative 32 with an α:β ratio of 3:1 (Scheme 6C), locking the glucuronic acid component into the 1C4 conformation (33) led to exclusive α selectivity (Scheme 6D).67,68 However, caution needs to be taken in extrapolating these results to the assembly of larger oligosaccharides. Thus, glycosylation of the L-idosyl-configured disaccharide derivative 36 by tetrasaccharide donor 35 led to hexasaccharide 37 as an inseparable anomeric mixture (Scheme 6E).69 The stereochemical outcome of the glycosylation reaction needs to be investigated individually, especially in the formation of large oligosaccharides.
Scheme 6.

Strategies for enhancing stereoselectivity in glycosylation.
d. Protecting-Group Strategy
In addition to their roles in dictating stereochemistry, protecting groups are widely used to control the location of sulfate groups. With the high level of functionality in heparin/HS oligosaccharides, and the large number of protecting groups employed, syntheses must be suitably designed to prevent the premature removal of a protecting group.
To establish protecting groups suitable for regioselective sulfation, the Hung group explored the possibility of synthesizing all 48 possible heparin/HS disaccharide structures (disaccharide derivatives 41 and 42), starting from eight monosaccharide building blocks (38–40) that are strategically protected.70 The benzoyl group was used to protect those hydroxyl groups to be sulfated, and benzyl ethers were employed as persistent protecting groups for hydroxyl groups that would remain free in the final oligosaccharide products. The TBDPS substituent temporarily masked the primary hydroxyl group on compound 39 to permit subsequent oxidation to glucuronic acid. The azido group could be selectively reduced by Staudinger reduction and then either acetylated or sulfated, while the benzyloxycarbonylamino (Cbz) group could be deprotected to generate the free amine upon the final hydrogenolysis step. This panel of 48 disaccharide derivatives (compounds 41 and 42) will be very useful for the assembly of heparin/HS libraries (Scheme 7).
Scheme 7.
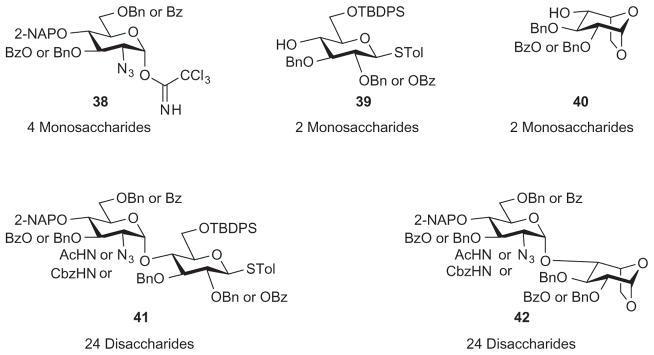
Synthesis of all potential heparin/HS disaccharides from eight monosaccharide precursors.
Instead of preparing multiple monosaccharides, Wei and coworkers synthesized the glucuronic acid-containing HS disaccharide 43 having each hydroxyl group orthogonally protected (Scheme 8). Each protecting group could be removed selectively without affecting others. The newly liberated hydroxyl group was sulfated, and other protecting groups were then removed to ensure that the sulfate groups were stable under each set of deprotection conditions.71 This strategy allowed the divergent synthesis of multiple sulfation patterns from a single backbone but required more synthetic steps to remove the various protecting groups remaining after sulfation. As the biologically active heparin/HS domains typically are pentasaccharides or longer, these protecting-group strategies need to be extended to the synthesis of longer oligosaccharides.
Scheme 8.

Disaccharide derivative 43 can be orthogonally deprotected for sulfation at various locations.
Sulfate groups have traditionally been installed after assembly of the oligosaccharide backbone. However, late-stage sulfation, especially with larger oligosaccharides, can be quite capricious and challenging. Low yields72 and incomplete reactions64,73 are common. As an alternative, the sulfate groups can be installed on building blocks as protected esters prior to glycosylation. Numerous sulfate esters have been developed,74–76 and Huang and coworkers investigated the utility of 2,2,2-trichloroethyl (TCE) sulfates57 as developed by the Taylor group. TCE sulfates are stable to common transformations encountered in oligosaccharide synthesis, and the deprotection conditions are very mild.77 An additional benefit of using TCE was that both sulfated and unsulfated building blocks can be derived from a common intermediate, thus increasing the efficiency of the overall process. For example, deprotection of the primary O-acetyl group in disaccharide derivative 51 followed by treatment with the sulfuryl imidazolium salt 52 provided the sulfate ester 53 (Scheme 9A).57 The disaccharide derivative 51 was also used for conversion into the nonsulfated acceptor 57 (Scheme 9B). The presence of sulfate ester groups in the building blocks did not significantly affect the glycosylation yield, as reaction of the sulfate ester donor 54 with the acceptor 57 gave tetrasaccharide derivative 58 in 82% yield (Scheme 9C). The sulfate ester-containing tetrasaccharide 59 also functioned as a competent acceptor, as it underwent glycosylation by donor 54 in 70% yield (Scheme 9D). The tetrasaccharide 59 was successfully deprotected, giving rise to the HS tetrasaccharide component 61 (Scheme 9E), demonstrating the compatibility of TCE sulfate esters in the synthesis of heparin/HS oligosaccharides.
Scheme 9.

Evaluation of TCE sulfate ester-containing donors and acceptors in glycosylation reactions.
With the foregoing general understanding of synthesis of heparin/HS components, the following sections focus on the recent development of strategies to form and extend the heparin/HS oligosaccharide backbone. The discussions are grouped according to the strategy utilized.
II. Linear Synthesis
1. Solution Phase
The linear approach is one of the earliest strategies in oligosaccharide synthesis and is the route employed by Nature to produce heparin/HS.13 Chemical glycosylation involves the activation of a donor, followed by nucleophilic attack of the activated donor on the acceptor to form a new glycosidic linkage. In the linear approach toward heparin/HS oligosaccharides, the protecting group on the 4-hydroxyl group at the nonreducing end of the newly formed disaccharide is selectively removed, leading to a new acceptor, which undergoes further glycosylation, extending the chain from the reducing end to the nonreducing end, and producing the heparin/HS oligosaccharide backbone (Scheme 10).
Scheme 10.
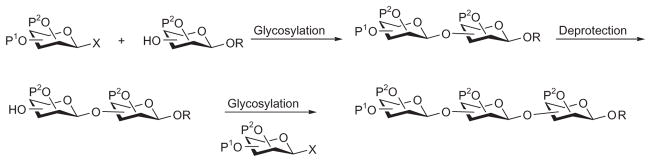
Linear synthesis of oligosaccharides from the reducing end.
Overall, the number of synthetic steps in linear synthesis is high because of the number of oligosaccharide intermediates generated and the deprotection step required after each glycosylation. Linear synthesis of oligosaccharides is therefore performed mainly for preparing shorter oligosaccharide sequences. Fügedi used the linear strategy to synthesize HS trisaccharides considered to be responsible for interactions of HS with the fibroblast growth factors FGF-1 and FGF-2.32,78 Glycosylation of acceptor 63 with thioglycoside 62, using the thiophilic promoter dimethyl (methylthio)sulfonium triflate (DMTST), followed by removal of the chloroacetyl protecting group with hydrazine dithiocarbonate (HDTC) furnished the disaccharide acceptor 64 (Scheme 11). A second round of DMTST-mediated glycosylation using donor 65 produced the trisaccharide 66. After removal of the benzoyl, the 4-methoxyphenyl (MP), and the t-butyl group, sulfation of the newly liberated hydroxyl groups was performed with the sulfur trioxide–pyridine complex in DMF. Hydrogenation and selective N-sulfation with the sulfur trioxide–trimethylamine complex under basic conditions furnished the final product 70.79 Following the same reaction sequence, except for reversing the steps of MPh-group removal and O-sulfation, generated the trisaccharide 71, which bore sulfation patterns different from those in compound 70 (Scheme 11).
Scheme 11.

A linear synthesis of the heparin/HS trisaccharides responsible for binding with the fibroblast growth factors FGF-1 and FGF-2.
Boons and coworkers used the linear approach to synthesize a trisaccharide, using the monosaccharide building blocks 72 and 73.52 Glycosylation of vinyl donor 72a with acceptor 73 was followed by removal of the p-methoxybenzyl (PMB) group at the nonreducing end by TFA, which generated the disaccharide acceptor 75 (Scheme 12). The trichloroacetimidate donor 72b was found to be superior to the corresponding vinyl donor 72a. The monosaccharide derivative 73 was benzoylated to produce vinyl donor 74, which glycosylated the acceptor 75 to furnish trisaccharide 76. Deprotection of 76 led to the unsulfated HS trisaccharide derivative 78. The synthesis, while linear, could be modified into a modular active–latent approach, as discussed in Section III.
Scheme 12.
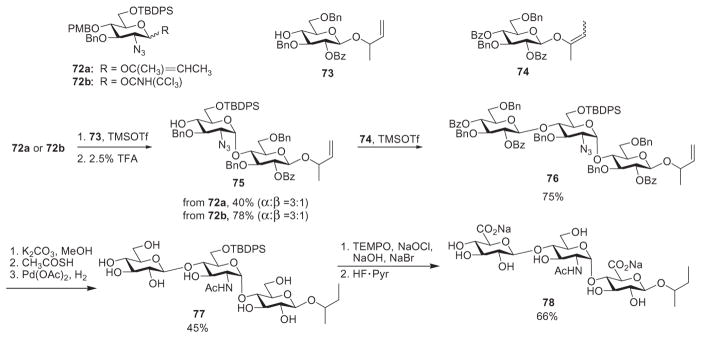
Linear synthesis and late-stage oxidation to generate trisaccharide 78.
2. Polymer-Supported Synthesis
The Holy Grail in oligosaccharide synthesis would be the availability of a general and fully automated system having the synthetic efficiency of the established automated systems for peptide synthesis. Toward this goal, the Seeberger group has adapted an automated peptide synthesizer for the synthesis of complex oligosaccharides.80,81 Thus far, the automated synthesis of heparin/HS oligosaccharides has not been achieved, because of the difficulties in translating solution-phase synthesis to high-yielding polymer-supported synthesis.
