

Synopsis
The cystic fibrosis transmembrane conductance regulator (CFTR) is expressed in the apical membrane of epithelial cells. Cell surface CFTR levels are regulated by endocytosis and recycling. A number of adaptor proteins including AP-2 (μ2) and Dab2 have been proposed to modulate CFTR internalization. We used small interfering RNA-mediated (siRNA) silencing of these adaptors to test their roles in the regulation of CFTR cell surface trafficking and stability in human airway epithelial cells. The results indicated that μ2 and Dab2 performed partially overlapping, but divergent functions. While μ2 depletion dramatically decreased CFTR endocytosis with little effect on CFTR protein half-life, Dab2 depletion increased CFTR half-life ~3-fold in addition to inhibiting CFTR endocytosis. Furthermore, Dab2 depletion inhibited CFTR trafficking from the sorting endosome to the recycling compartment as well as delivery of CFTR to the late endosome, thus providing a mechanistic explanation for increased CFTR expression and half-life. To test whether two E3 ligases were required for the endocytosis and/or down-regulation of surface CFTR, we siRNA-depleted CHIP and c-Cbl. We demonstrate that CHIP and c-Cbl depletion have no effect on CFTR endocytosis, but c-Cbl depletion modestly enhanced CFTR half-life. These results define a significant role for Dab2 both in the endocytosis and post endocytic fate of CFTR.
Keywords: CFTR, endocytosis, down-regulation, Dab2, AP-2, c-Cbl, CHIP
Introduction
Clathrin-mediated endocytosis is responsible for the internalization of a wide variety cell surface molecules including receptors, signaling molecules and channel proteins. Internalization of these proteins is mediated by cytoplasmic adaptor molecules that recruit the cell surface molecules into clathrin-coated pits by simultaneously binding to clathrin and to internalization signals in the cell surface protein's cytoplasmic domains (reviewed in [1]). The best-characterized adaptor protein is AP-2, a tetramer composed of α2, β2, μ2, and σ2 subunits [2]. AP-2 binds to some internalization signals via its μ2 subunit [3] and to clathrin via its β2 subunit [4], while the α2 subunit binds accessory proteins involved in coated pit assembly [5].
AP-2 is not the only adaptor that is important for internalization of cell surface molecules. Another example is Disabled-2 (Dab2), an adaptor that has been shown to facilitate the internalization of the low-density lipoprotein receptor (LDLR) [6]. The selective use of different adaptors is mediated at least in part by the different internalization signals found in the receptors. AP-2 recognizes a YXXΦ cytoplasmic tail signal, where X is any amino acid and Φ is a large hydrophobic residue as found in the transferrin receptor [7]; whereas Dab2 recognizes FXNPXY signals present in the LDLR [8]. It is now clear that multiple adaptor molecules are used selectively for different cell surface molecules, thus making the clathrin-mediated endocytosis much more complicated than originally envisioned (reviewed in [9]).
The cystic fibrosis transmembrane conductance regulator (CFTR) is a cyclic AMP (cAMP)-activated chloride channel expressed in airway, intestinal, and a number of other epithelial tissues [10–12]. CFTR is expressed on the apical surface of epithelia and its surface expression is regulated by endocytosis and recycling [13–15]. Efficient endocytosis of CFTR has been shown to require carboxy-terminal cytoplasmic tail internalization signals, Y1424DSI and L1430L [16–18]. Further, the tyrosine-based signal has been shown to interact with the μ2 subunit of AP-2 [19].
Other proteins have also been shown to be involved in CFTR endocytosis. These include myosin VI and c-Cbl (casitas B-lineage lymphoma proto-oncogene), an E3 ligase [20, 21]. Myosin VI, a motor traveling towards the minus end of the actin filaments, is targeted to clathrin-coated structures by its interactions with Dab2 [22]. Moreover, CFTR immunoprecipitation studies in human airway cells and intestinal epithelial cells have demonstrated that Dab2 co-immunoprecipitates in a complex with myosin VI and AP-2 [20, 23], suggesting that it is a CFTR binding partner in the clathrin-coated pit. In intestinal cells, CFTR was shown to interact directly with the α2 subunit of AP-2, but not Dab2, whereas Dab2 interacts directly with the α2 subunit of AP-2 [23]. Interestingly, Dab2 knockout mice have higher levels of CFTR expression in jejunum sections [23], suggesting that Dab2 regulates CFTR surface expression.
In the present studies, we wanted to determine the roles of AP-2 and Dab2 in human airway epithelial cells and define what their different functions were. The goal was to determine their relative importance in CFTR endocytosis, but more importantly, determine if either played a role in cell surface CFTR down-regulation. Using polarized human epithelial cells for the endocytosis assays, we found that siRNA knockdown (KD) of either the μ2 subunit of AP-2 or Dab2 dramatically inhibited CFTR endocytosis. In fact, both KDs had very similar inhibitory effects on CFTR endocytosis, indicating that both adaptors contribute to CFTR internalization. Analysis of CFTR protein half-life, however, revealed striking differences in CFTR stability in the different KDs. Depletion of AP-2 (μ2) had little effect on CFTR half-life, whereas Dab2 depletion increased the half-life ~3-fold. This suggested that while both adaptors are necessary for the endocytosis of CFTR, Dab2 is also a part of the post endocytic trafficking machinery that directs CFTR to the degradative pathway.
