

Abstract
Purpose
Selective inhibition of mutant BRAF by using class I RAF inhibitors in patients with metastatic melanoma has resulted in impressive clinical activity. However, there is also evidence that RAF inhibitors might induce carcinogenesis or promote tumor progression via stimulation of MAPK signaling in RAF wild-type cells. We analyzed melanocytic lesions arising under class I RAF inhibitor treatment for dignity, specific genetic mutations, or expression of signal transduction molecules.
Patients and Methods
In all, 22 cutaneous melanocytic lesions that had either developed or considerably changed in morphology in 19 patients undergoing treatment with selective BRAF inhibitors for BRAF-mutant metastatic melanoma at seven international melanoma centers within clinical trials in 2010 and 2011 were analyzed for mutations in BRAF and NRAS genes and immunohistologically assessed for expression of various signal transduction molecules in comparison with 22 common nevi of 21 patients with no history of BRAF inhibitor treatment.
Results
Twelve newly detected primary melanomas were confirmed in 11 patients within 27 weeks of selective BRAF blockade. In addition, 10 nevi developed of which nine were dysplastic. All melanocytic lesions were BRAF wild type. Explorations revealed that expression of cyclin D1 and pAKT was increased in newly developed primary melanomas compared with nevi (P = .01 and P = .03, respectively). There was no NRAS mutation in common nevi, but BRAF mutations were frequent.
Conclusion
Malignant melanocytic tumors might develop with increased frequency in patients treated with selective BRAF inhibitors supporting a mechanism of BRAF therapy–induced growth and tumorigenesis. Careful surveillance of melanocytic lesions in patients receiving class I RAF inhibitors seems warranted.
INTRODUCTION
Melanoma is an aggressive, therapy-resistant malignancy that is derived from melanocytes. In 2010, 68,130 new patients were estimated to have been diagnosed in the United States, with 8,700 melanoma-related deaths.1 Whereas melanomas diagnosed early can usually be cured surgically, patients with advanced metastatic disease have a 1-year survival rate of around 33%.2 Until recently, systemic therapies did not have a significant impact on clinical outcome. The anti-CTLA4 antibody ipilimumab was the first drug to demonstrate prolonged overall survival, and it was approved for the treatment of metastatic melanoma in the United States and Europe in 2011.3,4 However, response rates are low, and there is no reliable method to predict the subset of patients who will respond. Targeting activating mutations in the BRAF kinase gene, which occur in approximately 50% of melanomas, by selective class I RAF inhibitors induces dramatic clinical and radiographic responses in the majority of treated patients and has recently been shown to improve progression-free and overall survival.5–7 Class I RAF inhibitors include vemurafenib and GSK2118436 and are active against the activated form of the RAF kinases whereas class II RAF inhibitors, such as sorafenib, inhibit the resting conformation of the kinase, with low activity against BRAF V600E mutant cancer cell lines.8 BRAF mutations have also been reported in 40% to 70% of papillary thyroid carcinomas, 5% to 20% of colorectal carcinomas, 10% to 20% of cholangiosarcomas, and 1% to 5% of lung cancers,9 making BRAF a possible target in other tumors.
One frequently reported adverse effect of treatment with BRAF inhibitors is the development of squamous cell carcinomas (SCCs) and keratoacanthomas (KAs). In a large phase III study,6 26% of patients treated with a selective BRAF inhibitor developed at least one SCC or KA. The exact pathogenesis is unknown, but a significant proportion of these tumors harbored HRAS mutations.10 A paradoxic activation of the MAPK pathway has been postulated,11,12 and concern has been raised regarding carcinogenesis induction by this class of agent beyond the current observations of easily treated SCCs and KAs.8 The emergence of atypical melanocytic lesions has already been observed by others. Dalle et al13 reported on five BRAF wild-type primary melanomas and one dysplastic nevus in four patients undergoing selective BRAF inhibitor treatment. Chapman et al6,13 replied that another five cases were documented in 464 patients treated in phase II and III trials with a class I RAF inhibitor.
Herein, we report on 19 patients who developed 22 changing melanocytic lesions or secondary primary melanomas while undergoing treatment with class I RAF inhibitors. All tissue samples were analyzed for genetic mutations and expression of phosphorylated signaling molecules as well as cyclin D1 in an attempt to identify the underlying mechanism for their formation. The control group consisted of 22 common nevi from 21 patients with no history of treatment with BRAF inhibitors.
PATIENTS AND METHODS
Patients
All 19 patients from seven international melanoma centers were treated with class I RAF inhibitors as part of one of several phase I to phase III trials for metastatic melanoma at the time of lesion excision. Inclusion into study therapy as well as dosage of the BRAF inhibitor was defined as part of the relevant protocol. BRAF V600 mutation of the primary tumor had been confirmed in all patients as part of a central BRAF mutation analysis inside the studies. All patients underwent a full-body dermatology examination before initiation of study treatment, and there were no findings suggestive of malignant melanoma.
The 22 melanocytic lesions suggestive of malignant melanoma (changing moles) were excised in the 19 patients after informed consent was attained (Table 1 and Table 2). These lesions either were newly developed or had changed morphology considerably since the commencement of treatment with BRAF inhibitors.
Table 1.