To determine the influence of polymer supports on the synthesis of heparin/HS components, Martin-Lomas and coworkers evaluated the use of various polymer supports and linkers on the glycosylation process.82–84 Through a succinic acid linker, disaccharide derivative 79 was grafted onto the polystyrene resin ArgoGel™, which was then further functionalized, leading to the polymer-bound acceptor 82 (Scheme 13). Glycosylation of disaccharide derivative 82, mediated by TMSOTf, was performed using 3.7 equiv. of the disaccharide donor 80, and the excess of activated donor and reagent was removed after the reaction by multiple washes of the resin. As the reactivity of the polymer-bound acceptor was low, the glycosylation reaction was repeated two more times. The resin was then treated with hydrazine to cleave off the product, affording tetrasaccharide derivative 83 in 89% yield. This method was further extended to the synthesis of octasaccharide derivative 90 through successive iterations of deprotection and glycosylation. However, the overall yield of the octasaccharide was very low (~10%), presumably because of the low reactivity of the larger glycosyl acceptor caused by steric hindrance posed by the insoluble polymer.
Scheme 13.

Solid-supported synthesis of heparin oligomers.
To increase the flexibility of the polymer, a water-soluble polymer, monomethyl polyethylene glycol (MPEG), was tested as a support. The succinylated disaccharide derivative 81 was linked to MPEG in a manner similar to that employed with ArgoGel™ (Scheme 14). Following TMSOTf-catalyzed glycosylation with the imidate donor 80, the polymer was precipitated with diethyl ether and isolated by filtration. The glycosylation reaction was repeated three more times, and the tetrasaccharide derivative 92, released from the polymer support by hydrazinlolysis, was obtained in 20% overall yield from the polymer-bound disaccharide derivative 91. The authors proposed that the yield differences in using MPEG versus ArgoGel could be attributed to the inefficiency of MPEG precipitation, as small losses compounded could become significant over multiple steps of manipulation.
Scheme 14.
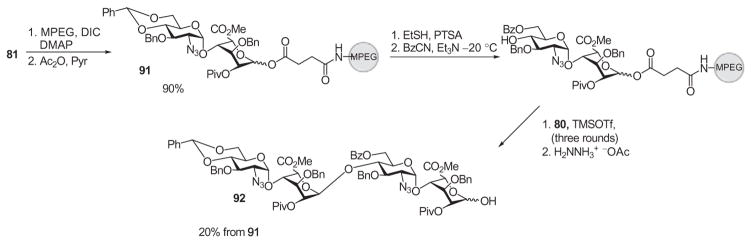
Use of the water-soluble polymer MPEG in synthesis of heparin/HS oligosaccharides.
One complication in using the anomeric position to link with the polymer is the production of anomeric mixtures upon release from the polymer. To avoid this, the 6-position of the glucosamine precursor was tested as the site of attachment to MPEG. However, conjugation to the polymer was only 40% effective, and with three rounds of glycosylation and subsequent detachment of the polymer, only 36% of the desired tetrasaccharide was obtained.83 The carboxylate position of iduronic acid was next evaluated as the site of attachment. This position was ideal for attachment of the polymer as it avoided blocking a potential sulfation site, and cleavage from the polymer support could afford an anomerically pure product. The disaccharide acceptor 93 was bound to the MPEG polymer through the carboxylate site of its iduronic residue, and this was subjected to glycosylation by disaccharide donor 80. Each backbone–elongation cycle consisted of four rounds of glycosylation (Scheme 15). After each glycosylation, to avoid the loss of desired product through incomplete MPEG precipitation, the nonconsumed acceptor was scavenged by carboxylic acid-functionalized, insoluble Merrifield resins, which were removed by simple filtration. Through this procedure, hexasaccharide product 94 was isolated in 37% overall yield from disaccharide derivative 93. One more round of elongation gave the octasaccharide derivative 95 in 26% overall yield from compound 93.83 Based on yields of product obtained, this route was more efficient than previous MPEG-supported synthesis.
Scheme 15.
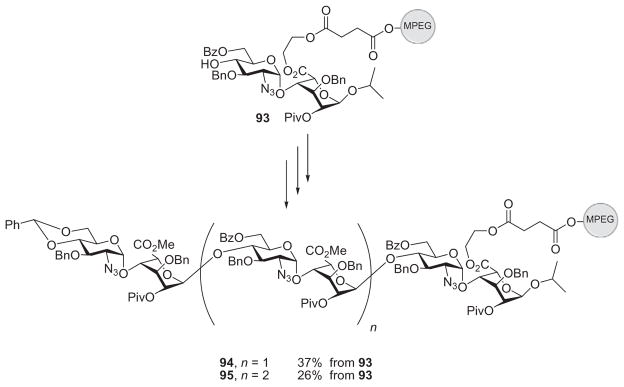
Soluble polymers anchored through the carboxylate group of the iduronic component.
In addition to the succinic acid linker, the Martin-Lomas group designed a novel linker that immobilized an idose-based acceptor (97) onto MPEG (Scheme 16). Five iterations of glycosylation of 97 by the imidate 96, followed by deprotection, gave the disaccharide derivative 99 in 82% yield. Further elongation of the chain by imidate 96 produced the polymer-bound trisaccharide, from which the polymer was cleaved off under basic conditions to yield oligosaccharide 100 bearing a protected amino group in the linker, in 53% yield. The free amino group could be released by hydrogenolysis, which is useful for bioconjugation or immobilization of the oligosaccharides onto glycan microarrays.
Scheme 16.

Protected amino linker used in conjunction with monosaccharide building blocks used in solid-supported synthesis of heparin/HS oligosaccharides.
In summary, the yields of heparin/HS oligosaccharide by glycosylation through polymer-supported synthesis decrease drastically as the chains grow longer. This is a serious challenge to any efforts at automation. Novel chemistry needs to be developed to significantly enhance the glycosylation yields on polymer support without resorting to the use of large excesses of donors. Until this becomes reality, solution-phase synthesis remains the preferred method for preparing complex heparin/HS oligosaccharides.
III. Active–Latent Glycosylation Strategy
The active–latent strategy is a solution-based method that builds oligosaccharides from the nonreducing end to the reducing end. In this approach, the acceptor carries a latent aglycon, which is inert to the conditions of glycosyl-donor activation. After glycosylation, the resultant oligosaccharide is transformed into an active donor for further chain elongation (Scheme 17).
Scheme 17.
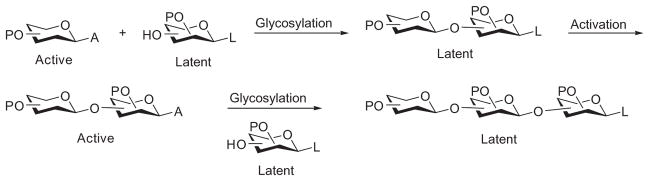
The active–latent glycosylation strategy.
Allyl glycosides are used widely for the active–latent glycosylation strategy. Inert to many of the conditions used for donor activation, allyl glycosides can be readily transformed into vinyl glycosides, which serve directly as active glycosyl donors in Lewis acid-catalyzed glycosylations. However, the vinyl donors typically give low yields in glycosylation reactions (see Scheme 12).85 To circumvent this problem, the vinyl aglycon can be cleaved to generate the hemiacetal, which can then be transformed into trichloroacetimidate donors, which are much more reactive.85–87
The Bonnaffé group developed an impressive synthesis of a heparin dodecamer by the active–latent strategy, using the allyl glycoside and glycosyl trichloroacetimidate combination.64 To improve the overall synthetic efficiencies, a PMB group was employed to protect the 4-position at the nonreducing end of the oligosaccharide intermediate. This substituent could be removed selectively to expose a free hydroxyl group for further elongation of the chain. In this synthesis, the PMB-derivatized latent allyl disaccharide 103 was first transformed into a trichloroacetimidate donor, 105, and the allyl disaccharide acceptor 104 (Scheme 18A).86 Glycosylation of acceptor 104 by imidate 105 generated the latent allyl tetrasaccharide, which was then modified to an active trichloroacetimidate donor 106 (Scheme 18B). The reaction of 106 with tetrasaccharide 107, followed by removal of the PMB group at the nonreducing end, and another round of glycosylation, furnished the dodecasaccharide 108 in 45% overall yield from the acceptor 107. After completion of the backbone, deprotection and sulfation were performed. O-Deacetylation by potassium carbonate, and reduction with 1,3-propanedithiol, followed by simultaneous O- and N-sulfation with the sulfur trioxide–pyridine complex gave the sulfated dodecamer. The simultaneous sulfation with pyridine–SO3 of the hydroxyl and amino groups did not proceed to completion. A second round of sulfation with pyridine–SO3 in basified water was necessary to complete the sulfation.64,73 Hydrolysis of the methyl esters, followed by hydrogenolysis, gave the fully deprotected dodecamer 109, which is the longest heparin oligosaccharide yet prepared by chemical synthesis.64
As two synthetic steps are needed to cleave the allyl groups necessitating the use of expensive transition-metal reagents and toxic mercury salts, silyl protecting groups provide attractive alternatives for masking the anomeric position for the active–latent strategy. Many successful syntheses have used a variety of silyl ethers, such as dimethylthexylsilyl (TDS), t-butyldimethylsilyl (TBDMS), and trimethylsilyl (TMS).37,68,73,84,88–91 As an example, glycosylation of the monosaccharide acceptor 111 by the trichloroacetimidate donor 110, followed by acetylation, generated the latent disaccharide derivative 112 (Scheme 19A). Removal of the anomeric TDS group from 112, followed by formation of the trichloroacetimidate, converted compound 112 into the active donor 113 (Scheme 19B).90 The acceptor 114 was prepared by acid-mediated removal of the 4,6-benzylidene acetal from disaccharide derivative 112 and selective benzoylation of the primary hydroxyl group. Glycosylation by donor 113 of acceptor 114 furnished the tetrasaccharide acceptor 115. The overall yield was only 40% because of the low glycosylation yield, with 44% of the starting disaccharide derivative 114 being recovered. Hexasaccharide derivative 117 was prepared by the reaction of tetrasaccharide derivative 115 with disaccharide donor 116. With the removal of its anomeric TDS group, the hexasaccharide derivative 117 was transformed into an active trichloroacetimidate donor 118, which upon reaction with methanol afforded the methyl glycoside 119 (Scheme 19D).
