

Abstract
Purpose
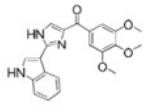
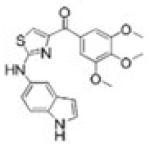
To evaluate the efficacy and oral activity of two promising indoles, (2-(1H-indol-3-yl)-1H-imidazol-4-yl)(3,4,5-trimethoxyphenyl)methanone [compound II] and (2-(1H-indol-5-ylamino)-thiazol-4-yl)(3,4,5-trimethoxyphenyl)methanone [compound IAT], in paclitaxel- and docetaxel-resistant tumor models in vitro and in vivo.
Methods
The in vitro drug-like properties, including potency, solubility, metabolic stability, and drug-drug interactions were examined for our two active compounds. An in vivo pharmacokinetic study and antitumor efficacy study were also completed to compare their efficacy with docetaxel.
Results
Both compounds bound to the colchicine-binding site on tubulin, and inhibited tubulin polymerization, resulting in highly potent cytotoxic activity in vitro. While the potency of paclitaxel and docetaxel was compromised in a multidrug-resistant cell line that overexpresses P-glycoprotein, the potency of compounds II and IATwas maintained. Both compounds had favorable drug-like properties, and acceptable oral bioavailability (21–50%) in mice, rats, and dogs. Tumor growth inhibition of greater than 100% was achieved when immunodeficient mice with rapidly growing paclitaxel-resistant prostate cancer cells were treated orally at doses of 3–30 mg/kg of II or IAT.
Conclusions
These studies highlight the potent and broad anticancer activity of two orally bioavailable compounds, offering significant pharmacologic advantage over existing drugs of this class for multidrug resistant or taxane-refractory cancers.
Keywords: paclitaxel resistant cancer, P-glycoprotein, pharmacokinetics, tubulin, xenograft
INTRODUCTION
Microtubules play important roles in cell mitosis and division (1–3). Some drugs targeting microtubules, such as the vinca alkaloids, the taxanes, and the epothilones, interrupt cell division, lead to apoptosis and are thus used for the treatment of cancer (4). Though their clinical efficacies are well established, patients almost invariably progress to refractory cancer wherein they fail to respond to additional therapy. One of the major problems facing this drug class is the multidrug resistance (MDR) developed by cancer cells due to over-expression of drug efflux pumps, most notably P-glycoprotein. In addition, the route of administration of these drugs is limited to intravenous infusion due to poor drug-like properties, such as extensive first-pass metabolism, affinity for P-glycoprotein, poor solubility, or low oral bioavailability. Ixabepilone, an epothilone that binds to the taxane-binding site on tubulin, was reported to be a poor substrate of P-glycoprotein, capable of maintaining potency in taxane-resistant cells (5). However, due to poor solubility, the formulation containing Cremophor® EL used for paclitaxel and docetaxel is still used for ixabepilone. Cremophor® EL itself is associated with hypersensitivity reactions (6) and patients need to be pre-treated with dexamethasone, clemastine, or ranitidine (7) to prevent unwanted side-effects. Oral administration of drugs is practical, convenient, and safer for patients with compromised immune function, as well as cost-effective. Although paclitaxel, docetaxel and ixabepilone suffer from poor oral bioavailability, they have been orally administered in phase I–II clinical trials in combination with P-glycoprotein inhibitors in attempts to achieve therapeutic exposure (7–10). However, limitations associated with the toxicity of the Cremophor EL vehicle used for oral administration, larger intersubject variation and the design of formulations with acceptable bioavailability have hampered further development of these drugs for oral use in cancer therapy.
Unlike drugs that bind to the taxane- or vinca alkaloid site on tubulin, drugs binding to a third site on tubulin, the colchicine-binding site, may provide a better opportunity to be developed for oral use due to their simpler chemical structures and favorable physicochemical properties. None of the colchicine-binding agents is approved for routine clinical use yet. However, some of them, such as ABT-751 (sulfonamide) (11–13), indibulin (14,15), and STX140 (16,17), are in preclinical or clinical development. These agents have shown the potential to be orally available and to circumvent P-glycoprotein-mediated drug resistance.
Combretastatin A4 phosphate (CA4P), a water-soluble prodrug of Combretastatin A4 (CA4), binds to the colchicine-binding site on tubulin, and has demonstrated activity in preclinical studies (18,19). CA4P is currently under clinical investigation in combination with other chemotherapy or radiation (20,21). Although larger patient studies will be needed to confirm its clinical activity, these early clinical trials suggest that the addition of CA4P to paclitaxel and carboplatin treatment may increase response rate in patients. Despite the fact that CA4P is a water-soluble small molecule, it is only administered via the intravenous route.
In the present study, we examined the in vitro and in vivo pharmacologic activity of two compounds, one with an imidazole linker and the other with an aminothiazole linker to a trimethoxy benzoyl ring, that arose from our studies to optimize the metabolic stability, permeability, and solubility of 4-substituted methoxy benzoyl-aryl-thiazoles and 2-arylbenzoyl imidazoles (22,23). Both were indoles with favorable pharmacokinetic properties, including satisfactory oral bioavailability, and circumvent P-glycoprotein-mediated drug resistance, exhibiting equal potency in PC-3 and paclitaxel-resistant PC-3 (PC-3/TxR) cell lines in vitro. Unlike docetaxel, treatment with the indoles caused tumor regression in PC-3/TxR xenografts. This study provides the first evidence that these compounds demonstrate great promise as the first members of a new class of orally bioavailable tubulin antagonists.
MATERIALS AND METHODS
Chemicals
(2-(1H-indol-3-yl)-1H-imidazol-4-yl)(3,4,5-trimethoxyphenyl)methanone [compound II] and (2-(1H-indol-5-ylamino)-thiazol-4-yl)(3,4,5-trimethoxyphenyl)methanone [compound IAT] were synthesized using methods similar to previously published procedures (22,24,25).
Cell Culture and Cytotoxicity Assays of Prostate Carcinoma and Glioma Cell Lines
The prostate cancer cell lines (LNCaP, PC-3, DU145, PPC-1) and glioma cell line (U87MG) were obtained from ATCC (American Type Culture Collection, Manassas,VA, USA). Prostate cancer cells were selected for these studies due to the prevalence of prostate cancer and the common use of docetaxel to treat this disease (26). The glioma cell line (U87MG) was used to examine the activity of the compounds in another type of cancer (i.e., glioma) and coordinate with the brain–blood barrier studies. Paclitaxel-resistant PC-3 (PC-3/TxR; a prostate cancer cell line that over-expresses P-glycoprotein) was a gift from Dr. Evan T. Keller at Department of Pathology, University of Michigan, Ann Arbor, Michigan (27). PC-3/TxR was employed as a MDR model. All cell lines were tested and authenticated by ATCC, and were immediately expanded and frozen such that they could be restarted every 2~3 months from a frozen vial of the same batch of cells. Cell culture supplies were purchased from Cellgro Mediatech (Herndon, VA, USA). Prostate cancer cell lines were maintained in RPMI 1640 media supplemented with 10% fetal bovine serum (FBS), while the glioma cancer cell line was maintained in Eagle's MEM media with 2 mML-glutamine and 10% FBS. The antiproliferative activity of the compounds of interest, paclitaxel, and docetaxel were tested in cell lines by sulforhodamine B (SRB) assay as previous described (28).