Summary of Clinicopathologic Characteristics of Benign and Malignant Melanocytic Samples
| Characteristic | Patient Group |
Exploratory P‡ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Primary Melanoma (newly developed)*† |
Nevi Removed During BRAF Inhibition Therapy*† |
Common Nevi*† |
||||||||
| No. | Median | Range | No. | Median | Range | No. | Median | Range | ||
| Total No. of patients | 11 | 8 | 21 | — | ||||||
| Female | 4 | 1 | 12 | .16 | ||||||
| Age at diagnosis, years | 51 | 22–77 | 48 | 44–66 | 45 | 20–76 | .35 | |||
| No. of weeks receiving selective BRAF inhibition therapy | 8 | 4–27 | 17.5 | 2–42 | — | — | ||||
| No. of suspected chronic sunlight-exposed tumor localizations§ | 5 | 2 | 5 | — | ||||||
| BRAF V600E | 0 | 0 | 8 | — | ||||||
| NRAS Q61K/Q61R | 1 | 2 | 0 | — | ||||||
| pERK | ||||||||||
| SI | 2 | 1–3 | 2 | 1–2 | 2 | 1–3 | .15 | |||
| NP | 3 | 2–3 | 2 | 1–3 | 2.5 | 1–3 | .19 | |||
| Score¶ | 5 | 4–6 | 4 | 3–5 | 4 | 3–6 | .07 | |||
| pAKT | ||||||||||
| SI | 3 | 2–3 | 2 | 2–3 | 2 | 1–3 | .16 | |||
| NP | 3 | 3–3 | 3 | 2–3 | 3 | 2–3 | .19 | |||
| Score¶ | 6 | 5–6 | 5 | 4–6 | 5 | 3–6 | .03∥ | |||
| IGF-1Rβ | ||||||||||
| SI | 3 | 2–3 | 2 | 2–3 | 2 | 2–3 | .13 | |||
| NP | 3 | 2–3 | 3 | 3–3 | 3 | 2–3 | .43 | |||
| Score¶ | 6 | 4–6 | 5 | 5–6 | 5 | 4–6 | .23 | |||
| Cyclin D1 | ||||||||||
| NP in the epidermis | 3 | 0–3 | 3 | 1–3 | 2 | 0–3 | .20 | |||
| NP in the dermis | 2 | 1–3 | 2 | 1–3 | 1 | 0–2 | .01∥ | |||
Abbreviations: IGF-1Rβ, insulin-like growth factor 1 receptor beta; NP, number of positive cells; pAKT, phospho-AKT; pERK, phospho-ERK; SI, staining intensity.
Results for only the first tumor considered (four patients with two lesions); results were similar (and claims identical) if the second tumor was considered.
Note that information on some characteristics was missing; the summary pertains to the available information only.
Two-sided from either generalized Fisher's exact test for 2 × 3 tables or the exact Kruskal-Wallis test; not calculated when group comparison was either not possible or when variation was too limited.
Summarizes face, forearm, hand, neck, right arm, upper arm, scalp, temple, cheek.
Score, sum of SI and NP.
Exploratory testing of the combined patients with primary melanoma (newly developed) and patients with nevi removed during BRAF inhibition compared with patients with common nevi by two-sided exact Wilcoxon and Mann-Whitney U tests resulted in 0.04 for the pAKT score and 0.01 for cyclin D NP in the dermis.
Table 2.
Clinicopathologic Characteristics of Benign and Malignant Melanocytic Samples for Each Patient
| Patient Group | Patient ID | Sex | Age at Diagnosis (Years) | Weeks Treated With Selective BRAF Inhibition | Tumor Thickness (mm) or Description | Tumor Localization | Pre-Existing Nevus | BRAF | NRAS | pERK |
pAKT |
IGF-1Rβ |
KI67NP | Cyclin D |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SI | NP | Score | SI | NP | Score | SI | NP | Score | NP in Epidermis | NP in Dermis | |||||||||||
| Primary melanoma (newly developed) | |||||||||||||||||||||
| 1 | F | 22 | 8 | 1.5 | Upper arm | Yes | wt | Q61R | 2 | 3 | 5 | 3 | 3 | 6 | 3 | 3 | 6 | < 5 | 3 | 3 | |
| 2 | F | 71 | 26 | 0.35 | Shoulder | Yes | wt | wt | 2 | 2 | 4 | 3 | 3 | 6 | 3 | 3 | 6 | N/A | 2 | N/A | |
| 2 | F | 71 | 26 | 0.70 | Back | Yes | wt | wt | 2 | 3 | 5 | 3 | 3 | 6 | 3 | 3 | 6 | 8 | 3 | 2 | |