Scheme 19.

Active–latent synthesis with silyl protecting groups.
Besides being compatible with the trichloroacetimidate donors, the silyl protecting group is robust and has also been applied with thioglycosides under various activating systems.37,59,60 The Boons group used this strategy to prepare disaccharide building blocks for their heparin/HS oligosaccharide synthesis (Scheme 20).59 Glycosylation of 1-thioglycoside donor 120 with the TDS-protected acceptor 121 formed the latent disaccharide 122. After oxidation and protecting group manipulation, the TDS group in 122 was removed and the resulting hemiacetal was converted into the trichloroacetimidate disaccharide donor 123. Eight disaccharide building blocks were prepared in this manner and were used to construct a panel of 11 heparin/HS tetrasaccharides and 1 hexasaccharide having different backbone structures and sulfation patterns. These tetrasaccharides were used to probe the important structural features of HS for inhibiting β-secretase, a protease considered to be involved in the development of Alzheimer’s disease.
Scheme 20.
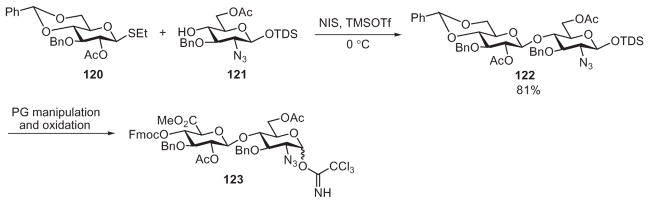
Synthesis of one of the eight disaccharide building blocks used by Boons and coworkers to prepare a library of heparin oligosaccharides.
In addition to allyl and silyl groups, other functionalities, including isopropylidene acetals35,69,72 and 1,6-anhydro sugars, have been used to mask the anomeric position of the latent glycosyl donors. The 1,6-anhydro sugars are advantageous to use as they do not require another selectively removable protecting group for the anomeric position. Hung and coworkers developed rapid routes of access to such 1,6-anhy-dro-L-idose building blocks as compound 125.35,36 Glycosylation of anhydro derivative 125 by the glycosyl trichloroacetimidate donor 124 furnished disaccharide 126 (72% yield, α:β = 5.5:1, Scheme 21A). To activate this latent disaccharide, the 1,6-anhydro ring of the α anomer of 126 was cleaved by Cu(OTf)2-catalyzed acetolysis, and the newly installed acetyl group at O-6 was exchanged for the more selectively cleavable levulinoyl ester, followed by formation of the trichloroacetimidate (Scheme 21B). The resulting disaccharide donor was condensed with the glucosaminide precursor 128, generating trisaccharide derivative 129. The 2-naphthyl-substituted trisaccharide 129 was selectively deprotected to expose the 4-hydroxyl group at the nonreducing end, where it was glycosylated by the disaccharide donor 127. Repetition of these deprotection and glycosylation sequences two more times led to formation of the HS nonasaccharide derivative 132.72 The active–latent strategy, coupled with the use of a selectively removable protecting group, such as 2-naphthyl, at the 4-hydroxyl group at the nonreducing end, provides additional versatility in comparison to the linear strategy, as oligosaccharides can be built up from both the nonreducing and reducing end. However, multiple synthetic manipulations are still needed on the oligosaccharide intermediates to activate the latent donor.
Scheme 21.

The use of the 1,6-anhydro sugars in latent–active strategy.
IV. Selective Activation
To decrease the number of steps required for modification of intermediates, as is encountered in the latent–active strategy, the selective-activation method utilizes donors and acceptors having different types of activable aglycons. Upon selective activation of the donor and glycosylation of the acceptor, the resulting disaccharide can be used directly as a donor under a new set of activation conditions, without the need for manipulation of the intermediate (Scheme 22). The most common pairs of glycosyl building blocks in selective-activation methods are glycosyl trichloroacetimidates and thioglycosides, since thioglycosides are stable under the acidic conditions encountered in trichloroacetimidate activation.55,90,92,93 The selective-activation method can often be combined with the active–latent strategy within a single synthetic operation.
Scheme 22.

Glycosylation strategy employing selective activation.
In the preparation of two heparin/HS tetrasaccharides, Yu and coworkers used the active–latent approach to produce disaccharide building blocks and selective activation for extension of the backbone. Glycosylation of the 1,6-anhydro acceptor 134 by the ethyl 1-thio-L-idoside donor 133 was performed with NIS and AgOTf (Scheme 23).68,90 To convert disaccharide derivative 135 into an active donor, the anhydro ring was opened, followed by protecting-group adjustment, an oxidative manipulation at C-6, and formation of the trichloroacetimidate disaccharide donor 136. Donor 136 was selectively activated by TMSOTf, with the thioglycoside 137 serving as acceptor, leading to the trisaccharide derivative 138. Without further synthetic manipulation, trisaccharide 138 was activated by the thiophilic promoter 1-benzenesulfinylpiperidine (BSP) and Tf2O, which glycosylated monosaccharide 139 to produce tetrasaccharide derivative 140.
Scheme 23.

Synthesis utilizing the selective activation of trichloroacetimidate donors in the presence of thioglycoside acceptors.
Instead of glycosyl trichloroacetimidates and thioglycosides, van der Marel and coworkers explored the utility of free glycoses (glycosyl hemiacetals) and thioglycosides in a selective glycosylation approach toward the pentasaccharide derivative 148, a fully protected precursor of the heparin backbone. The hemiacetal 141 was selectively activated in the absence of the glycosylthio acceptor 142, utilizing the pre-activation strategy, where the donor was treated with the diphenyl sulfoxide and Tf2O promoter system developed by the Gin laboratory.94 Upon complete activation, the acceptor 142 was added to the reaction mixture to yield disaccharide derivative 143 (Scheme 24). To extend the chain, the 1,6-anhydro acceptor 134 was glycosylated with disaccharide derivative 143, producing trisaccharide derivative 144 as a latent donor. The 1,6-anhydro bridge was then opened under acidic conditions to create a trisaccharide hemiacetal donor 145, which after selective activation was coupled to the glycosylthio acceptor 137. The resultant tetrasaccharide thioglycoside 146 reacted with the reducing-end acceptor 147 to complete the synthesis.66 Aided by the 1,6-anhydro ring as a masked hemiacetal, thioglycosides and glycosyl hemiacetals proved to be very effective partners for the selective glycosylation approach. Only one manipulation of an intermediate aglycon was required for preparation of the pentasaccharide derivative 148.
Scheme 24.

Congruent use of hemiacetals and thioglycosides.
Although the selective-activation strategy improves synthetic efficiency, it requires two different types of glycosyl donor. To simplify the overall synthetic design, it is desirable for a single type of glycosyl donor to be employed throughout the synthesis, avoiding the need for modification of the aglycon leaving group of the intermediate oligosaccharides. Toward this goal, two chemoselective strategies have been developed. These are the reactivity-based, armed–disarmed method and the reactivity-independent, pre-activation-based method.
V. Reactivity-Based Chemoselective Glycosylation
In reactivity-based, armed–disarmed glycosylation strategy, glycosyl building blocks, typically thioglycosides, are designed to have different reactivities at the anomeric position. When a mixture of a more-reactive donor (armed) and an acceptor having lower anomeric reactivity (disarmed) is subjected to a limiting amount of promoter, the more-reactive, armed donor is activated preferentially, and this donor glycosylates the acceptor (Scheme 25). The resulting oligosaccharide can function directly as a donor by using the same conditions for further glycosylation with a thioglycoside acceptor that has even lower anomeric reactivity. With suitable design, the anomeric reactivities of various building blocks can be sufficiently different so as to enable multiple glycosylation reactions sequentially in one vessel without the need for purification of the oligosaccharide intermediates.
Scheme 25.
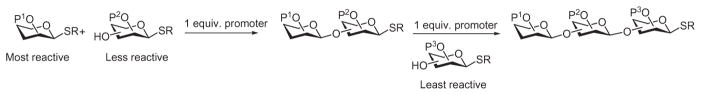
The armed–disarmed strategy for chemoselective glycosylation relies on differences in anomeric reactivities of the building blocks.
To achieve the required differentiation of anomeric reactivity, the electronic property or conformational rigidity of the donor can be tuned by strategically placing suitable protecting groups on the glycon ring95–97 or by modification of the aglycon.98–100 Reactivities can be quantified as relative reactivity values (RRVs), with the reactivity of p-tolyl 2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside toward methanol acceptor being set as 1.0.97
Wong and coworkers conducted the synthesis of heparin/HS oligosaccharides via the reactivity-based approach. Four monosaccharide building blocks (149–152) were prepared and their RRVs measured (Scheme 26A).101 As the glucoside building block 151 was 30 times more reactive than the acceptor glucosamine precursor 150, chemoselective activation of 151 was achieved in preference to 150, leading to disaccharide derivative 153 in excellent (89%) yield (Scheme 26B). Manipulation of protecting groups and oxidation at C-6 of the glucosyl component furnished the new disaccharide building block 154 having a RRV of 18.3. Chemoselective glycosylation of 154 by the azidoglucose donor 149 (RRV = 53.7) was therefore feasible (Scheme 26C) to give the corresponding trisaccharide derivative. The latter was coupled to O-4 of the disaccharide acceptor 155, and more promoter was added to the reaction mixture. This led to the formation in one vessel of the fully protected HS pentasaccharide precursor 156 in 20% overall yield for the two steps. The modest net yield was most probably attributable to the small reactivity differential between donor 149 and disaccharide 154.
Scheme 26.