Competitive Mass Spectrometry Binding Assay
Competitive mass spectrometry binding studies were conducted as previously described (29). Colchicine (1.2 μM) was incubated with porcine brain tubulin (1.0 mg/mL) in the incubation buffer [80 mM piperazine-N,N'-bis(2-ethanesulfonic acid) (PIPES), 2.0 mM magnesium chloride (MgCl2), 0.5 mM ethylene glycol tetraacetic acid (EGTA), pH 6.9] at 37°C for 1 h. Varying concentrations (0.2–200 μM) of podophyllotoxin (positive control), compound II, compound IAT, and vinblastine (negative control) were used to compete with the binding of colchicine to tubulin. After 1 h incubation, the filtrate was obtained using an ultrafiltration method (microconcentrator) (Microcon, Bedford, MA) with a molecular cutoff size of 30 kDa. The ability of the compounds of interest to inhibit the binding of each ligand was expressed as a percentage of control binding in the absence of any competitor. Each experiment was performed in triplicate.
In Vitro Microtubule Polymerization Assay
Porcine brain tubulin (0.4 mg) (Cytoskeleton, Denver, CO) was mixed with 5 μM of the compounds of interest or vehicle [Dimethyl sulfoxide (DMSO)] and incubated in 100 μL of buffer [80 mM PIPES, 2.0 mM MgCl2, 0.5 mM EGTA, pH 6.9 and 1 mM guanosine-5'-triphosphate (GTP)]. The absorbance at 340 nm wavelength was monitored every min for 15 min (SYNERGY 4 Microplate Reader, Bio-Tek Instruments, Winooski, VT). The spectro-photometer was maintained at 37 °C for tubulin polymerization.
Metabolic Stability
Metabolic stability studies were conducted by incubating the compounds of interest (0.5 μM) in a total reaction volume of 1 mL containing 1 mg/mL microsomal protein in reaction buffer [0.2 M of phosphate buffer solution (pH 7.4), 1.3 mM nicotinamide adenine dinucleotide phosphate (NADP+), 3.3 mM glucose-6-phosphate, and 0.4 U/mL glucose-6-phosphate dehydrogenase] at 37°C in a shaking water bath. Pooled human liver microsomes were utilized to examine metabolic stability. The NADPH regenerating system (solution A and B) was obtained from BD Biosciences (Bedford, MA). The total DMSO concentration in the reaction solution was approximately 0.5% (v/v). Aliquots (100 μL) from the reaction mixtures used to determine metabolic stability were sampled at 5, 10, 20, 30, 60, and 90 min. Acetonitrile (150 μL) containing 200 nM of the internal standard (23) was added to quench the reaction and to precipitate the proteins. Samples were then centrifuged at 4,000g for 15 min at room temperature, and the supernatant was analyzed directly by liquid chromatography tandem mass spectrometry (LC-MS/MS).
Permeability in Caco-2 Assay
1.2×104 Caco-2 cells/well were seeded in 24-well PET (polyethylene terephthalate) transwell plates (BD Bioscience, San Jose, CA) and maintained for 21 days in Dulbecco's Modified Eagle's Medium (DMEM) medium with 20% FBS to allow the cells to mature in terms of transporter expression (30,31). Compounds II, IAT, or propranolol (10 μM) was added to the donor chamber for 2 h. Aliquots (100 μL) of medium from both donor and receiver chambers were mixed with 100 μL acetonitrile containing 200 nM internal standard. Concentrations were determined by LC-MS/MS, and used to calculate permeability values (Papp). The Papp was calculated using the following equation: Papp 0 (Vr/C1) (1/S) (C2/t), where Vr is the volume of medium in the receiver chamber, C1 is the measured concentration of the test compound in the donor chamber at t=0, S is the surface area of monolayer, C2 is the measured concentration of the test compound in the receiver chamber after 2 h incubation, and t is the incubation time (2 h). Propranolol was used as a control in this assay.
Aqueous Solubility
The solubility of compounds II ad IAT was determined by Multiscreen Solubility Filter Plate (Millipore Corporate, Billerica, MA) coupled with LC-MS/MS. Briefly, 198 μL of phosphate buffered saline (PBS) buffer (pH 7.4) was loaded into a 96-well plate, and 2 μL of 10 mM test compounds (in DMSO) was dispensed and mixed with gentle shaking (200–300 rpm) for 1.5 h at room temperature (N=3). The plate was centrifuged at 800g for 10 min, and the filtrate was used to determine its concentration and solubility of test compound by LC-MS/MS methods.
CYP Enzyme Specific Assays
Five CYP enzyme inhibition assays were performed, and analyzed by LC-MS/MS. Briefly, for CYP2D6, CYP2C9, CYP1A2, and CYP2C19, 0.1 mg/mL microsomal protein was incubated with their specific substrates in 0.1 M potassium phosphate buffer (pH 7.4) at 37°C, while 0.05 mg/mL microsomal protein was used for CYP3A4 assays. The substrates, testosterone (50 μM), dextromethorphan (7 μM), (S)-mephenytoin (80 μM), diclofenac (7 μM), and phenacetin (100 μM), were used for CYP 3A4, 2D6, 2 C19, 2 C9, 1A2 inhibition assays, respectively. Various concentrations ranging 0.04–50 μM of compound II or IAT were examined in these assays. Positive controls used for CYP 3A4, 2D6, 2 C19, 2 C9, and 1A2 inhibition assays were ketoconazole (0.0009–1 μM), quinindine (0.0009–1 μM), ticlopidine (0.019–20 μM), sulfaphenazole (0.019–20 μM), and furafylline (0.04–50 μM), respectively.