| 3 | M | 62 | 7 | 1.57 | Back | Yes | wt | wt | 3 | 3 | 6 | 2 | 3 | 5 | 3 | 3 | 6 | 24 | 3 | 2 | |
| 4 | F | 40 | 19 | 0.45 | Scalp | No | wt | wt | 2 | 3 | 5 | 2 | 3 | 5 | 3 | 3 | 6 | < 5 | N/A | ||
| 5 | M | 51 | 8 | In situ | Shoulder | Yes | wt | wt | 3 | 3 | 6 | 3 | 3 | 6 | N/A | N/A | N/A | 11 | 3 | 2 | |
| 6 | M | 64 | 6 | 0.65 | Scalp | No | wt | wt | 3 | 3 | 6 | 3 | 3 | 6 | 3 | 3 | 6 | 15 | 3 | 3 | |
| 7 | M | 45 | 10 | 0.30 | Right arm | Yes | wt | wt | 2 | 2 | 4 | 2 | 3 | 5 | 2 | 2 | 4 | N/A | 3 | 1 | |
| 8 | M | 46 | 4 | In situ | Back | Yes | wt | wt | 3 | 3 | 6 | 3 | 3 | 6 | 3 | 3 | 6 | N/A | 3 | 1 | |
| 9 | M | 71 | 27 | In situ | Temple | N/A | wt | wt | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | |
| 10 | F | 77 | 8 | In situ | Thigh | Yes | wt | wt | 1 | 3 | 4 | 3 | 3 | 6 | 3 | 3 | 6 | < 5 | 2 | 1 | |
| 11 | M | 47 | 18 | In situ | Thigh | Yes | wt | wt | 2 | 3 | 5 | 2 | 3 | 5 | 2 | 3 | 5 | N/A | 0 | N/A | |
| Nevi removed during BRAF inhibition | |||||||||||||||||||||
| 12 | M | 46 | 18 | Dysplastic | Breast | N/A | wt | Q61R | 1 | 2 | 3 | 2 | 2 | 4 | 2 | 3 | 5 | < 5 | N/A | N/A | |
| 12 | M | 46 | 33 | Dysplastic | Thigh | N/A | wt | wt | 2 | 3 | 5 | 2 | 2 | 4 | 2 | 3 | 5 | N/A | 2 | N/A | |
| 13 | F | 44 | 11 | Dysplastic | Abdomen | N/A | wt | wt | 2 | 3 | 5 | 2 | 3 | 5 | 3 | 3 | 6 | N/A | 3 | N/A | |
| 14 | M | 47 | 2 | Common | Shoulder | N/A | wt | wt | 2 | 3 | 5 | 3 | 3 | 6 | 2 | 3 | 5 | 24 | N/A | 3 | |
| 15 | M | 48 | 42 | Dysplastic | Upper arm | N/A | wt | wt | 2 | 1 | 3 | 2 | 3 | 5 | 2 | 3 | 5 | N/A | 1 | N/A | |
| 16 | M | 46 | 10 | Dysplastic | Back | N/A | wt | wt | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | |
| 17 | M | 63 | 33 | Dysplastic | Thigh | N/A | wt | Q61K | 2 | 3 | 5 | 3 | 3 | 6 | 2 | 3 | 5 | < 5 | 3 | 1 | |
| 18 | M | 66 | 13 | Dysplastic | Back | N/A | wt | wt | 2 | 2 | 4 | 2 | 3 | 5 | 3 | 3 | 6 | N/A | 3 | N/A | |
| 19 | M | 58 | 22 | Dysplastic | Hand | N/A | wt | wt | 2 | 2 | 4 | 3 | 3 | 6 | 2 | 3 | 5 | N/A | 1 | N/A | |
| 19 | M | 58 | 22 | Dysplastic | Hand | N/A | N/A | N/A | 2 | 2 | 4 | 3 | 3 | 6 | 3 | 3 | 6 | N/A | 1 | N/A | |
| Common nevi | |||||||||||||||||||||
| 20 | M | 74 | 0 | Common | Gluteal | N/A | wt | wt | 3 | 3 | 6 | 2 | 3 | 5 | 3 | 3 | 6 | <5 | 2 | 1 | |
| 21 | M | 67 | 0 | Dysplastic | Back | N/A | wt | wt | 2 | 1 | 3 | 2 | 2 | 4 | 2 | 2 | 4 | N/A | 1 | 1 | |
| 22 | F | 37 | 0 | Dysplastic | Abdomen | N/A | V600E | wt | 3 | 3 | 6 | 3 | 3 | 6 | 3 | 3 | 6 | < 5 | 3 | 1 | |
| 23 | F | 45 | 0 | Dysplastic | Face | N/A | wt | wt | 2 | 3 | 5 | 3 | 3 | 6 | 2 | 3 | 5 | N/A | 3 | N/A | |
| 24 | F | 45 | 0 | Common | Decollete | N/A | V600E | wt | 2 | 3 | 5 | 3 | 2 | 5 | 2 | 3 | 5 | < 5 | 3 | 1 | |
| 25 | M | 50 | 0 | Common | Shoulder | N/A | wt | wt | 2 | 3 | 5 | 2 | 3 | 5 | 3 | 3 | 6 | < 5 | 1 | 0 | |
| 26 | M | 76 | 0 | Dysplastic | Back | N/A | wt | wt | 2 | 2 | 4 | 3 | 2 | 5 | 2 | 3 | 5 | N/A | 2 | N/A | |
| 27 | F | 48 | 0 | Dysplastic | Abdomen | N/A | wt | wt | 2 | 2 | 4 | 2 | 2 | 4 | 2 | 3 | 5 | N/A | 2 | N/A | |
| 28 | M | 68 | 0 | Common | Forearm | N/A | V600E | wt | 2 | 3 | 5 | 2 | 3 | 5 | 2 | 3 | 5 | < 5 | N/A | 2 | |
| 29 | F | 44 | 0 | Common | Back | N/A | wt | wt | 2 | 3 | 5 | 2 | 3 | 5 | 2 | 2 | 4 | 5 | 2 | 1 | |
| 30 | F | 36 | 0 | Common | Face | N/A | V600E | wt | 2 | 3 | 5 | 2 | 3 | 5 | 3 | 3 | 6 | 0 | N/A | 1 | |
| 31 | F | 36 | 0 | Common | Neck | N/A | V600E | wt | 1 | 3 | 4 | 2 | 3 | 5 | 3 | 3 | 6 | < 5 | N/A | 1 | |