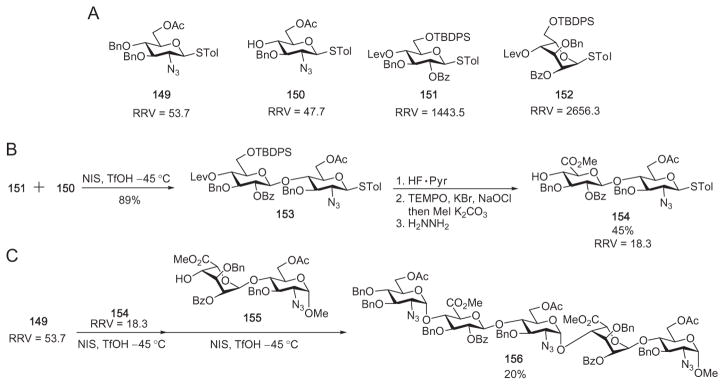
(A) Monosaccharide building blocks used in Wong’s synthesis of heparin components; (B) preparation of disaccharide building block 154; (C) one-pot synthesis of heparin pentasaccharide precursor 156 by the armed–disarmed strategy.
The RRVs provide general guidance toward the selection of building blocks. However, the RRVs are quantified with reference to methanol as the acceptor, and these values can change according to the structure of the acceptor and the reaction conditions.102 Accordingly, caution needs to be exercised in relying solely on RRVs to predict the outcome of a reaction. Furthermore, applying the reactivity-based method to the synthesis of longer heparin/HS oligosaccharides could be challenging because the polymeric nature of heparin/HS would require the same glycosyl units to have greatly differing reactivities according to their location in the backbone. The building blocks at the nonreducing end should have higher reactivities than those situated toward the reducing end. This challenge can be overcome by the reactivity-independent, pre-activation-based chemoselective strategy for glycosylation.
VI. Reactivity-Independent, Pre-Activation-Based, Chemoselective Glycosylation
The aforementioned glycosylation strategies rely on differences in anomeric reactivity. The acceptor either cannot be activated, as in the case of linear, active–latent, and selective-activation methods, or has a much lower reactivity than the donor in the armed–disarmed reactivity-based approach. The underlying cause for this is the fact that the glycosyl donor and acceptor are both present in the reaction mixture when the promoter is added. Thus, the anomeric reactivities of donors and acceptors must be differentiated to achieve selective activation of the donor. To overcome this limitation, the pre-activation strategy was developed, wherein the donor is activated by a promoter to generate a reactive intermediate in the absence of an acceptor (Scheme 27). The acceptor is then added to react with the reactive intermediate and form a new glycosidic bond. Activation of the donor in the absence of the acceptor allows the acceptor to carry the same aglycon group as the donor, negating the need for reactivity tuning. The prerequisite for pre-activation is that the promoter used must be in stoichiometric amount to avoid activation of the acceptor or product, and any side-products from activation of the donor must not be nucleophilic. Several types of glycosyl donor have been used in the pre-activation scheme, and these include hemiacetals,94 glycals,103 selenoglycosides,104 and thioglycosides.105,106
Scheme 27.
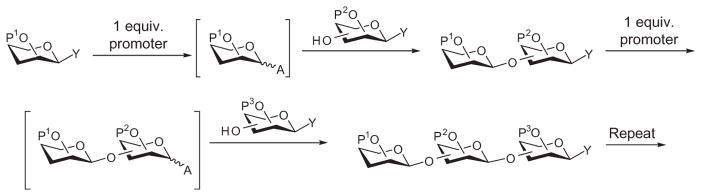
Pre-activation-based strategy for glycosylation.
Huang and coworkers synthesized a library of 12 heparin/HS hexasaccharides by the reactivity-independent, pre-activation-based strategy. This synthesis employed thioglycoside modules and the powerful promoter p-toluenesulfenyl triflate (p-TolSOTf), which was generated in situ from p-toluenesulfenyl chloride (p-TolSCl) and AgOTf.106 To simplify the preparation of building blocks, a divergent approach was designed. Starting from three monosaccharide building blocks, two disaccharide derivatives (162 and 163) were prepared (Scheme 28A). These compounds were then divergently modified, leading to six disaccharide modules (164–169, Scheme 28B and C).58 To assemble the hexasaccharide, disaccharide donor 166 was pre-activated with p-TolSCl and AgOTf at −78 °C (Table I). Upon complete activation, the bifunctional 1-thioglycoside acceptor 165 was added to the reaction mixture. The reactive intermediate generated through activation of the donor glycosylated the acceptor 165, producing a tetrasaccharide. As this tetrasaccharide product already bore an arylthio aglycon, it was activated directly with another equivalent of the promoter and allowed to react with acceptor 167 in the same reaction flask. Hexasaccharide 170 was obtained from this reaction in 54% yield in less than 5 h. Since this synthesis did not require adjustment of the aglycon structure or purification of the intermediate tetrasaccharide, the efficiency of the glycosidic assembly was greatly enhanced.
Scheme 28.

Divergent synthesis of the building blocks needed for the assembly of a hexasaccharide library.
Table I.
One-Pot Preparation of Heparin/HS Hexasaccharides
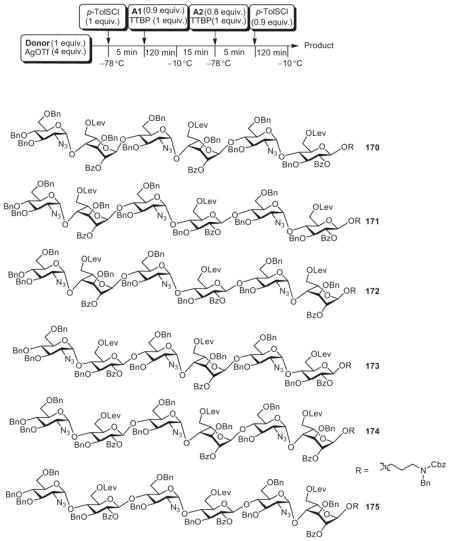
| ||||
|---|---|---|---|---|
| Donor | Acceptor 1 | Acceptor 2 | Product | Yield (%) |
| 166 | 165 | 167 | 170 | 54 |
| 166 | 168 | 167 | 171 | 59 |
| 166 | 168 | 164 | 172 | 58 |
| 169 | 165 | 167 | 173 | 62 |
| 169 | 165 | 164 | 174 | 57 |
| 169 | 168 | 164 | 175 | 50 |
As the pre-activation method does not require the glycosyl donor to have higher anomeric reactivities than the glycosyl acceptor, the disaccharide building blocks 164–169 could be used in a combinatorial fashion to prepare a library of oligosaccharides (Table I).58 For example, substituting compound 165 by 168 and then following the same reaction scheme as in the preparation of hexasaccharide 170, hexasaccharide derivative 171 was formed in 59% yield in a one-pot process. By mixing the disaccharide building blocks 164–169, six hexasaccharides having systematically varied and precisely controlled backbone structures were produced in 50–62% yields within a few hours (Table I). These hexasaccharides were then deprotected and subsequently sulfated, creating a set of 12 heparin/HS hexasaccharides, which were used to decipher structure–activity relationships in the binding of fibroblast growth factor-2 to heparin.
In summary, as discussed up to this point, chemical synthesis has been the major path for access to synthetically pure heparin/HS oligomers. Given the length and difficulties in chemical synthesis, several groups have begun to explore the potential of enzymatic synthesis and its integration with chemical methods.
VII. Chemoenzymatic Synthesis
In Nature, the biosynthesis of heparin/HS is performed by multiple enzymes in the Golgi apparatus. Assembly of the HS backbone by glycosyltransferases is followed by such enzymatic modifications as N-deacetylase/N-sulfotransferase (NDST) for removal of the N-acetyl group and subsequent N-sulfation, C5-epimerase for isomerization of the uronic acid, and three types of O-sulfotransferases, namely 2-OST for sulfating O-2 of IdoA, 3-OST for sulfating O-3 of GlcN, and 6-OST for sulfating O-6 of GlcN. The enzymatic modification of the HS backbone is typically incomplete and thus leads to a wide range of structural variations in naturally occurring heparin and HS.
In order to develop a laboratory synthesis of a pentasaccharide exhibiting strong binding with ATIII, the Rosenberg group explored the enzymatic approach.107 The backbone of their oligosaccharide was obtained from “heparosan,” a polysaccharide from the Escherichia coli K5 capsule composed of disaccharide repeating units of [→4)-α-D-GlcNAc-(1→4)-β-D-GlcA-(1→]. The synthesis of pentasaccharide 178 started with N-sulfation of the “heparosan” by incubation with NDST2 and the sulfate-group donor 3′-phospho-5′-adenylyl sulfate (PAPS, Scheme 29). Following N-sulfation, the polymer was depolymerized by heparitinase, and hexasaccharide 176 was isolated by HPLC from the resulting mixture. Sequential epimerization and O-2 sulfation of hexasaccharide 176 by C5-epimerase and 2-OST1, followed by sulfation at O-6, provided the sulfated hexasaccharide 177. Removal of the unsaturated uronic acid by Δ4,5-glycosiduronase with subsequent 3-OST-catalyzed sulfation at O-3 produced pentasaccharide 178, a compound having anticoagulant activity. While this synthesis was groundbreaking, the product 178 was isolated in only microgram quantity and with an overall yield of 1.1%.107 The low yield was presumably due to the difficulties in purification, particularly in the isolation of hexasaccharide 176 from the complex mixture that arose from cleavage by heparitinase.108–110 Another obstacle was the low yields of the enzymes expressed from a baculovirus system.
Scheme 29.

Enzymatic synthesis of pentasaccharide 178 from “heparosan.”