Pharmacokinetic Study
Male ICR mice (N=3 or 4 per group) 6–8 weeks of age were purchased from Harlan Inc. (Indianapolis, IN), and used to examine the pharmacokinetics (PK) of compound II or IAT. Both chemicals were formulated in DMSO/Polyethylene glycol 300 (PEG300), 1/9, v/v, and Tween80/DMSO/H2O, 2/2/6, v/v/v for intravenous bolus (i.v., 10 mg/kg) and oral (p.o., 20 mg/kg) administration, respectively. Dosing volume for i.v. was 50 μL via tail vein, while the volume for p.o. was 200 μL through oral gavage. For i.v. administration, blood samples were collected at 2, 5, 15, and 30 min, 1, 2, 4, 8, 16, and 24 h after administration. For p.o. administration, blood samples were collected at 0.5, 1, 1.5, 2, 3, 4, 8, 16, and 24 h after administration. Plasma samples were prepared by centrifuging the blood samples at 8,000g for 5 min. All plasma samples were stored immediately at −80°C until analyzed.
Female Sprague–Dawley rats (N=3 or 4 per group) were purchased from Harlan Inc. (Indianapolis, IN). Rat thoracic jugular vein catheters were purchased from Braintree Scientific Inc. (Braintree, MA). All animals were fed prior to dosing. Dosing volumes for intravenous bolus (i.v.) and oral (p.o.) solutions were 2 and 4 mL/kg, respectively. Compound II or IAT was administered i.v. into the thoracic jugular vein at a dose of 5 mg/kg (in DMSO/PEG300, 1/9, v/v). The dose in rats was chosen to be one-half of the dose in mice based onbody surface area. Catheters were flushed with 1 mL of heparinized saline after i.v. bolus. An equal volume of heparinized saline was injected to replace the removed blood, and blood samples (250 μL) were collected via the jugular vein catheter at 10, 20, 30 min, and 1, 2, 4, 8, 12, 24 h. Compounds II and IAT were also given (p.o.) by oral gavage at 10 mg/kg (in Tween80/DMSO/H2O, 2/2/6, v/v/v) to evaluate their oral bioavailability. All blood samples (250 μL) after oral administration were collected via the jugular vein catheter at 30, 60, 90 min, 120 min, 150 min, 180 min, 210 min, 240 min, and 8, 12, 24 h. Heparinized syringes and vials were prepared prior to blood collection.
Female beagle dogs weighing about 10 kg were used in this study. The dogs (N=4) were given a single intravenous dose of compound II or IAT (2 mg/kg, in DMSO/PFG300, 1/9, v/v), in a dosing volume of 0.2 mL/kg. Blood was drawn at 10, 20, 30 min, and 1, 2, 4, 8, 12, 24, 48, 96 h. For p.o. administration (N=4), the dogs (N=4) were given a single oral dose of compound IAT (5 mg/kg, in Tween80/DMSO/H2O, 2/2/6, v/v/v) in a dosing volume of 1 mL/kg. We selected an oral dose level (5 mg/kg) in dogs that was one-fourth of the dose in mice and slightly higher than would be needed to correct for differences in body surface area (i.e., one sixth of the dose in mice) due to in vitro studies with liver microsomes from these species indicating less metabolic stability of the compounds in dogs (data not shown). Blood was drawn at 20, 40, 60, 80, 100, 120, 150, 180, 210 min and 4, 8, 12, 24, 48, 96 h.
A protein precipitation method was used for sample preparation. An aliquot (200 μL) of acetonitrile (ACN) containing the internal standard was added to 100 μL of plasma and then was thoroughly vortexed for 15 s. After centrifugation, the supernatant was analyzed by LC-MS/MS. The pharmacokinetic parameters were determined using noncompartmental analysis (WinNonlin 6.0, Pharsight Corporation, Mountain View, CA)
Brain Penetration Study
Plasma and brain tissue were collected after a single dose oral administration (20 mg/kg) of compounds II and IAT, and single intraperitoneal administration (10 mg/kg) of docetaxel from nude mice. All three chemicals were formulated in Tween80/DMSO/H2O, 2/2/6, v/v/v. At the indicated time points (1 h and 4 h) after dosing, blood and brain tissue was collected from nude mice. Plasma was prepared as previously described and stored at −80°C until analyzed.
Brain tissue samples were individually ground to a powder with a Bessman tissue pulverizer (Spectrum Laboratories, Rancho Dominquez, CA) (32). The pulverizer was pre-cooled for 1 min in liquid nitrogen. Approximately 50 mg of tissue was placed on the pulverizer, and the whole apparatus was cooled in liquid nitrogen for 1 min and then the tissue was ground to a fine powder. The powder was immediately transferred to a sample vial, vortexed with 4 volumes of water, and then 10 volumes of acetonitrile containing the internal standard were added for extraction. After centrifugation, the supernatant was analyzed by LC-MS/MS to determine their brain and plasma concentrations.
PC-3 and Paclitaxel-Resistant PC-3 (PC-3/TxR) Tumor Xenograft Studies
PC-3 or PC-3/TxR cells (108 per mL) were prepared in growth media containing 10% FBS and mixed with high concentration, phenol red-free Matrigel (BD Biosciences, San Jose, CA) at 1:1 ratio. Tumors were established by injecting 100 μL of the mixture (5×106 cells per animal) subcutaneously (s.c.) into the flank of 6–8-week-old male athymic nude mice. The length and width of tumors were measured and the tumor volume (mm3) was calculated by the formula, π/6 × L × W2, where length (L) and width (W) were determined in mm. When the tumor volumes reached about 150–300 mm3, the animals were treated with an intravenous formulation [Tween80/ethanol/saline (7.5/7.5/85)] or oral formulation [Tween80/DMSO/H2O (2/2/6)]. Docetaxel (10 or 20 mg/kg) was intravenously dosed on day 1 and day 9 in both PC-3 and PC-3/TxR xenograft models while compound II (6.7 mg/kg) was dosed orally (qd, five times a week) in PC-3/TxR xenograft model. In another PC-3/TxR xenograft study, compound II (3.3 mg/kg) was dosed twice a day (b.i.d.) for the first four days in the first week, and then the schedule was changed to once daily, five days a week during week 2–4 due to toxicity. While compound IAT (10 and 30 mg/kg) was orally dosed b.i.d. on mice, five times a week for four weeks, a higher dose of compound II (10 mg/kg) was also examined in PC-3/TxR xenografts, with every other day (q2d) treatments.
RESULTS
Compound II and IAT Exhibit Potent Cytotoxicity in Cancer Cells, Including Multidrug-Resistant Cells
The ability of compound II and IAT to inhibit the growth of cancer cell lines was evaluated using SRB assay (Table I). Both compounds inhibited the growth of several human cancer cell lines, including one glioma and five prostate cancer cell lines, with IC50 values in the low nanomolar range. Compound II exhibited 1.7~4.3 fold higher potency than compound IAT in these cell lines. The PC-3/TxR cell line that over-expressed P-glycoprotein was used to examine the ability of compounds II and IAT to maintain activity in paclitaxel-refractory cells in vitro (Table I). Compounds II and IAT were equipotent against parents PC-3 and drug resistant PC-3/TxR cells, whereas paclitaxel and docetaxel exhibited relative resistance value of 85- and 15-fold, respectively. These data indicate that both compounds II and IAT circumvent P-glycoprotein-mediated drug resistance consistent with our previous lead compound, 4-(3,4,5-trimethoxybenzoyl)-2-phenyl-thiazole (SMART-H) (28).