| 32 | F | 50 | 0 | Common | Thigh | N/A | wt | wt | N/A | N/A | N/A | 1 | 2 | 3 | 3 | 3 | 6 | 0 | 1 | 1 | |
| 33 | F | 43 | 0 | Common | Chest | N/A | V600E | wt | 1 | 2 | 3 | 2 | 3 | 5 | 3 | 3 | 6 | < 5 | N/A | 1 | |
| 34 | F | 61 | 0 | Dysplastic | Chest | N/A | wt | wt | 2 | 1 | 3 | 2 | 3 | 5 | 2 | 3 | 5 | < 5 | 2 | 1 | |
| 35 | F | 61 | 0 | Dysplastic | Chest | N/A | V600E | wt | 1 | 2 | 3 | 2 | 3 | 5 | 2 | 3 | 5 | < 5 | 0 | 0 | |
| 36 | M | 45 | 0 | Dysplastic | Shoulder | N/A | wt | wt | 2 | 1 | 3 | N/A | N/A | N/A | 2 | 2 | 4 | 0 | 0 | 0 | |
| 37 | M | 45 | 0 | Dysplastic | Back | N/A | wt | wt | 2 | 1 | 3 | 2 | 3 | 5 | 2 | 2 | 4 | 0 | 1 | 1 | |
| 38 | F | 40 | 0 | Dysplastic | Thigh | N/A | wt | wt | 2 | 2 | 4 | 2 | 3 | 5 | 2 | 2 | 4 | < 5 | 3 | 1 | |
| 39 | M | 44 | 0 | Dysplastic | Back | N/A | wt | wt | 2 | 2 | 4 | 2 | 3 | 5 | 3 | 3 | 6 | 8 | N/A | N/A | |
| 40 | M | 20 | 0 | Dermal | Cheek | N/A | V600E | wt | 2 | 3 | 5 | 3 | 3 | 6 | 3 | 3 | 6 | < 5 | N/A | 1 | |
| 40 | M | 20 | 0 | Dysplastic | Chest | N/A | wt | wt | 2 | 3 | 5 | 3 | 3 | 6 | 3 | 3 | 6 | < 5 | 3 | 1 | |
Abbreviations: ID, identification; IGF-1Rβ, insulin-like growth factor 1 receptor beta; N/A, not applicable; NP, number of positive cells; pAKT, phospho-AKT; pERK, phospho-ERK; SI, staining intensity; wt, wild type.
In addition, 22 common nevi from 21 patients with no history of malignant melanoma or any cancer treatment including BRAF inhibitor therapy, were identified in our paraffin archives and were analyzed similarly (Tables 1 and 2). Patients from the control group had similar age and no obvious differences in lesion location distributions when compared with the patients in the other groups (Table 1).
Statistics
Standard descriptive statistics were used to summarize the patient characteristics (Table 1) and patient-specific information (Table 2). Characteristics of the three patient groups were compared in an exploratory fashion by using exact test statistics (to address the small number) for cross-tables or nonparametric Kruskal-Wallis tests. Because of the exploratory approach and the small sample size, we applied no correction for multiple testing and used a nominal significance level of α = .05 (two-sided) to indicate exploratory group differences.
Procedures
Histology.
All tissue samples were embedded in paraffin, and conventional histology with hematoxylin and eosin staining and immunhistochemistry staining for melan-A and HMB-45 was performed. Diagnosis of primary melanoma was made by the local pathologist(s), was submitted for central review, and was confirmed in each case independently by a least one experienced dermatopathologist.
Immunohistochemistry.
Immunohistochemistry was performed for phospho-ERK (pERK), phospho-AKT (pAKT), insulin-like growth factor 1 receptor beta (IGF-1Rβ), and platelet-derived growth factor receptor beta (PDGF-Rβ). Sections (4 μm) were mounted on superfrost slides and processed according to the manufacturer's instructions. Antibodies were purchased and diluted as follows: phospho-p44/42 MAPK (ERK1/2, 1:100), phospho-AKT (Ser473, 1:20), IGF-1Rβ (1:600), and PDGF-Rβ (1:50; Cell Signaling Technology, Danvers, CT). Immunohistochemistry of cyclin D1 (1:20; DCS Innovative Diagnostik-Systeme, Hamburg, Germany) was done by using an automated staining system. As a negative control, sections omitting the first antibody were stained.
Scoring of immunohistologic stains.