The Liu and Linhardt groups took a different approach for the chemoenzymatic synthesis of heparin/HS oligosaccharides. Instead of relying on the difficult isolation of hexasaccharide 176 from the complex mixture of degradation products resulting from the action of heparitinase on “heparosan,” they obtained gram quantities of disaccharide 179 through the complete digestion of “heparosan” by nitrous acid.109,111 To elongate the chain, two bacterial glycosyltransferases, heparan synthase-2 (PmHS2)112 and the N-acetylglucosaminyl transferase (KfiA) of E. coli,113 were used to transfer GlcA and GlcNAc, respectively (Scheme 30). All of the enzymes for backbone modification, including C5-epimerase, NDST2, and the O-sulfotransferases, were expressed in large quantities in the E. coli system. The conversion of the N-acetyl group to N-sulfate is difficult because of the stability of the acetamido group and the low activity of the N-deacetylase. To overcome this, Liu, Linhardt, and coworkers took advantage of the broad substrate specificity of KfiA by incorporating N-trifluoro-protected glucosamine (GlcNTFA) into the backbone where N-sulfation is desired.114,115 Treatment of disaccharide 179 with the glycosyl donor UDP-GlcNTFA and the transferase KfiA, followed by UDP-GlcA and transferase pmHS2, provided tetrasaccharide 180 in 75% yield. An additional round of elongation with both monosaccharides, followed by removal of the TFA protecting groups with triethylamine, and subsequent N-sulfation by N-sulfotransferase (NST) furnished the N-sulfated hexasaccharide 181. Following the addition of another GlcNTFA group, epimerization and sulfation at O-2 were performed in one flask with 2-OST and C5-epimerase to yield the heptasaccharide 182. The location of enzymatic modification was controlled by the substrate structure. As the C5-epimerase causes GlcA to be modified only when flanked by N-sulfated glucosamine groups, the GlcA component closer to the reducing end in 181 alone was epimerized and O-2 sulfated. The last TFA protecting group in 182 was removed with triethylamine, and the product was incubated with NST and PAPS, then PAPS, 6-OST-1, 6-OST-3, and finally PAPS, and 3-OST1 in sequential reactions to provide the final heptasaccharide 183, which had anticoagulant activity similar to that of the FDA-approved pentasaccharide fondaparinux.114 In an analogous manner, 49 mg of the heptasaccharide 184 was prepared with an overall yield of 38% from the disaccharide 179. This work has laid a great foundation for future gram-scale preparation of heparin/HS oligosaccharides.115 Extending the chemoenzymatic strategy to preparation of fondaparinux will provide an attractive alternative complementing the current complex chemical synthesis of this important molecule.
Scheme 30.

The chemoenzymatic synthesis of heparin heptasaccharides 183 and 184.
VIII. Future Outlook
The past decade has seen tremendous advancements in the production of Heparin/HS oligosaccharides. In addition to the more traditional target-oriented synthesis, efforts are being directed toward generating an array of oligosaccharides having diverse patterns of sulfation. In chemical synthesis, multiple strategies have been developed to expedite the glyco-assembly process. Methods are now available for access to tens of oligosaccharides to construct a sample library. However, challenges remain in decreasing the number of synthetic steps required for preparation of building blocks, as well as for establishing a robust method to perform multiple sulfations simultaneously. The enzymatic synthesis of compound 184 at the 49 mg scale is an impressive accomplishment. The substrate specificities of the enzymes may possibly limit the total number of structures that can be generated. Ongoing research has suggested that enzymatic modification can be integrated with chemical synthesis.116 The combination of the regiospecificity of enzymatic reactions with the flexibility of chemical synthesis can significantly expand our overall synthetic capability, which in turn can greatly aid in the efforts to decipher the exciting biological functions of heparin and HS.
Acknowledgments
We thank the National Institute of General Medical Sciences, NIH (R01GM 72667), and the National Science Foundation (CHE 1111550) for generous financial support of our work.
Abbreviations
- 2-NAP
2-naphthyl
- 2-OST
2-O-sulfotransferase
- 3-OST
3-O-sulfotransferase
- 6-OST
6-O-sulfotransferase
- Ac
acetyl
- AgOTf
silver triflate
- All
allyl
- ATIII
antithrombin III
- AZMB
2-(azidomethyl)benzoyl
- BAIB
iodobenzene diacetate
- Bn
benzyl
- BSP
1-benzenesulfinylpiperidine
- Bz
benzoyl
- C5-epi
C5-epimerase
- CAN
ceric ammonium nitrate
- Cbz
benzyloxycarbonyl
- CSA
camphorsulfonic acid
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DCC
N,N′-dicyclohexylcarbodiimide
- DDQ
2,3-dichloro-5,6-dicyanobenzoquinone
- DIAD
diisopropylazodicarboxylate
- DIC
N,N′-diisopropylcarbodiimide
- DMAP
4-dimethylaminopyridine
- DMTST
dimethyl (methylthio)sulfonium triflate
- EDC
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide
- Et3N
triethylamine
- FGF
fibroblast growth factor
- Fmoc
fluorenylmethoxycarbonyl
- GAG
glycosaminoglycan
- D-GlcA
D-glucuronic acid
- D-GlcN
D-glucosamine
- GlcNAc
N-acetyl-D-glucosamine
- HDTC
hydrazine dithiocarbonate
- HOBT
hydroxybenzotriazole
- HS
heparan sulfate
- L-IdoA
L-iduronic acid
- KfiA
N-acetyl-glucosaminyl transferase
- Lev
levulinoyl
- MP
4-methoxyphenyl
- MPEG
monomethyl polyethylene glycol
- NDST
N-deacetylase/N-sulfotransferase
- NIS
N-iodosuccinimide
- NST
N-sulfotransferase
- PAPS
3′-phospho-5′-adenylyl sulfate
- PEG
polyethylene glycol
- Piv
pivaloyl
- PMB
p-methoxybenzyl ether
- PmHS2
heparan synthase-2
- p-TolSCl
p-toluenesulfenyl chloride
- p-TolSOTf
p-toluenesulfenyl triflate
- Pyr
pyridine
- RRV
relative reactivity value
- SEM
2-(trimethylsilyl)ethoxymethyl
- TBDMS
t-butyldimethylsilyl
- TBDMSOTf
t-butyldimethylsilyl triflate
- TBDPS
t-butyldiphenylsilyl
- TCE
2,2,2-trichloroethyl
- TDS
dimethylthexylsilyl
- TEMPO
2,2,6,6-tetramethylpiperidin-1-oxyl
- Tf2O
triflic anhydride
- TFA
trifluoroacetic acid
- TMS
trimethylsilyl
- TMSOTf
trimethylsilyl triflate
- Tol
tolyl
- p-TsOH
p-toluenesulfonic acid
- TTBP
2,4,6-tri-t-butylpyrimidine
- UDP
uridine 5′-diphosphate
References
- 1.Linhardt RJ. Heparin: An important drug enters its seventh decade. Chem Ind. 1991:45–47. [Google Scholar]
- 2.Cifonelli JA, Dorfman A. Uronic acid of heparin. Biochem Biophys Res Commun. 1962;7:41–45. doi: 10.1016/0006-291x(62)90141-9. [DOI] [PubMed] [Google Scholar]
- 3.Hook M, Bjork I, Hopwood J, Lindahl U. Anticoagulant activity of heparin: Separation of high-activity and low-activity heparin species by affinity chromatography on immobilized antithrombin. FEBS Lett. 1976;66:90–93. doi: 10.1016/0014-5793(76)80592-3. [DOI] [PubMed] [Google Scholar]
- 4.Damus PS, Hicks M, Rosenberg RD. Anticoagulant action of heparin. Nature. 1973;246:355–357. doi: 10.1038/246355a0. [DOI] [PubMed] [Google Scholar]
- 5.Gandhi NS, Mancera RL. The structure of glycosaminoglycans and their interactions with proteins. Chem Biol Drug Des. 2008;72:455–482. doi: 10.1111/j.1747-0285.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- 6.Seeberger PH, Werz DB. Synthesis and medical applications of oligosaccharides. Nature. 2007;446:1046–1051. doi: 10.1038/nature05819. [DOI] [PubMed] [Google Scholar]
- 7.Parish CR. The role of heparan sulphate in inflammation. Nat Rev Immunol. 2006;6:633–643. doi: 10.1038/nri1918. [DOI] [PubMed] [Google Scholar]
- 8.Powell AK, Yates EA, Fernig DG, Turnbull JE. Interactions of heparin/heparan sulfate with proteins: Appraisal of structural factors and experimental approaches. Glycobiology. 2004;14:17R–30R. doi: 10.1093/glycob/cwh051. [DOI] [PubMed] [Google Scholar]
- 9.Sasisekharan R, Shriver Z, Venkataraman G, Narayanasami U. Roles of heparan-sulphate glycosaminoglycans in cancer. Nat Rev Cancer. 2002;2:521–528. doi: 10.1038/nrc842. [DOI] [PubMed] [Google Scholar]
- 10.Liu D, Shriver Z, Qi Y, Venkataraman G, Sasisekharan R. Dynamic regulation of tumor growth and metastasis by heparan sulfate glycosaminoglycans. Semin Thromb Hemost. 2002;28:67–78. doi: 10.1055/s-2002-20565. [DOI] [PubMed] [Google Scholar]
- 11.Casu B, Lindahl U. Structure and biological interactions of heparin and heparan sulfate. Adv Carbohydr Chem Biochem. 2001;57:159–206. doi: 10.1016/s0065-2318(01)57017-1. [DOI] [PubMed] [Google Scholar]