Table I.
Cytotoxicity Data of Compounds II and IAT. IC50 Values (Mean ± SD) were Determined after 96 h Treatment (N=3). Paclitaxel was used as a Positive Control. Data in Parentheses Indicated Resistance Factor when Compared IC50 Values in PC-3and PC-3/TxR. NR=Not Reported
| Cell line | Type | Cytotoxicity [IC50 values, mean ± SD nM] | |||
|---|---|---|---|---|---|
| II | IAT | Paditaxel | Docetaxel | ||
|
| |||||
|
|
|
|
||
| PC-3 | Prostate | 5.2±0.2 | 16±15 | 0.6±0.05 | 1.2±0.1 |
| PC-3/TxR | Prostate | 2.1±0.1 (0.4) | 67±0.5 (0.4) | 51±2.3 (85) | 18±0.7 (15) |
| LNCaP | Prostate | 12±0.1 | 27±0.6 | 1.7±0.2 | 4.7±1.3a |
| Du-145 | Prostate | 17±0.2 | 38±0.6 | 5.1±0.1 | 5.2±1.0a |
| PPC-1 | Prostate | 21±0.1 | 36±0.4 | 23±0.8 | 2.7±1.0a |
| U87MG | Glioma | 10±1.6 | 22±30 | NR | NR |
Data previously reported in Ahn et al., MCT 9:2959, 2010 (33)
Compounds II and IAT Bind to the Colchicine-Binding Site on Tubulin, Inhibit Tubulin Polymerization, and Induce Cancer Cell Apoptosis
A competitive mass spectrometry binding assay was developed to study the interactions of small molecule inhibitors with tubulin, and was applied to examine our lead compound, SMART-H, which selectively binds to the colchicine site on tubulin (29). Compounds II and IAT compounds competed effectively with colchicine for tubulin binding (Fig. 1a), with potency similar to podophyllotoxin. Vinblastine, the negative control, did not inhibit colchicine binding to tubulin, successfully demonstrating the specificity of this competitive mass spectrometry binding assay.
Fig. 1.

Compounds II and IAT bind to the colchicine-binding site on tubulin and inhibit tubulin polymerization. (a) Competitive mass spectrometry binding. Tubulin (1 mg/mL) and colchicine (1.2 μM) were incubated with various concentrations of podophylltoxin, vinblastine, II, and IAT. N=3; mean ± SD. Podophylltoxin and vinblastine were used as positive and negative controls, respectively. (b) Effect on tubulin polymerization. Tubulin (0.4 mg) was exposed to test compounds (5 μM). Colchicine was used as positive control. (c, d) Ability of II and IAT to enhance cytoplasmic DNA-Histone complex formation (apoptosis) at 24 h in PC-3 (c) and PC-3/TxR (d) cells (N=3); mean ± SD. Docetaxel was used as positive control.
Porcine brain tubulin (> 97% pure) was incubated with II or IAT (5 μM) to test their effects on tubulin polymerization (Fig. 1b). Compounds II and IAT inhibited tubulin polymerization by 47 and 40% at 15 min, respectively. Colchicine at 5 μM was used as a positive control and inhibited tubulin polymerization by 32%. These data suggested that both II and IAT inhibit tubulin polymerization better than colchicine and indicated that these compounds bind the same site as SMART compounds (28).
PC-3 and PC-3/TxR cells were exposed to 0.8–600 nmol/L of compound II, IAT or docetaxel for 24 h. Both compound II and IAT were equally potent in their ability to induce cell apoptosis in PC-3 (Fig. 1c) and PC-3/TxR (Fig. 1d) within 24 h as measured by DNA histone complex formation. Although docetaxel potently induced cell apoptosis in PC-3 cells, it was substantially weaker in PC-3/TxR cells due to the over-expression of P-glycoprotein.
Compounds II and IAT Exhibited Favorable Drug-Like Properties
The drug-like properties of II and IAT, such as metabolic stability, permeability, aqueous solubility, and drug-drug interactions, were examined (Table II). Compound II exhibited greater metabolic stability and aqueous solubility than IAT. Both compounds exhibited more than adequate permeability values, suggesting that they would be amenable to oral administration. In addition, both compounds showed high IC50 values (in the micromolar range) during CYP enzyme inhibition assays, suggesting that they will not cause CYP-mediated drug-drug interactions.
Table II.
Drug-like Properties of Compounds II and IAT Metabolic Stability, Permeability, Solubility, and Potential Drug-drug Interactions were Evaluated. Each Value Represents the Mean from Duplicate Studies
| Measurement | Units | II | IAT | positive controls (mean) |
|---|---|---|---|---|
| Metabolic stability | ||||
| half-life in human liver microsomes | min | >60 | 28 | Verapamil (12) |
| Permeability | ||||
| Papp(A→B) in CaCO-2 assay | 10−6 cm/s | 36 | 99 | Propranolol (19) |
| Aqueous solubility | μg/ml | >75 | 19 | SMART-H (1.1) |
| Drug-drug interactions | ||||
| IC50 value for Cyp3A4 (substrate: Testosterone) | μM | 20 | 5.5 | Ketoconazole (0.02) |
| IC50 value for Cyp2D6 (substrate: Dextromethorhan) | μM | >50 | 34 | Quinindine (0.1) |
| IC50 value for Cyp2C19 (substrate: (S)-mephennytion) | μM | 6.6 | 5.3 | Ticlopidine (0.37) |
| IC50 value for Cyp2C9 (substrate: Diclofenac) | μM | 17 | 4.9 | Sulfaphenazole (0.5) |
| IC50 value for Cyp1CA2 (substrate: Phenacetin) | μM | 9.2 | 8.1 | Furafylline (2.2) |
Pharmacokinetic Studies in Mice, Rats and Dogs
The pharmacokinetic parameters of compounds II and IAT after single intravenous or oral doses in ICR mice, Sprague–Dawley rats, and Beagle dogs are summarized in Table III. Compound II exhibited low clearance in mice and rats, suggesting that it exhibited prolonged metabolic stability and minimal first-pass hepatic metabolism in these species. In addition, II had a moderate volume of distribution in mice and rats, suggesting that it is widely distributed in tissues. Surprisingly, the total clearance of compound II in dogs was high. Two abundant metabolites in dog plasma, a hydroxylated metabolite and an unknown metabolite with +34 m/z than the parent (data not shown), were observed, which were consistent with those found in dog liver microsomes. In addition, abundant metabolites were observed when compound II was incubated with dog liver microsomes, but not in mouse, rat or human liver microsomes (data not shown). Nevertheless, compound II showed acceptable oral bioavailability of 21%, 36%, and 50% in rats, mice, and dogs, respectively. Compound IAT had low systemic clearance in rats and moderate clearance in mice and dogs. Similar to II, compound IAT exhibited moderate volume of distribution in these species. Compound IAT had comparable oral bioavailability among the three species (24%~36%).