Histology slides were assessed independently by two experienced dermatopathologists who were blinded to the previous treatment by BRAF inhibitors.
pERK and pAKT may be localized in the nucleus or can be detected in cytoplasm; thus, both cytoplasmic and nuclear immunostaining were considered.14 Slides were analyzed with regard to the number and percentage of tumor cells stained (NP), the staining intensity, and the staining pattern. For pAKT, the NP scoring system was 0, 0%; 1, 1% to 33%; 2, 34% to 66%; and 3, 67% to 100%.15 Staining intensity was scored in comparison with the internal control: 0, negative; 1, lower; 2, equal to the internal control; and 3, stronger than the internal control. The sum of the two tissue scores was taken as the final score: 0, negative; 1 to 2, weak; 3 to 4, moderate; 5 to 6, strong.15 For pERK and IGF-Rβ, scoring of the percentage of positive tumor cells (NP) was as follows: 0, ≤ 1%; 1, more than 1% to ≤ 20%; 2, more than 20% to ≤ 50%; and 3, more than 50%.14 Sum scores were used for final scoring as described for pAKT. Endothelia of peritumoral vessels served as an internal control for pERK,14 keratinocytes of the outer root sheath for pAKT,16 and basal keratinocytes (in which IGF-1Rβ is predominantly expressed in normal adult skin) for IGF-1Rβ.17 Only cells with nuclear staining of cyclin D1 were considered positive. The following scoring was used: 0, 0%; 1, 1% to 19%; 2, 20% to 49%; and 3, ≥ 50%.18 Epidermal- and dermal-located staining were analyzed separately (Table 1).
Detection of gene mutations in NRAS and BRAF by PCR.
Tumor tissue genotyping was carried out by using standardized protocols. The top slides of the paraffin blocks were stained with hematoxylin and eosin and were reviewed by at least two pathologists. The following five slides were used for DNA extraction. Before extracting DNA, normal tissue was macroscopically dissected. Genomic DNA was isolated by using the QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer's instructions. Nested polymerase chain reaction (PCR) was performed to amplify BRAF exon 15 and NRAS exons 1 and 2 as previously described.19 The PCR products were purified by using QIAquick PCR Purification Kit (QIAGEN) and then sequenced (Eurofins MWG Operon; Ebersberg, Germany).
RESULTS
Clinical Description
Demographics for patients are summarized in Table 1, and patient-specific information is provided in Table 2. The diagnosis of all melanocytic lesions was confirmed by two central experienced dermatopathologists. In 11 patients, seven invasive and five in situ melanomas developed over a time period of 4 to 27 weeks (median, 8 weeks) after initiation of treatment with a BRAF inhibitor. Six primary melanomas were detected and removed within the first 8 weeks of treatment. We could not detect evidence for a correlation between tumor thickness and the duration of exposure. Instead, new melanomas (five of 12) developed more often at sites of previous high sun exposure compared with common nevi (five of 22; Tables 1 and 2). Ten nevi, of which nine were classified as dysplastic, had emerged or demonstrated significant morphologic changes within 2 to 42 weeks (median, 17.5 weeks) after initiation of BRAF inhibitor therapy in eight patients (Table 1).
Genotyping of BRAF and NRAS Mutations
None of the 12 newly emerged primary melanomas carried a detectable BRAF V600 mutation. However, an NRAS mutation was detected in one melanoma. Similarly, an NRAS mutation was detected in two of 10 nevi removed during treatment with a BRAF inhibitor, but none of the nevi demonstrated a BRAF mutation. This is in contrast to eight of 22 common nevi excised from patients with no melanoma in whom a BRAF mutation was detected by PCR. No NRAS mutation at amino acid position 12, 13, or 61 was found in the control group of common nevi (Table 1).
Immunohistochemistry of pERK, pAKT, IGF-1Rβ, and Cyclin D1
A moderate expression of pERK was observed in untreated nevi and in nevi removed during the course of treatment but was upregulated on exposure to therapy with selective BRAF inhibitors in newly developed melanomas (strong expression; Table 2 and Figs 1B, 2B, and 3B). The difference was not significant (Table 1). However, this may be due to a small sample size. In patient 1, a cutaneous satellite metastasis that was removed 15 months before initiation of the BRAF inhibitor therapy was available (Fig 4); pERK expression (Fig 4B) was scarce in comparison with the melanoma that had developed under BRAF inhibitor therapy (Fig 1B).
Fig 1.
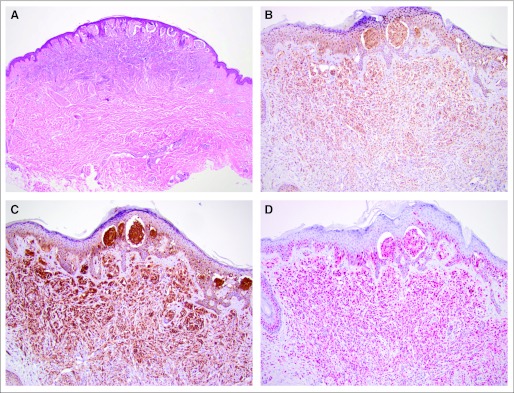
Newly developed melanoma on selective BRAF inhibition (patient 1). (A) Hematoxylin and eosin stain (×40), and immunohistologic staining (×100) of (B) phospho-ERK, (C) phospho-AKT, and (D) cyclin D1.
Fig 2.
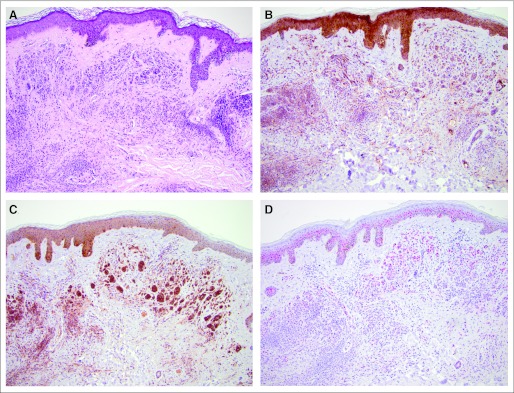
Nevus removed during BRAF inhibitor therapy (patient 17). (A) Hematoxylin and eosin stain (×100), and immunohistologic staining (×100) of (B) phospho-ERK, (C) phospho-AKT, and (D) cyclin D1.