- 12.Sanderson RD. Heparan sulfate proteoglycans in invasion and metastasis. Semin Cell Dev Biol. 2001;12:89–98. doi: 10.1006/scdb.2000.0241. [DOI] [PubMed] [Google Scholar]
- 13.Casu B. Structure and biological activity of heparin. Adv Carbohydr Chem Biochem. 1985;43:51–134. doi: 10.1016/s0065-2318(08)60067-0. [DOI] [PubMed] [Google Scholar]
- 14.Esko JD, Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest. 2001;108:169–173. doi: 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gallagher JT, Walker A. Molecular distinctions between heparan sulfate and heparin. Analysis of sulfation patterns indicates that heparan sulfate and heparin are separate families of N-sulfated polysaccharides. Biochem J. 1985;230:665–674. doi: 10.1042/bj2300665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindahl U, Kjellen L. Heparin or heparan sulfate—What is the difference? Thromb Haemostasis. 1991;66:44–48. [PubMed] [Google Scholar]
- 17.Lam LH, Silbert JE, Rosenberg RD. The separation of active and inactive forms of heparin. Biochem Biophys Res Commun. 1976;69:570–577. doi: 10.1016/0006-291x(76)90558-1. [DOI] [PubMed] [Google Scholar]
- 18.Lindahl U, Baeckstroem G, Hoeoek M, Thunberg L, Fransson LA, Linker A. Structure of the antithrombin-binding site in heparin. Proc Natl Acad Sci U S A. 1979;76:3198–3202. doi: 10.1073/pnas.76.7.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenberg RD, Lam L. Correlation between structure and function of heparin. Proc Natl Acad Sci U S A. 1979;76:1218–1222. doi: 10.1073/pnas.76.3.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petitou M, Casu B, Lindahl U. 1976–1983, a critical period in the history of heparin: The discovery of the antithrombin binding site. Biochimie. 2003;85:83–89. doi: 10.1016/s0300-9084(03)00078-6. [DOI] [PubMed] [Google Scholar]
- 21.Petitou M, van Boeckel CAA. A synthetic antithrombin III binding pentasaccharide is now a drug! What comes next? Angew Chem Int Ed. 2004;43:3118–3133. doi: 10.1002/anie.200300640. [DOI] [PubMed] [Google Scholar]
- 22.Petitou M, Duchaussoy P, Lederman I, Choay J, Sinaÿ P. Binding of heparin to antithrombin III: A chemical proof of the critical role played by a 3-sulfated 2-amino-2-deoxy-D-glucose residue. Carbohydr Res. 1988;179:163–172. doi: 10.1016/0008-6215(88)84116-8. [DOI] [PubMed] [Google Scholar]
- 23.Ishihara M, Kariya Y, Kikuchi H, Minamisawa T, Yoshida K. Importance of 2-O-sulfate groups of uronate residues in heparin for activation of FGF-1 and FGF-2. J Biochem. 1997;121:345–349. doi: 10.1093/oxfordjournals.jbchem.a021593. [DOI] [PubMed] [Google Scholar]
- 24.Ishihara M, Takano R, Kanda T, Hayashi K, Hara S, Kikuchi H, Yoshida K. Importance of 6-O-sulfate groups of glucosamine residues in heparin for activation of FGF-1 and FGF-2. J Biochem. 1995;118:1255–1260. doi: 10.1093/oxfordjournals.jbchem.a125015. [DOI] [PubMed] [Google Scholar]
- 25.Noti C, Seeberger PH. Chemical approaches to define the structure-activity relationship of heparin-like glycosaminoglycans. Chem Biol. 2005;12:731–756. doi: 10.1016/j.chembiol.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 26.Noti C, Seeberger PH. Synthetic approach to define structure-activity relationship of heparin and heparan sulfate. In: Garg HG, Linhardt RJ, Hales CA, editors. Chemistry and Biology of Heparin and Heparan Sulfate. Elsevier; Oxford: 2005. pp. 79–142. [DOI] [PubMed] [Google Scholar]
- 27.Codée JDC, Overkleeft HS, van der Marel GA, van Boeckel CAA. The synthesis of well-defined heparin and heparan sulfate fragments. Drug Discov Today Technol. 2004;1:317–326. doi: 10.1016/j.ddtec.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 28.Karst NA, Linhardt RJ. Recent chemical and enzymatic approaches to the synthesis of glycosaminoglycan oligosaccharides. Curr Med Chem. 2003;10:1993–2031. doi: 10.2174/0929867033456891. [DOI] [PubMed] [Google Scholar]
- 29.Poletti L, Lay L. Chemical contributions to understanding heparin activity: Synthesis of related sulfated oligosaccharides. Eur J Org Chem. 2003:2999–3024. [Google Scholar]
- 30.Sinaÿ P, Jacquinet JC, Petitou M, Duchaussoy P, Lederman I, Choay J, Torri G. Total synthesis of a heparin pentasaccharide fragment having high affinity for antithrombin III. Carbohydr Res. 1984;132:C5–C9. [Google Scholar]
- 31.Van Boeckel CAA, Beetz T, Vos JN, de Jong AJM, van Aelst SF, van den Bosch RH, Mertens JMR, van der Vlugt FA. Synthesis of a pentasaccharide corresponding to the antithrombin III binding fragment of heparin. J Carbohydr Chem. 1985;4:293–321. [Google Scholar]
- 32.Tatai J, Osztrovszky G, Kajtar-Peredy M, Fügedi P. An efficient synthesis of L-idose and L-iduronic acid thioglycosides and their use for the synthesis of heparin oligosaccharides. Carbohydr Res. 2008;343:596–606. doi: 10.1016/j.carres.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 33.Hansen SU, Barath M, Salameh BAB, Pritchard RG, Stimpson WT, Gardiner JM, Jayson GC. Scalable synthesis of L-iduronic acid derivatives via stereocontrolled cyanohydrin reaction for synthesis of heparin-related disaccharides. Org Lett. 2009;11:4528–4531. doi: 10.1021/ol901723m. [DOI] [PubMed] [Google Scholar]
- 34.Dilhas A, Bonnaffe D. Efficient selective preparation of methyl-1,2,4-tri-O-acetyl-3-O-benzyl-β-L-idopyranuronate from methyl 3-O-benzyl-L-iduronate. Carbohydr Res. 2003;338:681–686. doi: 10.1016/s0008-6215(02)00525-6. [DOI] [PubMed] [Google Scholar]
- 35.Hung SC, Thopate SR, Chi FC, Chang SW, Lee JC, Wang CC, Wen YS. 1,6-Anhydro-β-L-hexopyranoses as potent synthons in the synthesis of the disaccharide units of bleomycin A2 and heparin. J Am Chem Soc. 2001;123:3153–3154. doi: 10.1021/ja003508a. [DOI] [PubMed] [Google Scholar]
- 36.Hung SC, Puranik R, Chi FC. Novel synthesis of 1,2:3,5-di-O-isopropylidene-β-L-idofuranoside and its derivatives at C6. Tetrahedron Lett. 2000;41:77–80. [Google Scholar]
- 37.Saito A, Wakao M, Deguchi H, Mawatari A, Sobel M, Suda Y. Toward the assembly of heparin and heparan sulfate oligosaccharide libraries: Efficient synthesis of uronic acid and disaccharide building blocks. Tetrahedron. 2010;66:3951–3962. doi: 10.1016/j.tet.2010.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adibekian A, Bindschaedler P, Timmer MSM, Noti C, Schuetzenmeister N, Seeberger PH. De Novo synthesis of uronic acid building blocks for assembly of heparin oligosaccharides. Chem Eur J. 2007;13:4510–4522. doi: 10.1002/chem.200700141. [DOI] [PubMed] [Google Scholar]
- 39.Timmer MSM, Adibekian A, Seeberger PH. Short de novo synthesis of fully functionalized uronic acid monosaccharides. Angew Chem Int Ed. 2005;44:7605–7607. doi: 10.1002/anie.200502742. [DOI] [PubMed] [Google Scholar]
- 40.Yu HN, Furukawa J, Ikeda T, Wong CH. Novel efficient routes to heparin monosaccharides and disaccharides achieved via regio- and stereoselective glycosidation. Org Lett. 2004;6:723–726. doi: 10.1021/ol036390m. [DOI] [PubMed] [Google Scholar]
- 41.Gavard O, Hersant Y, Alais J, Duverger V, Dilhas A, Bascou A, Bonnaffe D. Efficient preparation of three building blocks for the synthesis of heparan sulfate fragments: Towards the combinatorial synthesis of oligosaccharides from hypervariable regions. Eur J Org Chem. 2003:3603–3620. [Google Scholar]
- 42.Ke W, Whitfield DM, Gill M, Larocque S, Yu SH. A short route to L-iduronic acid building blocks for the syntheses of heparin-like disaccharides. Tetrahedron Lett. 2003;44:7767–7770. [Google Scholar]
- 43.Schell P, Orgueira HA, Roehrig S, Seeberger PH. Synthesis and transformations of D-glucuronic and L-iduronic acid glycals. Tetrahedron Lett. 2001;42:3811–3814. [Google Scholar]
- 44.Lubineau A, Gavard O, Alais J, Bonnaffe D. New accesses to L-iduronyl synthons. Tetrahedron Lett. 2000;41:307–311. [Google Scholar]
- 45.Fernandez R, Martin-Zamora E, Pareja C, Lassaletta JM. Stereoselective nucleophilic formylation and cyanation of α-alkoxy- and α-aminoaldehydes. J Org Chem. 2001;66:5201–5207. doi: 10.1021/jo015711+. [DOI] [PubMed] [Google Scholar]
- 46.Huang L, Huang X. Highly efficient syntheses of hyaluronic acid oligosaccharides. Chem Eur J. 2007;13:529–540. doi: 10.1002/chem.200601090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang L, Teumelsan N, Huang X. A facile method for oxidation of primary alcohols to carboxylic acids and its application in glycosaminoglycan syntheses. Chem Eur J. 2006;12:5246–5252. doi: 10.1002/chem.200600290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Palmacci ER, Seeberger PH. Toward the modular synthesis of glycosaminoglycans: Synthesis of hyaluronic acid disaccharide building blocks using a periodic acid oxidation. Tetrahedron. 2004;60:7755–7766. [Google Scholar]