Table III.
Pharmacokinetic Parameters of Compounds II and IAT in Mice, Rats, and Dogs
| II |
IAT |
|||
|---|---|---|---|---|
| IV | PO | IV | PO | |
| Mouse PK (N=3) | ||||
| Dose, mg/kg | 10 | 20 | 10 | 20 |
| Clearance, mL/min/kg | 19 | NR | 40 | NR |
| Vss, L/kg | 2.9 | NR | 1.3 | NR |
| t1/2, min | 101 | 339 | 46 | 126 |
| AUC, min*μg/ml | 540 | 384 | 249 | 171 |
| Cmax, ng/ml | 4800 | 1560 | 7739 | 1253 |
| F, % | 36% | 34% | ||
| Rat PK (N=3) | ||||
| Dose, mg/kg | 5 | 10 | 5 | 10 |
| Clearance, mL/min/kg | 905±2.3 | NR | 10±1.4 | NR |
| Vss, L/kg | 1.8±0.2 | NR | 1.0±0.1 | NR |
| t1/2, min | 139±24 | 206±12 | 73±5.0 | 350±214 |
| AUC, min*μg/mL | 553±143 | 233±134 | 509±73 | 246±163 |
| Cmax, ng/mL | 3672±519 | 999±445 | 4609±55 | 757±520 |
| F, % | 21% | 24% | ||
| Dog PK (N=4) | ||||
| Dose, mg/kg | 2 | 5 | 2 | 5 |
| Clearance, mL/min/kg | 109±29 | NR | 15±3.2 | NR |
| Vss, L/kg | 94±95 | NR | 0.9±0.2 | NR |
| t1/2, min | 2757±1573 | 1695±439 | 82±15 | 191±9.0 |
| AUC, min*μg/mL | 18.5±4.7 | 23.1±11.3 | 141±30 | 128±154 |
| Cmax, ng/mL | 400±118 | 210±133 | 2552±576 | 862±1010 |
| F, % | 50% | 36% | ||
Brain Penetration of Compounds II and IAT in Nude Mice
The ratios of whole brain to plasma concentrations of compound II and IAT were determined and compared to docetaxel in the nude mice (Table IV). Compound IAT exhibited a greater brain penetration than compound II and docetaxel. Compound II achieved slightly greater brain/plasma concentration ratios than docetaxel at both 1 and 4 h, while the IAT concentrations in brain reached 14–19% of plasma concentrations at 1 h and 4 h, respectively, showing a 3.2-fold higher brain/plasma ratio at both 1 h and 4 h compared to docetaxel.
Table IV.
Brain–Blood Barrier (BBB) Studies of Compounds II and IAT Brain and Plasma Concentrations were Determined in Nude Mice at 1 and 4 h after Administration of Docetaxel (i.p., 10 mg/kg), II (p.o., 20 mg/kg), and IAT (p.o., 20 mg/kg). Each Value Represents the Mean ± SD from 3 Nude Mice
| Measurement | Docetaxel |
II |
IAT |
|||
|---|---|---|---|---|---|---|
| 1 hr | 4 hr | 1 hr | 4 hr | 1 hr | 4 hr | |
| Brain (ng/mL) | 33±14 | 20±9 | 124±108 | 49±32 | 180±44 | 73±18 |
| Plasma (ng/mL) | 768±92 | 345±94 | 2058±1252 | 570±438 | 1669±867 | 380±32 |
| Brain/plasma (%) | 4.4±2.0 | 6.0±2.9 | 5.4±1.9 | 8.9±1.7 | 14±7.9 | 19±3.1 |
Compounds II an IAT Inhibit Paclitaxel-Resistant Prostate (PC-3/TxR) Xenograft Growth
Parental PC-3 and paclitaxel-resistant prostate cancer PC-3 (PC-3/TxR) cells were inoculated into nude mice and the tumor volumes were allowed to reach about 150~300 mm3. Docetaxel (10 or 20 mg/kg), an anticancer drug approved for clinical use in advanced prostate cancer, was used for comparison. PC-3/TxR tumor xenografts grew rapidly and reached tumor volumes of 1500–2500 mm3 by the end of the study. Although intravenously administered docetaxel (10 and 20 mg/kg) demonstrated in vivo anticancer activity in both models (Fig. 2a, b), the tumor growth inhibition (TGI) effect decreased from 84% TGI in PC-3 tumors to 14% TGI in PC-3/TxR tumors when intravenously dosed at 10 mg/kg (Table V). At the higher dose (20 mg/kg), docetaxel elicited partial regression (>100% TGI) of PC-3 tumors, but only 56% TGI in PC-3/TxR tumors. The effectiveness of docetaxel in PC-3/TxR tumors was decreased when compared to that in PC-3 tumors, suggesting that the efficacy was impaired by P-glycoprotein-mediated drug resistance. These results were consistent with our in vitro cytotoxicity and apoptosis data. In contrast to the lack of efficacy of docetaxel in PC-3/TxR tumors, compound II (6.7 mg/kg) was dosed orally and demonstrated more than 100% TGI without an effect on body weight (Fig. 2b and Table V). In addition, 2 out of 4 nude mice bearing PC-3/TxR tumors treated with compound II were tumor free on day 19.
Fig. 2.

In vivo anticancer efficacy. (a) Nude mice bearing PC-3 tumors were treated with docetaxel (i.v., 10 or 20 mg/kg) on day 1 and 9 (N=5–6). Bars, SE. (b) Nude mice bearing PC-3/TxR tumors were treated with docetaxel (i.v., 10 or 20 mg/kg) on day 1 and 9, II treatments (p.o., 6.7 mg/kg) once daily, five days a week. (N=4–5). Bars, SE. (c) Nude mice bearing PC-3/TxR tumors were treated with II (p.o., 3.3 mg/kg) twice a day for the first four consecutive days in the first week, and then dosed once daily, five days a week for week 2–4 (N=7), while IAT treatments (p.o., 10 or 30 mg/kg) were twice daily, five days a week for four weeks (N=7). Bars, SE. (d) Nude mice bearing PC-3/TxR tumors were treated with II (p.o., 10 mg/kg) three times a week for four weeks (N5). Bars, SE.