Fig 3.
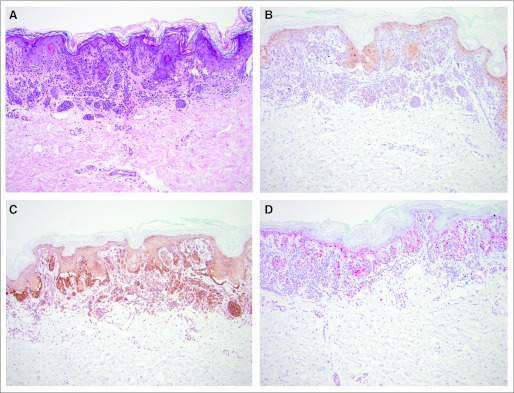
Nevus without exposure to BRAF inhibitor (patient 38). (A) Hematoxylin and eosin stain (×100), and immunohistologic staining (×100) of (B) phospho-ERK, (C) phospho-AKT, and (D) cyclin D1.
Fig 4.
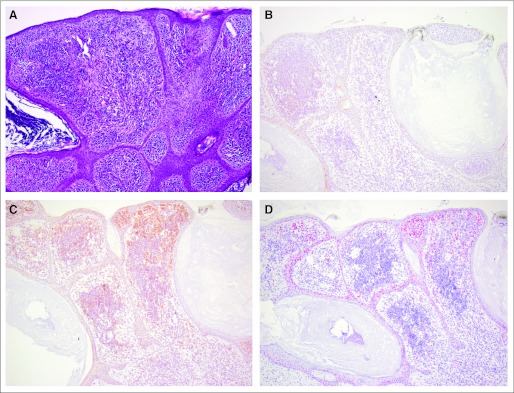
Cutaneous satellite metastasis in patient 1 removed 15 months before BRAF inhibitor therapy. (A) Hematoxylin and eosin stain (×100), and immunohistologic staining (×100) of (B) phospho-ERK, (C) phospho-AKT, and (D) cyclin D1.
pAKT was highly expressed and changed only slightly in all benign and malignant lesions (Table 2 and Figs 1C, 2C, and 3 C). The total overall score in the statistical exploratory analysis was significantly different (P = .03), suggesting a modulation with exposure to mutant BRAF inhibition (Table 1).
PDGF-Rβ expression was not detectable in newly developed nevi and melanomas, regardless of exposure to selective BRAF inhibitors (data not shown). IGF-1Rβ expression was high in all lesions and only slightly stronger in melanoma cells than in both nevus groups (Table 2).
Cyclin D1 expression in melanocytic cells located in the epidermis or dermis was generally stronger in malignant cells (Fig 1D), with average to low expression in nevi from patients treated or not treated with BRAF inhibitor, including untreated melanoma metastases (Figs 2D, 3D, and 4D). Whereas there was no significant difference in cells located in the epidermis, expression of cyclin D1 in melanocytic cells located in the dermis was significantly higher in tumor cells (P = .01).
DISCUSSION
BRAF-mutant melanoma displays features of oncogene addiction in vitro.20 Emerging data indicate that high-activity mutations lock BRAF in an active state, providing constitutive oncogenic signaling through MEK, a kinase downstream of BRAF in the mitogen-activated protein kinase (MAPK) signaling pathway.8 The impressive tumor response rates in clinical trials of selective class I RAF inhibitors in patients with advanced melanoma5–7 provides definitive clinical evidence of the role of BRAF in maintaining oncogene addiction in advanced melanoma progression.
Whereas primary resistance to selective BRAF inhibitors is low, secondary resistance is observed in the majority of all patients undergoing therapy with single-agent BRAF inhibitors. Various mechanisms of primary and secondary resistance and resistance development of melanoma to BRAF blockade have been recently described, including CRAF upregulation and co-occurrence of BRAF mutation and RAS activation,21–24 flexible switching among the three RAF isoforms,25 secondary mutations in NRAS,26 increased expression of the cancer Osaka thyroid (MAP3K8),27 or the upregulation of receptor tyrosine kinases such as PDGF-Rβ26 or IGF-1Rβ.25 In tumor biopsies of patients with newly developed progressive disease while being treated with BRAF inhibitors, ERK was found to be upregulated while pAKT levels were high.28 In vitro studies confirmed that recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy.29
In RAS-mutated tumors harboring the BRAF wild type, inhibitor binding induces RAF dimerization (CRAF-BRAF, CRAF-CRAF complexes), transactivates the drug-free promoter, and thereby activates the MEK-ERK pathway.21,23,30 Furthermore, a paradoxic activation of the MAPK pathway in normal BRAF wild-type cells has been described.21,23,30 The induction of SCCs and KAs is possibly induced by similar mechanisms.10,30
Here we describe, for the first time (to the best of our knowledge), a systematic approach to analyzing newly developing primary cutaneous melanomas in patients undergoing treatment with class I RAF inhibitors for BRAF V600–mutant metastatic melanoma. The rate of secondary melanomas emerging under therapy is notable, given the expected exploration of BRAF inhibitors as a treatment option in the adjuvant situation in the near future as well as in other tumor entities. In our series, most of the melanomas developed within a few weeks of treatment and were detected at an early clinical stage. Although detailed prospective skin examinations have generally not been performed in clinical trials of patients with advanced melanoma, the number of melanocytic lesions identified in our series would appear to be higher than the reported absence of such lesions in clinical trials of investigational agents in patients with advanced melanoma. We currently do not know the exact frequency of newly developing melanomas during selective BRAF blockade. The frequency of newly developing or changing moles is at least 10-fold lower than the emergence of cutaneous SCC or KA, on the basis of internal statistics within the treating centers. However, since participating centers were selected because they had observed a melanoma during BRAF inhibitor therapy, this may still lead to a highly biased assumption. Whether there is a predominance of malignant melanocytic lesions occurring in previously sun-exposed areas needs to be explored in larger data sets. In comparison with nevi removed during treatment with BRAF inhibitors as well as common melanocytic nevi identified in a healthy and untreated control group, expression of dermal cyclin D1 and pAKT was increased in malignant lesions. Furthermore, pERK scores demonstrated a tendency toward increases in newly arisen melanomas as would also be expected in other malignancies. Activation of MEK-ERK signaling may represent one mechanism to promote the growth of the pre-existing melanocytic lesions in our patients, but upregulation of other signaling pathways (eg, PI3K/AKT) may also play a role.