- 49.La Ferla B, Lay L, Guerrini M, Poletti L, Panza L, Russo G. Synthesis of disaccharidic subunits of a new series of heparin related oligosaccharides. Tetrahedron. 1999;55:9867–9880. [Google Scholar]
- 50.Tabeur C, Mallet JM, Bono F, Herbert JM, Petitou M, Sinaÿ P. Oligosaccharides corresponding to the regular sequence of heparin: Chemical synthesis and interaction with FGF-2. Bioorg Med Chem. 1999;7:2003–2012. doi: 10.1016/s0968-0896(99)00113-3. [DOI] [PubMed] [Google Scholar]
- 51.Kovensky J, Duchaussoy P, Bono F, Salmivirta M, Sizun P, Herbert JM, Petitou M, Sinaÿ P. A synthetic heparan sulfate pentasaccharide, exclusively containing L-iduronic acid, displays higher affinity for FGF-2 than its D-glucuronic acid-containing isomers. Bioorg Med Chem. 1999;7:1567–1580. doi: 10.1016/s0968-0896(99)00106-6. [DOI] [PubMed] [Google Scholar]
- 52.Haller M, Boons GJ. Towards a modular approach for heparin synthesis. J Chem Soc Perkin Trans. 2001;1:814–822. [Google Scholar]
- 53.de Nooy AEJ, Besemer AC, van Bekkum H. Highly selective nitroxyl radical-mediated oxidation of primary alcohol groups in water-soluble glucans. Carbohydr Res. 1995;269:89–96. [Google Scholar]
- 54.Davis NJ, Flitsch SL. Selective oxidation of monosaccharide derivatives to uronic acids. Tetrahedron Lett. 1993;34:1181–1184. [Google Scholar]
- 55.Hu YP, Lin SY, Huang CY, Zulueta MML, Liu JY, Chang W, Hung SC. Synthesis of 3-O-sulfonated heparan sulfate octasaccharides that inhibit the herpes simplex virus type 1 host-cell interaction. Nat Chem. 2011;3:557–563. doi: 10.1038/nchem.1073. [DOI] [PubMed] [Google Scholar]
- 56.van den Bos LJ, Codée JDC, van der Toorn JC, Boltje TJ, van Boom JH, Overkleeft HS, van der Marel GA. Thioglycuronides: Synthesis and application in the assembly of acidic oligosaccharides. Org Lett. 2004;6:2165–2168. doi: 10.1021/ol049380+. [DOI] [PubMed] [Google Scholar]
- 57.Tiruchinapally G, Yin Z, El-Dakdouki M, Wang Z, Huang X. Divergent heparin oligosaccharide synthesis with pre-installed sulfate esters. Chem Eur J. 2011;17:10106–10112. doi: 10.1002/chem.201101108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Z, Xu Y, Yang B, Tiruchinapally G, Sun B, Liu R, Dulaney S, Liu J, Huang X. Preactivation-based, one-pot combinatorial synthesis of heparin-like hexasaccharides for the analysis of heparin-protein interactions. Chem Eur J. 2010;16:8365–8375. doi: 10.1002/chem.201000987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arungundram S, Al-Mafraji K, Asong J, Leach FE, III, Amster IJ, Venot A, Turnbull JE, Boons GJ. Modular synthesis of heparan sulfate oligosaccharides for structure-activity relationship studies. J Am Chem Soc. 2009;131:17394–17405. doi: 10.1021/ja907358k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen J, Zhou Y, Chen C, Xu W, Yu B. Synthesis of a tetrasaccharide substrate of heparanase. Carbohydr Res. 2008;343:2853–2862. doi: 10.1016/j.carres.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 61.Walvoort MTC, de Witte W, van Dijk J, Dinkelaar J, Lodder G, Overkleeft HS, Codée JDC, van der Marel GA. Mannopyranosyl uronic acid donor reactivity. Org Lett. 2011;13:4360–4363. doi: 10.1021/ol2016862. [DOI] [PubMed] [Google Scholar]
- 62.Zeng Y, Wang Z, Whitfield D, Huang X. Installation of electron-donating protective groups, a strategy for glycosylating unreactive thioglycosyl acceptors using the preactivation-based glycosylation method. J Org Chem. 2008;73:7952–7962. doi: 10.1021/jo801462r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dilhas A, Bonnaffe D. PhBCl2 promoted reductive opening of 2′,4′-O-p-methoxybenzylidene: New regioselective differentiation of position 2′ and 4′ of α-L-iduronyl moieties in disaccharide building blocks. Tetrahedron Lett. 2004;45:3643–3645. [Google Scholar]
- 64.Baleux F, Loureiro-Morais L, Hersant Y, Clayette P, Arenzana-Seisdedos F, Bonnaffe D, Lortat-Jacob H. A synthetic CD4-heparan sulfate glycoconjugate inhibits CCR5 and CXCR4 HIV-1 attachment and entry. Nat Chem Biol. 2009;5:743–748. doi: 10.1038/nchembio.207. [DOI] [PubMed] [Google Scholar]
- 65.Paulsen H. Progress in the selective chemical synthesis of complex oligosaccharides. Angew Chem Int Ed. 1982;21:155–173. [Google Scholar]
- 66.Codée JDC, Stubba B, Schiattarella M, Overkleeft HS, van Boeckel CAA, van Boom JH, van der Marel GA. A modular strategy toward the synthesis of heparin-like oligosaccharides using monomeric building blocks in a sequential glycosylation strategy. J Am Chem Soc. 2005;127:3767–3773. doi: 10.1021/ja045613g. [DOI] [PubMed] [Google Scholar]
- 67.Orgueira HA, Bartolozzi A, Schell P, Litjens REJN, Palmacci ER, Seeberger PH. Modular synthesis of heparin oligosaccharides. Chem Eur J. 2003;9:140–169. doi: 10.1002/chem.200390009. [DOI] [PubMed] [Google Scholar]
- 68.Orgueira HA, Bartolozzi A, Schell P, Seeberger PH. Conformational locking of the glycosyl acceptor for stereocontrol in the key step in the synthesis of heparin. Angew Chem Int Ed. 2002;41:2128–2131. [PubMed] [Google Scholar]
- 69.Lohman GJS, Seeberger PH. A stereochemical surprise at the late stage of the synthesis of fully N-differentiated heparin oligosaccharides containing amino, acetamido, and N-sulfonate groups. J Org Chem. 2004;69:4081–4093. doi: 10.1021/jo035732z. [DOI] [PubMed] [Google Scholar]
- 70.Lu LD, Shie CR, Kulkarni SS, Pan GR, Lu XA, Hung SC. Synthesis of 48 disaccharide building blocks for the assembly of a heparin and heparan sulfate oligosaccharide library. Org Lett. 2006;8:5995–5998. doi: 10.1021/ol062464t. [DOI] [PubMed] [Google Scholar]
- 71.Fan RH, Achkar J, Hernandez-Torres JM, Wei A. Orthogonal sulfation strategy for synthetic heparan sulfate ligands. Org Lett. 2005;7:5095–5098. doi: 10.1021/ol052130o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee JC, Lu XA, Kulkarni SS, Wen YS, Hung SC. Synthesis of heparin oligosaccharides. J Am Chem Soc. 2004;126:476–477. doi: 10.1021/ja038244h. [DOI] [PubMed] [Google Scholar]
- 73.Noti C, de Paz JL, Polito L, Seeberger PH. Preparation and use of microarrays containing synthetic heparin oligosaccharides for the rapid analysis of heparin-protein interactions. Chem Eur J. 2006;12:8664–8686. doi: 10.1002/chem.200601103. [DOI] [PubMed] [Google Scholar]
- 74.Huibers M, Manuzi A, Rutjes FPJT, van Delft FL. A sulfitylation-oxidation protocol for the preparation of sulfates. J Org Chem. 2006;71:7473–7476. doi: 10.1021/jo060404v. [DOI] [PubMed] [Google Scholar]
- 75.Simpson LS, Widlanski TS. A comprehensive approach to the synthesis of sulfate esters. J Am Chem Soc. 2006;128:1605–1610. doi: 10.1021/ja056086j. [DOI] [PubMed] [Google Scholar]
- 76.Karst NA, Islam TF, Linhardt RJ. Sulfo-protected hexosamine monosaccharides: Potentially versatile building blocks for glycosaminoglycan synthesis. Org Lett. 2003;5:4839–4842. doi: 10.1021/ol035882w. [DOI] [PubMed] [Google Scholar]
- 77.Ingram LJ, Taylor SD. Introduction of 2,2,2-trichloroethyl-protected sulfates into monosaccharides with a sulfuryl imidazolium salt and application to the synthesis of sulfated carbohydrates. Angew Chem Int Ed. 2006;45:3503–3506. doi: 10.1002/anie.200600153. [DOI] [PubMed] [Google Scholar]
- 78.Kreuger J, Salmivirta M, Sturiale L, Gimenez-Gallego G, Lindahl U. Sequence analysis of heparan sulfate epitopes with graded affinities for fibroblast growth factors 1 and 2. J Biol Chem. 2001;276:30744–30752. doi: 10.1074/jbc.M102628200. [DOI] [PubMed] [Google Scholar]
- 79.Tatai J, Fügedi P. Synthesis of the putative minimal FGF binding motif heparan sulfate trisaccharides by an orthogonal protecting group strategy. Tetrahedron. 2008;64:9865–9873. [Google Scholar]
- 80.Weishaupt M, Eller S, Seeberger PH. Solid phase synthesis of oligosaccharides. Methods Enzymol. 2010;478:463–484. doi: 10.1016/S0076-6879(10)78022-8. [DOI] [PubMed] [Google Scholar]
- 81.Plante OJ, Palmacci ER, Seeberger PH. Automated solid-phase synthesis of oligosaccharides. Science. 2001;291:1523–1527. doi: 10.1126/science.1057324. [DOI] [PubMed] [Google Scholar]
- 82.Czechura P, Guedes N, Kopitzki S, Vazquez N, Martin-Lomas M, Reichardt NC. A new linker for solid-phase synthesis of heparan sulfate precursors by sequential assembly of monosaccharide building blocks. Chem Commun. 2011;47:2390–2392. doi: 10.1039/c0cc04686h. [DOI] [PubMed] [Google Scholar]