Table V.
Antitumor Activity of Compounds II and IAT Versus Concomitantly Evaluated Docetaxel in vivo
| Dosing Schedule | End point | Number End/Start | Body weight (g) |
Tumor size (mm3) |
TGI (%) | |||
|---|---|---|---|---|---|---|---|---|
| Start | End | Start | End | |||||
| PC-3 xenograft | ||||||||
| Vehicle_IV | day 1and 9 | day 19 | 6/6 | 30±2 | 32±4 | 271±83 | 875±292 | – |
| Docetaxel_IV_10mpk | day 1and 9 | day 19 | 5/5 | 29±2 | 24±2 | 247±49 | 341±101 | 84 |
| Docetaxel_IV_20mpk | day 1and 9 | day 19 | 5/5 | 28±3 | 24±3 | 243±68 | 172±62 | >100 |
| PC-3TxR xenograft | ||||||||
| Vehicle_IV | day 1and 9 | day 19 | 5/5 | 33±1 | 26±5 | 171±57 | 2061±858 | – |
| Docetaxel_IV_10mpk | day 1and 9 | day 19 | 4/4 | 31±2 | 25±2 | 143±20 | 1774±183 | 14 |
| Docetaxel_IV_20mpk | day 1and 9 | day 19 | 4/4 | 30±1 | 25±4 | 170±86 | 999±905 | 56 |
| II_PO_6.7mpk | qd×5/wa | day 19 | 4/4 | 33±3 | 34±3 | 172±69 | 126±100 | >100 |
| Vehicle_PO | b.i.d×5/wb | day 26 | 6/7 | 30±2 | 25±2 | 156±30 | 2591±1423 | – |
| IAT_PO_10mpk | b.i.d×5/w | day 26 | 7/7 | 29±2 | 26±3 | 143±44 | 1152±43 | 59 |
| IAT_PO_30mpk | b.i.d×5/w | day 26 | 7/7 | 29±3 | 30±2 | 134±34 | 101±19 | >100 |
| II_PO_3.3mpkd | qd×5/w | day 26 | 7/7 | 29±2 | 30±2 | 139±44 | 214±172 | 97 |
| Vehicle_PO | q2d×3/wc | day 29 | 5/5 | 24±2 | 21±1 | 299±40 | 1521±580 | – |
| II_PO_10mpk | q2d×3/w | day 29 | 5/5 | 24±2 | 28±2 | 294±156 | 237±103 | >100 |
qd × 5/w = one administration given daily consecutively for five times a week
b.i.d. × 5/w = twice administration given daily consecutively for five times a week
q2d × 3/w = every other day administration for three times a week
A b.i.d. × 4/w schedule was used for the first week in this group, and qd × 5/w for weeks 2–4
The PC-3/TxR xenograft model was further utilized to evaluate the efficacies of compound II and IAT when administered using different dosing schedules. The maximal tolerated doses (body weight loss >20%) of II was 10 mg/kg when orally dosed once daily for 4 days; or at 3.3 mg/kg when administered twice daily (b.i.d.) for 5 days (data not shown). As shown in Fig. 2c, 3.3 mg/kg of II was administered b.i.d. for the first four consecutive days in the first week, and the schedule was then changed to once daily on week 2 and 4. Partial regression was obtained during days 4–19. The TGI was 97% and one of the seven mice was tumor free on day 26. A higher dose (10 mg/kg) with lower dosing frequency (q2d) of compound II (Fig. 2d) elicited partial tumor regression during days 13–29, suggesting that alternative dosing regimens successfully inhibit PC-3/TxR xenograft growth. Compound IAT was orally administered to nude mice at a dose of 10 or 30 mg/kg b.i.d., five times a week between weeks 1 and 4 (Fig. 2c). The TGI value was 59% for mice that received 10 mg/kg IAT, while partial regression (>100% TGI) from day 19 to the termination of the study (day 26) was observed in animals treated with the higher dose (30 mg/kg) of IAT. Some mice in vehicle group had lower body weights at the endpoint, in part, due to muscle wasting and/or cancer cachexia. On the contrary, mice treated with Compound II (3.3 mg/kg) or IAT (30 mg/kg) gained weight (Table V), suggesting that these optimized doses of II or IAT were well-tolerated.
DISCUSSION
Docetaxel and paclitaxel are widely used in the treatment of various malignancies, but require intravenous administration due to low passive absorption and/or high active efflux (P-glycoprotein) if administered orally. Although cyclosporine has been used in an attempt to increase the oral activity of taxanes, it also causes undesirable side effects and can result in variable pharmacokinetics and exposure (7). The goal of this study was to develop highly potent tubulin antagonists with efficacy comparable to intravenously administered docetaxel, but with good oral bioavailabililty.
A successful lead optimization for oral use requires careful consideration of various drug-like properties, including metabolic stability, aqueous solubility, drug-drug interactions, permeability, in addition to potency and efficacy. The poor oral bioavailability of our previous compound, SMART-H, was mainly due to its low solubility (23). In this investigation, the solubility was improved by introducing an imidazole ring (compound II) or an amino-linker (compound IAT) (22,24). Both compounds exhibited favorable drug-like properties and had 21–50% oral bioavailability in mice, rats, and dogs. Most importantly, both compounds maintained anti-proliferative potency in the low nanomolar range against cancer cell lines, including an P-glycoprotein-overexpressing cell line. Altogether, these properties support the conclusion that both II and IAT may be potent and orally available tubulin antagonists.
We tested compounds II and IAT against a multidrug-resistant cell line (PC-3/TxR) that over-expresses P-glycoprotein and demonstrated that they had equipotent anti-proliferative effects in PC-3 (parent) and PC-3/TxR (MDR-positive) cells, suggesting that they effectively inhibited tumor cell proliferation in a P-glycoprotein-independent manner. In contrast, docetaxel, a p-glycoprotein substrate showed a 15-fold difference when comparing its effects in these two cell lines. Docetaxel effectively reduced the tumor burden of nude mice bearing PC-3 tumor but was less effective in mice bearing PC-3/TxR xenografts, suggesting that the compromised antitumor efficacy was associated with p-glycoprotein-mediated drug resistance. The potency and efficacy of compounds II and IAT were effective in the PC-3/TxR xenografts. Partial regression or TGI % values exceeding 90% were achieved after 4 weeks of treatment with II (3.3–10 mg/kg) and IAT (30 mg/kg). In general, anticancer compounds are considered to be “moderately active” when they achieve a TGI % value of 60% (or %T/C value of 40%, TGI%=100%-%T/C value), and considered to have “good activity” when TGI values approach 90% (28). As such, II and IAT can be classified as very active in this paclitaxel- or docetaxel-refractory animal model, indicating that they circumvent P-glycoprotein-mediated drug resistance.