BRAF mutations are known to be present in approximately 79% of acquired nevi,31 whereas NRAS or HRAS mutations occur less commonly and are primarily found in congenital nevi and Spitz nevi, respectively.32 Importantly, overexpression of BRAF V600E in melanocytes has been shown to induce melanocyte senescence.33 However, no BRAF mutation was found in any of the 22 melanocytic lesions removed during exposure to BRAF inhibitors in our series, which is consistent with the model of BRAF inhibitor–induced proliferation of cells containing other genetic events. Thus, changes in (pre-existent) melanocytic lesions were not caused by secondary resistance to BRAF inhibitor but probably were due to paradoxic activation of the MAPK pathway10,11 resulting in upregulation of cyclin D1.
These findings shed light on a new and important potential adverse event associated with BRAF inhibitors. Our observations suggest that melanocytic cells bearing or acquiring oncogenic RAS are at increased risk of developing secondary melanoma. Additional mechanisms may also be of clinical relevance since an NRAS mutation was detected in only one melanoma and in two of the nevi of patients treated with BRAF inhibitors. Several other mechanisms conferring resistance to BRAF inhibitors have been described34–36 but could not be explored in our samples because of the limited tissue resources. Further investigations and confirmatory evaluations in larger cohorts are needed to fully understand the underlying mechanisms of the potentially melanoma-inducing effect of selective BRAF inhibitors. Since pERK, pAKT, and cyclin D1 expression can also play a role in the development of SCC, these molecules should be investigated in SCC lesions that developed during treatment with BRAF inhibitors. A careful and regular skin examination would be of importance for all patients receiving BRAF inhibitor therapy.
Acknowledgment
We thank the staff of the Institute for Pathology (K. Schmid, MD) for performing the cyclin D1 immunohistochemistry.
Footnotes
Supported by the Bangerter Rhyner Foundation (R.D.).
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: Mario E. Lacouture, GlaxoSmithKline, Roche, Genentech, OSI Pharmaceuticals, Bayer Pharmaceuticals, Onyx Pharmaceuticals (C); Axel Hauschild, Roche, GlaxoSmithKline (C); Reinhard Dummer, Roche, AstraZeneca Deutschland, Novartis, GlaxoSmithKline (C); Georgina V. Long, GlaxoSmithKline, Roche (C); Grant McArthur, Roche (U), GlaxoSmithKline (U); Dirk Schadendorf, Roche, GlaxoSmithKline, Amgen, AstraZeneca Deutschland, Morphotek, Merck Sharp & Dohme (C) Stock Ownership: None Honoraria: Lisa Zimmer, Roche; Axel Hauschild, Roche, GlaxoSmithKline; Georgina V. Long, Roche Research Funding: Mario E. Lacouture, Bayer Pharmaceuticals; Axel Hauschild, Roche Expert Testimony: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Lisa Zimmer, Uwe Hillen, Dirk Schadendorf
Financial support: Dirk Schadendorf
Administrative support: Dirk Schadendorf
Provision of study materials or patients: Lisa Zimmer, Mario E. Lacouture, Klaus Busam, Richard D. Carvajal, Friederike Egberts, Axel Hauschild, Mohammed Kashani-Sabet, Simone M. Goldinger, Reinhard Dummer, Georgina V. Long, Grant McArthur, Dirk Schadendorf
Collection and assembly of data: Lisa Zimmer, Uwe Hillen, Mario E. Lacouture, Klaus Busam, Richard D. Carvajal, Friederike Egberts, Axel Hauschild, Mohammed Kashani-Sabet, Simone M. Goldinger, Reinhard Dummer, Georgina V. Long, Grant McArthur, Antje Sucker, Dirk Schadendorf
Data analysis and interpretation: Lisa Zimmer, Uwe Hillen, Elisabeth Livingstone, Mario E. Lacouture, Klaus Busam, Richard D. Carvajal, Georgina V. Long, Grant McArthur, André Scherag, Dirk Schadendorf
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.American Cancer Society. Atlanta, Georgia: 2010. Cancer Facts & Figures 2010. http://www.cancer.org/downloads/STT/Cancer_Facts_and_Figures_2010.pdf. [Google Scholar]
- 2.Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 5.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kefford R, Arkenau H, Brown MP, et al. Phase I/II study of GSK2118436, a selective inhibitor of oncogenic mutant BRAF kinase, in patients with metastatic melanoma and other solid tumors. J Clin Oncol. 2010;28:611s. suppl; abstr 8503. [Google Scholar]
- 8.Ribas A, Flaherty KT. BRAF targeted therapy changes the treatment paradigm in melanoma. Nat Rev Clin Oncol. 2011;8:426–433. doi: 10.1038/nrclinonc.2011.69. [DOI] [PubMed] [Google Scholar]