- 83.Ojeda R, Terenti O, de Paz JL, Martin-Lomas M. Synthesis of heparin-like oligosaccharides on polymer supports. Glycoconj J. 2004;21:179–195. doi: 10.1023/B:GLYC.0000045091.18392.a8. [DOI] [PubMed] [Google Scholar]
- 84.de Paz JL, Angulo J, Lassaletta JM, Nieto PM, Redondo-Horcajo M, Lozano RM, Gimenez-Gallego G, Martin-Lomas M. The activation of fibroblast growth factors by heparin: Synthesis, structure, and biological activity of heparin-like oligosaccharides. Chembiochem. 2001;2:673–685. doi: 10.1002/1439-7633(20010903)2:9<673::AID-CBIC673>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 85.Prabhu A, Venot A, Boons GJ. New set of orthogonal protecting groups for the modular synthesis of heparan sulfate fragments. Org Lett. 2003;5:4975–4978. doi: 10.1021/ol0359261. [DOI] [PubMed] [Google Scholar]
- 86.Lubineau A, Lortat-Jacob H, Gavard O, Sarrazin S, Bonnaffe D. Synthesis of tailor-made glycoconjugate mimetics of heparan sulfate that bind IFN-γ in the nanomolar range. Chem Eur J. 2004;10:4265–4282. doi: 10.1002/chem.200306063. [DOI] [PubMed] [Google Scholar]
- 87.Poletti L, Fleischer M, Vogel C, Guerrini M, Torri G, Lay L. A rational approach to heparin-related fragments—Synthesis of differently sulfated tetrasaccharides as potential ligands for fibroblast growth factors. Eur J Org Chem. 2001:2727–2734. [Google Scholar]
- 88.de Paz JL, Martin-Lomas M. Synthesis and biological evaluation of a heparin-like hexasaccharide with the structural motifs for binding to FGF and FGFR. Eur J Org Chem. 2005:1849–1858. [Google Scholar]
- 89.Lucas R, Angulo J, Nieto PM, Martin-Lomas M. Synthesis and structural study of two new heparin-like hexasaccharides. Org Biomol Chem. 2003;1:2253–2266. doi: 10.1039/b303115b. [DOI] [PubMed] [Google Scholar]
- 90.de Paz JL, Ojeda R, Reichardt N, Martin-Lomas M. Some key experimental features of a modular synthesis of heparin-like oligosaccharides. Eur J Org Chem. 2003:3308–3324. [Google Scholar]
- 91.Ojeda R, Angulo J, Nieto PM, Martin-Lomas M. The activation of fibroblast growth factors by heparin: Synthesis and structural study of rationally modified heparin-like oligosaccharides. Can J Chem. 2002;80:917–936. [Google Scholar]
- 92.Daragics K, Fügedi P. Synthesis of glycosaminoglycan oligosaccharides. Part 5: Synthesis of a putative heparan sulfate tetrasaccharide antigen involved in prion diseases. Tetrahedron. 2010;66:8036–8046. [Google Scholar]
- 93.Cole CL, Hansen SU, Barath M, Rushton G, Gardiner JM, Avizienyte E, Jayson GC. Synthetic heparan sulfate oligosaccharides inhibit endothelial cell functions essential for angiogenesis. PLoS One. 2010;5:e11644. doi: 10.1371/journal.pone.0011644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nguyen HM, Poole JL, Gin DY. Chemoselective iterative dehydrative glycosylation. Angew Chem Int Ed. 2001;40:414–417. doi: 10.1002/1521-3773(20010119)40:2<414::AID-ANIE414>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 95.McDonnell C, Lopez O, Murphy P, Bolanos JGF, Hazell R, Bols M. Conformational effects on glycoside reactivity: Study of the high reactive conformer of glucose. J Am Chem Soc. 2004;126:12374–12385. doi: 10.1021/ja047476t. [DOI] [PubMed] [Google Scholar]
- 96.Jensen HH, Nordstrom LU, Bols M. The disarming effect of the 4,6-acetal group on glycoside reactivity: Torsional or electronic? J Am Chem Soc. 2004;126:9205–9213. doi: 10.1021/ja047578j. [DOI] [PubMed] [Google Scholar]
- 97.Zhang Z, Ollmann IR, Ye XS, Wischnat R, Baasov T, Wong CH. Programmable one-pot oligosaccharide synthesis. J Am Chem Soc. 1999;121:734–753. [Google Scholar]
- 98.Huang L, Wang Z, Huang X. One-pot oligosaccharide synthesis: Reactivity tuning by post-synthetic modification of aglycon. Chem Commun. 2004:1960–1961. doi: 10.1039/b405886k. [DOI] [PubMed] [Google Scholar]
- 99.Pornsuriyasak P, Gangadharmath UB, Rath NP, Demchenko AV. A novel strategy for oligosaccharide synthesis via temporarily deactivated S-thiazolyl glycosides as glycosyl acceptors. Org Lett. 2004;6:4515–4518. doi: 10.1021/ol048043y. [DOI] [PubMed] [Google Scholar]
- 100.Lahmann M, Oscarson S. Investigation of the reactivity difference between thioglycoside donors with variant aglycon parts. Can J Chem. 2002;80:889–893. [Google Scholar]
- 101.Polat T, Wong CH. Anomeric reactivity-based one-pot synthesis of heparin-like oligosaccharides. J Am Chem Soc. 2007;129:12795–12800. doi: 10.1021/ja073098r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li X, Huang L, Hu X, Huang X. Thio-arylglycosides with various aglycon para-substituents: A probe for studying chemical glycosylation reactions. Org Biomol Chem. 2009;7:117–127. doi: 10.1039/b813048e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Park TK, Kim IJ, Hu S, Bilodeau MT, Randolph JT, Kwon O, Danishefsky SJ. Total synthesis and proof of structure of a human breast tumor (Globo-H) antigen. J Am Chem Soc. 1996;118:11488–11500. [Google Scholar]
- 104.Yamago S, Yamada T, Hara O, Ito H, Mino Y, Yoshida Ji. A new, iterative strategy of oligosaccharide synthesis based on highly reactive β-bromoglycosides derived from selenoglycosides. Org Lett. 2001;3:3867–3870. doi: 10.1021/ol016713j. [DOI] [PubMed] [Google Scholar]
- 105.Yamago S, Yamada T, Maruyama T, Yoshida J. Iterative glycosylation of 2-deoxy-2-amino-thioglycosides and its application to the combinatorial synthesis of linear oligo-glucosamines. Angew Chem Int Ed. 2004;43:2145–2148. doi: 10.1002/anie.200353552. [DOI] [PubMed] [Google Scholar]
- 106.Huang X, Huang L, Wang H, Ye XS. Iterative one-pot synthesis of oligosaccharides. Angew Chem Int Ed. 2004;43:5221–5224. doi: 10.1002/anie.200460176. [DOI] [PubMed] [Google Scholar]
- 107.Kuberan B, Lech MZ, Beeler DL, Wu ZL, Rosenberg RD. Enzymatic synthesis of antithrombin III-binding heparan sulfate pentasaccharide. Nat Biotechnol. 2003;21:1343–1346. doi: 10.1038/nbt885. [DOI] [PubMed] [Google Scholar]
- 108.Chen J, Jones CL, Liu J. Using an enzymatic combinatorial approach to identify anticoagulant heparan sulfate structures with differing sulfation patterns. Chem Biol. 2007;14:986–993. doi: 10.1016/j.chembiol.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen J, Avci FY, Munoz EM, McDowell LM, Chen M, Pedersen LC, Zhang L, Linhardt RJ, Liu J. Enzymatic redesigning of biologically active heparan sulfate. J Biol Chem. 2005;280:42817–42825. doi: 10.1074/jbc.M504338200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lindahl U, Li JP, Kusche-Gullberg M, Salmivirta M, Alaranta S, Veromaa T, Emeis J, Roberts I, Taylor C, Oreste P, Zoppetti G, Naggi A, Torri G, Casu B. Generation of “Neoheparin” from E. coli K5 capsular polysaccharide. J Med Chem. 2005;48:349–352. doi: 10.1021/jm049812m. [DOI] [PubMed] [Google Scholar]
- 111.Wang Z, Ly M, Zhang F, Zhong W, Suen A, Hickey AM, Dordick JS, Linhardt RJ. E. coli K5 fermentation and the preparation of heparosan, a bioengineered heparin precursor. Biotechnol Bioeng. 2010;107:964–973. doi: 10.1002/bit.22898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Griffiths G, Cook NJ, Gottfridson E, Lind T, Lidholt K, Roberts IS. Characterization of the glycosyltransferase enzyme from the Escherichia coli K5 capsule gene cluster and identification and characterization of the glucuronyl active site. J Biol Chem. 1998;273:11752–11757. doi: 10.1074/jbc.273.19.11752. [DOI] [PubMed] [Google Scholar]
- 113.Hodson N, Griffiths G, Cook N, Pourhossein M, Gottfridson E, Lind T, Lidholt K, Roberts IS. Identification that KfiA, a protein essential for the biosynthesis of the Escherichia coli K5 capsular polysaccharide, is an α-UDP-GlcNAc glycosyltransferase. The formation of a membrane-associated K5 biosynthetic complex requires KfiA, KfiB, and KfiC. J Biol Chem. 2000;275:27311–27315. doi: 10.1074/jbc.M004426200. [DOI] [PubMed] [Google Scholar]
- 114.Xu Y, Masuko S, Takieddin M, Xu H, Liu R, Jing J, Mousa SA, Linhardt RJ, Liu J. Chemoenzymatic synthesis of homogeneous ultralow molecular weight heparins. Science. 2011;334:498–501. doi: 10.1126/science.1207478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu R, Xu Y, Chen M, Weiwer M, Zhou X, Bridges AS, De APL, Zhang Q, Linhardt RJ, Liu J. Chemoenzymatic design of heparan sulfate oligosaccharides. J Biol Chem. 2010;285:34240–34249. doi: 10.1074/jbc.M110.159152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Xu Y, Wang Z, Liu R, Bridges AS, Huang X, Liu J. Directing the biological activities of heparan sulfate oligosaccharides using a chemoenzymatic approach. Glycobiology. 2012;22:96–106. doi: 10.1093/glycob/cwr109. [DOI] [PMC free article] [PubMed] [Google Scholar]