Interestingly, although compound II exhibited 4.3-fold higher potency than IAT in the PC-3/TxR cell line, approximate 15-fold higher doses were needed for IAT compared to II to achieve more than 90% TGI in vivo (Fig. 2c). It appears that three main properties, potency, oral exposure (the area under the plasma concentration-time curve, AUC) in mice, and tumor penetration, contributed to this difference. Compound II demonstrated about 4.3-fold, 2.2-fold, and 2.6-fold greater potency, oral exposure, and tumor penetration, respectively than compound IAT. In addition, tumor/plasma concentration ratios (N=3) at 1 h were 113 ± 70 and 43 ± 33% for II (3.3 mg/kg) and IAT (30 mg/kg), respectively, when administered orally.
Brain concentrations of II and IAT were also examined since both compounds exhibited high permeability and a lack of dependence on P-glycoprotein. Compound IAT (20 mg/kg) had brain/plasma concentration ratios of 14% and 19%, respectively, at 1 and 4 h after oral dosing suggesting that it readily crossed the brain–blood barrier. In addition, brain/plasma concentration ratios remained relatively constant for II and IAT over time, suggesting that brain concentrations were in the same pharmacokinetic compartment as plasma and that II and IAT will not accumulate in the brain, perhaps reducing the possibility for neurotoxicity or unwanted side effects. We performed neurotoxicity studies for our previous lead compound (SMART-H), and did not find any signs of obvious neurotoxicity (28). We plan to conduct similar studies with compounds II and IAT to test this hypothesis. We also plan to conduct studies using orthotopic glioma xenografts as a first step toward determining the efficacy, if any, of these compounds for gliomas.
In summary, we have demonstrated that compounds II and IAT are potent, orally active tubulin antagonists. They bind to the colchicine-binding site on tubulin, inhibit tubulin polymerization, induce cell apoptosis, and retain potency in P-glycoprotein-overexpressing cell lines. Based on the CYP inhibition studies, they are unlikely to cause or be affected by drug-drug interactions involving the predominant CYP enzymes. Both compounds exhibit favorable drug-like and pharmacokinetic properties, making the oral route feasible for anticancer therapy and offering several potential benefits, including cost-effectiveness and convenience for patients. Unlike docetaxel, both compounds are very active in paclitaxel- or docetaxel-refractory tumors in vivo. These results suggest that compounds II and IAT may have therapeutic potential for oral use in paclitaxel- or docetaxel-refractory cancer.
ACKNOWLEDGMENTS AND DISCLOSURES
This work was partially supported by the NIH/NCI [Grant R01CA148706-01A1]. CML, YL, JC, SA, WL, DDM and JTD are inventors on patents related to these compounds and may receive royalties if commercialized.
REFERENCES
- 1.Jordan A, Hadfield JA, Lawrence NJ, McGown AT. Tubulin as a target for anticancer drugs: agents which interact with the mitotic spindle. Med Res Rev. 1998;18(4):259–96. doi: 10.1002/(sici)1098-1128(199807)18:4<259::aid-med3>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 2.Desai A, Mitchison TJ. Microtubule polymerization dynamics. Annu Rev Cell Dev Biol. 1997;13:83–117. doi: 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- 3.Jordan MA, Wilson L. In: The Role of Microtubules in Cell Biology, Neurobiology, and Oncology. Fojo T, editor. Humana Press; Totowa: 2008. pp. 47–81. [Google Scholar]
- 4.Jordan MA, Kamath K. How do microtubule-targeted drugs work? An overview. Curr Cancer Drug Targets. 2007;7(8):730–42. doi: 10.2174/156800907783220417. [DOI] [PubMed] [Google Scholar]
- 5.Denduluri N, Swain S. Ixabepilone: clinical role in metastatic breast cancer. Clin Breast Cancer. 2011;11(3):139–45. doi: 10.1016/j.clbc.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 6.Gelderblom H, Verweij J, Nooter K, Sparreboom A, Cremophor EL. The drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37(13):1590–8. doi: 10.1016/s0959-8049(01)00171-x. [DOI] [PubMed] [Google Scholar]
- 7.Koolen SL, Beijnen JH, Schellens JH. Intravenous-to-oral switch in anticancer chemotherapy: a focus on docetaxel and paclitaxel. Clin Pharmacol Ther. 2010;87(1):126–9. doi: 10.1038/clpt.2009.233. [DOI] [PubMed] [Google Scholar]
- 8.Malingre MM, Beijnen JH, Schellens JH. Oral delivery of taxanes. Invest New Drugs. 2001;19(2):155–62. doi: 10.1023/a:1010635000879. [DOI] [PubMed] [Google Scholar]
- 9.Malingre MM, Beijnen JH, Rosing H, Koopman FJ, van Tellingen O, Duchin K. A phase I and pharmacokinetic study of bi-daily dosing of oral paclitaxel in combination with cyclosporin A. Cancer Chemother Pharmacol. 2001;47(4):347–54. doi: 10.1007/s002800000226. [DOI] [PubMed] [Google Scholar]
- 10.Cheng KL, Bradley T, Budman DR. Novel microtubule-targeting agents - the epothilones. Biologics. 2008;2(4):789–811. doi: 10.2147/btt.s3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fox E, Maris JM, Cohn SL, Goodspeed W, Goodwin A, Kromplewski M, et al. Pharmacokinetics of orally administered ABT-751 in children with neuroblastoma and other solid tumors. Cancer Chemother Pharmacol. 2010;66(4):737–43. doi: 10.1007/s00280-009-1218-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Michels J, Ellard SL, Le L, Kollmannsberger C, Murray N, Tomlinson Guns ES, et al. A phase IB study of ABT-751 in combination with docetaxel in patients with advanced castration-resistant prostate cancer. Ann Oncol. 2010;21(2):305–11. doi: 10.1093/annonc/mdp311. [DOI] [PubMed] [Google Scholar]
- 13.Mauer AM, Cohen EE, Ma PC, Kozloff MF, Schwartzberg L, Coates AI, et al. A phase II study of ABT-751 in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2008;3(6):631–6. doi: 10.1097/JTO.0b013e318174e01f. [DOI] [PubMed] [Google Scholar]