- 9.Flaherty KT, McArthur G. BRAF, a target in melanoma: Implications for solid tumor drug development. Cancer. 2010;116:4902–4913. doi: 10.1002/cncr.25261. [DOI] [PubMed] [Google Scholar]
- 10.Oberholzer PA, Kee D, Dziunycz P, et al. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J Clin Oncol. 2012;30:316–321. doi: 10.1200/JCO.2011.36.7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cichowski K, Jänne PA. Drug discovery: Inhibitors that activate. Nature. 2010;464:358–359. doi: 10.1038/464358a. [DOI] [PubMed] [Google Scholar]
- 12.Pratilas CA, Solit DB. Targeting the mitogen-activated protein kinase pathway: Physiological feedback and drug response. Clin Cancer Res. 2010;16:3329–3334. doi: 10.1158/1078-0432.CCR-09-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dalle S, Poulalhon N, Thomas L. Vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;365:1448–1449. doi: 10.1056/NEJMc1108651. author reply 1450. [DOI] [PubMed] [Google Scholar]
- 14.Venesio T, Chiorino G, Balsamo A, et al. In melanocytic lesions the fraction of BRAF V600E alleles is associated with sun exposure but unrelated to ERK phosphorylation. Mod Pathol. 2008;21:716–726. doi: 10.1038/modpathol.2008.41. [DOI] [PubMed] [Google Scholar]
- 15.Dai DL, Martinka M, Li G. Prognostic significance of activated Akt expression in melanoma: A clinicopathologic study of 292 cases. J Clin Oncol. 2005;23:1473–1482. doi: 10.1200/JCO.2005.07.168. [DOI] [PubMed] [Google Scholar]
- 16.Williams R, Baker AF, Ihle NT, et al. The skin and hair as surrogate tissues for measuring the target effect of inhibitors of phosphoinositide-3-kinase signaling. Cancer Chemother Pharmacol. 2006;58:444–450. doi: 10.1007/s00280-006-0190-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodak E, Gottlieb AB, Anzilotti M, et al. The insulin-like growth factor 1 receptor is expressed by epithelial cells with proliferative potential in human epidermis and skin appendages: Correlation of increased expression with epidermal hyperplasia. J Invest Dermatol. 1996;106:564–570. doi: 10.1111/1523-1747.ep12344044. [DOI] [PubMed] [Google Scholar]
- 18.Ramirez JA, Guitart J, Rao MS, et al. Cyclin D1 expression in melanocytic lesions of the skin. Ann Diagn Pathol. 2005;9:185–188. doi: 10.1016/j.anndiagpath.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 19.Houben R, Becker JC, Kappel A, et al. Constitutive activation of the Ras-Raf signaling pathway in metastatic melanoma is associated with poor prognosis. J Carcinog. 2004;3:6. doi: 10.1186/1477-3163-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma SV, Settleman J. Oncogene addiction: Setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007;21:3214–3231. doi: 10.1101/gad.1609907. [DOI] [PubMed] [Google Scholar]
- 21.Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 22.Poulikakos PI, Rosen N. Mutant BRAF melanomas: Dependence and resistance. Cancer Cell. 2011;19:11–15. doi: 10.1016/j.ccr.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 23.Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montagut C, Sharma SV, Shioda T, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–4861. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McArthur GA, Ribas A, Chapman PB, et al. Molecular analyses from a phase I trial of vemurafenib to study mechanism of action (MOA) and resistance in repeated biopsies from BRAF mutation-positive metastatic melanoma patients (pts) J Clin Oncol. 2011;29(suppl):526s. abstr 8502. [Google Scholar]
- 29.Paraiso KH, Fedorenko IV, Cantini LP, et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer. 2010;102:1724–1730. doi: 10.1038/sj.bjc.6605714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pollock PM, Harper UL, Hansen KS, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 32.Bauer J, Curtin JA, Pinkel D, et al. Congenital melanocytic nevi frequently harbor NRAS mutations but no BRAF mutations. J Invest Dermatol. 2007;127:179–182. doi: 10.1038/sj.jid.5700490. [DOI] [PubMed] [Google Scholar]
- 33.Dhomen N, Reis-Filho JS, da Rocha Dias S, et al. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009;15:294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 34.Shao Y, Aplin AE. Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res. 2010;70:6670–6681. doi: 10.1158/0008-5472.CAN-09-4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paraiso KH, Xiang Y, Rebecca VW, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71:2750–2760. doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagle N, Emery C, Berger MF, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–3096. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]