- 14.Oostendorp RL, Witteveen PO, Schwartz B, Vainchtein LD, Schot M, Nol A, et al. Dose-finding and pharmacokinetic study of orally administered indibulin (D-24851) to patients with advanced solid tumors. Invest New Drugs. 2010;28(2):163–70. doi: 10.1007/s10637-009-9244-6. [DOI] [PubMed] [Google Scholar]
- 15.Bacher G, Nickel B, Emig P, Vanhoefer U, Seeber S, Shandra A, et al. D-24851, a novel synthetic microtubule inhibitor, exerts curative antitumoral activity in vivo, shows efficacy toward multidrug-resistant tumor cells, and lacks neurotoxicity. Cancer Res. 2001;61(1):392–9. [PubMed] [Google Scholar]
- 16.Foster PA, Stengel C, Ali T, Leese MP, Potter BV, Reed MJ, et al. A comparison of two orally bioavailable anti-cancer agents, IRC-110160 and STX140. Anticancer Res. 2008;28(3A):1483–91. [PubMed] [Google Scholar]
- 17.Day JM, Foster PA, Tutill HJ, Newman SP, Ho YT, Leese MP, et al. BCRP expression does not result in resistance to STX140 in vivo, despite the increased expression of BCRP in A2780 cells in vitro after long-term STX140 exposure. Br J Cancer. 2009;100(3):476–86. doi: 10.1038/sj.bjc.6604873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marrelli M, Conforti F, Statti GA, Cachet X, Michel S, Tillequin F, et al. Biological potential and structure-activity relationships of most recently developed vascular disrupting agents: an overview of new derivatives of natural combretastatin a-4. Curr Med Chem. 2011;18(20):3035–81. doi: 10.2174/092986711796391642. [DOI] [PubMed] [Google Scholar]
- 19.Chaplin DJ, Hill SA. The development of combretastatin A4 phosphate as a vascular targeting agent. Int J Radiat Oncol Biol Phys. 2002;54(5):1491–6. doi: 10.1016/s0360-3016(02)03924-x. [DOI] [PubMed] [Google Scholar]
- 20.Ng QS, Mandeville H, Goh V, Alonzi R, Milner J, Carnell D, et al. Phase Ib trial of radiotherapy in combination with combretastatin-A4-phosphate in patients with non-small-cell lung cancer, prostate adenocarcinoma, and squamous cell carcinoma of the head and neck. Ann Oncol. 2012;23(1):231–7. doi: 10.1093/annonc/mdr332. [DOI] [PubMed] [Google Scholar]
- 21.Zweifel M, Jayson GC, Reed NS, Osborne R, Hassan B, Ledermann J, et al. Phase II trial of combretastatin A4 phosphate, carboplatin, and paclitaxel in patients with platinum-resistant ovarian cancer. Ann Oncol. 2011;22(9):2036–41. doi: 10.1093/annonc/mdq708. [DOI] [PubMed] [Google Scholar]
- 22.Lu Y, Li CM, Wang Z, Chen J, Mohler ML, Li W, et al. Design, Synthesis, and SAR Studies of 4-Substituted Methoxylbenzoylaryl-thiazoles Analogues as Potent and Orally Bioavailable Anti-cancer Agents. J Med Chem. 2011;54(13):4678–93. doi: 10.1021/jm2003427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li CM, Chen J, Lu Y, Narayanan R, Parke DN, Li W, et al. Pharmacokinetic Optimization of 4-Substituted Methoxybenzoyl-Aryl-Thiazole (SMART) and 2-Aryl-4-Benzoyl-Imidazole (ABI) for Improving Oral Bioavailability. Drug Metab Dispos. 2011;30(10):1833–9. doi: 10.1124/dmd.110.036616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, Wang Z, Li CM, Lu Y, Vaddady PK, Meibohm B, et al. Discovery of novel 2-aryl-4-benzoyl-imidazoles targeting the colchicines binding site in tubulin as potential anticancer agents. J Med Chem. 2010;53(20):7414–27. doi: 10.1021/jm100884b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, Li CM, Wang J, Ahn S, Wang Z, Lu Y, et al. Synthesis and antiproliferative activity of novel 2-aryl-4-benzoyl-imidazole derivatives targeting tubulin polymerization. Bioorg Med Chem. 2011;19(16):4782–95. doi: 10.1016/j.bmc.2011.06.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogunbiyi OJ. Impact of health system challenges on prostate cancer control: health care experiences in Nigeria. Infect Agent Cancer. 2011;6(Suppl 2):S5. doi: 10.1186/1750-9378-6-S2-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takeda M, Mizokami A, Mamiya K, Li YQ, Zhang J, Keller ET, et al. The establishment of two paclitaxel-resistant prostate cancer cell lines and the mechanisms of paclitaxel resistance with two cell lines. Prostate. 2007;67(9):955–67. doi: 10.1002/pros.20581. [DOI] [PubMed] [Google Scholar]
- 28.Li CM, Wang Z, Lu Y, Ahn S, Narayanan R, Kearbey JD, et al. Biological activity of 4-substituted methoxybenzoyl-aryl-thiazole: an active microtubule inhibitor. Cancer Res. 2011;71(1):216–24. doi: 10.1158/0008-5472.CAN-10-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li CM, Lu Y, Ahn S, Narayanan R, Miller DD, Dalton JT. Competitive mass spectrometry binding assay for characterization of three binding sites of tubulin. J Mass Spectrom. 2010;45(10):1160–6. doi: 10.1002/jms.1804. [DOI] [PubMed] [Google Scholar]
- 30.Behrens I, Kissel T. Do cell culture conditions influence the carrier-mediated transport of peptides in Caco-2 cell monolayers? Eur J Pharm Sci. 2003;19(5):433–42. doi: 10.1016/s0928-0987(03)00146-5. [DOI] [PubMed] [Google Scholar]
- 31.Natoli M, Leoni BD, D'Agnano I, D'Onofrio M, Brandi R, Arisi I, et al. Cell growing density affects the structural and functional properties of Caco-2 differentiated monolayer. J Cell Physiol. 2011;226(6):1531–43. doi: 10.1002/jcp.22487. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen L, Zhong WZ, Painter CL, Zhang C, Rahavendran SV, Shen Z. Quantitative analysis of PD 0332991 in xenograft mouse tumor tissue by a 96-well supported liquid extraction format and liquid chromatography/mass spectrometry. J Pharm Biomed Anal. 2010;53(3):228–34. doi: 10.1016/j.jpba.2010.02.031. [DOI] [PubMed] [Google Scholar]
- 33.Ahn S, Duke CB, 3rd, Barrett CM, Hwang DJ, Li CM, Miller DD, et al. I-387, a novel antimitotic indole, displays a potent in vitro and in vivo antitumor activity with less neurotoxicity. Mol Cancer Ther. 2010;9(11):2859–68. doi: 10.1158/1535-7163.MCT-10-0399. [DOI] [PubMed] [Google Scholar]