Experimental Procedures
Cell culture
CFBE41o-WT (expressing WT-CFTR) cells were cultured as described previously [24]. For the functional analysis in Ussing chambers and endocytosis assays using polarized monolayer, CFBE41o-WT cells were seeded onto 12-mm Transwell filters (Costar, Corning) and cultured on an air-liquid interface for 5 days before analysis as described previously [25].
Antibodies and chemicals
Anti-CFTR, mouse monoclonal (clone 24-1 (ATCC) and MM13-4 (Millipore)) and rabbit polyclonal (anti- NBD1) antibodies were used as described previously [24]. Mouse anti-AP50/μ2 and anti-EEA1 antibodies were from BD Transduction Laboratories. Rabbit polyclonal antibody to mannose-6-phosphate receptor and c-Cbl were purchased from Abcam. Rabbit polyclonal antibody against Dab2 was from Santa Cruz Biotechnology. Rabbit polyclonal, anti-CHIP antibody was from Thermo Scientific. Polyclonal anti-actin antibody was purchased from Sigma-Aldrich. Alexa Fluor 488-labeled goat anti-mouse IgG antibody, Alexa Fluor 594-labeled goat anti-rabbit IgG antibody, and Alexa Fluor 594-conjugated human transferrin were from Invitrogen. HRP-labeled goat anti-mouse IgG antibody and HRP-labeled goat, anti-rabbit IgG antibody were from Bio-Rad Laboratories. The SuperSignal West Pico chemiluminescence substrate was from Pierce Chemical Co. All other chemicals were from Sigma-Aldrich or Fisher Scientific.
Western blotting
CFTR, μ2, Dab2, c-Cbl and CHIP protein levels in control or siRNA-depleted samples were determined as described previously [25]. Briefly, cells were lysed in RIPA buffer (50 mM Tris-HCl, pH 8.0, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 150 mM NaCl and Complete Protease Inhibitor (Roche)). Proteins were separated by SDS/PAGE and transferred onto PVDF membranes (Bio-Rad). CFTR was detected with a monoclonal antibody (MM13-4, 1:500 dilution; Millipore Corporation). Dab2 and μ2 were detected with a polyclonal anti-Dab2 antibody (1:200 dilution; Santa Cruz Biotechnology) and a monoclonal anti-μ2 antibody (1:100 dilution; BD Transduction Lab), respectively. c-Cbl and CHIP were examined using antibodies against c-Cbl (1:500 dilution; Abcam, Inc.) and CHIP (1:200 dilution, Thermo Scientific). HRP-labeled secondary antibodies and Super Signal West Pico chemiluminescence substrate (Pierce) were used for visualization of specific protein bands. Densitometry was performed using Image J software (NIH).
Co-Immunoprecipitation of CFTR and Dab2
CFTR and Dab2 were co-immunoprecipitated from CFBE41o-WT cells expressing wild-type CFTR. CFBE41o- cells with no detectable CFTR expression were tested as a negative control. Briefly, cells were lysed in a buffer containing 150 mM NaCl, 50 mM Tris, pH 8.0, 1% IGEPAL (Sigma) and Complete Protease Inhibitor mixture (Roche). After centrifugation at 15,000 × g for 15 min, the soluble fraction (supernatant) was incubated with Protein G-Agarose beads (Roche) pre-loaded with anti-CFTR (24–1) or anti-Dab2 antibodies at 4°C for 2 h. Proteins were eluted in Laemmli sample buffer and separated by SDS PAGE followed by Western transfer to PVDF membranes as described previously [26]. CFTR was then detected with anti-CFTR antibody (MM13-4) in the anti-Dab2 precipitated samples and Dab2 was detected in the anti-CFTR precipitated samples using anti-Dab2 antibody.
Immunofluorescence microscopy
Indirect immunofluorescence microscopy was performed as described previously [26]. Briefly, cells were grown on glass coverslips, fixed in 4% paraformaldehyde/PBS and permeabilized with 0.1% Triton X-100/PBS for 5 min, washed three times for 2 min each with PBS, and then blocked with 2.5% goat serum/PBS. Cells were incubated with primary antibodies diluted in blocking solution for 2 h. Following washing steps, the secondary antibodies (1:500) were applied and incubated 45 min and mounted with Vectashield/DAPI (Vector Labs). All incubations and washing steps were at room temperature unless stated otherwise. Microscopy was performed using Leitz epifluorescence microscope equipped with a step motor, filter-wheel assembly (Ludl Electronics Products) and an 83,000-filter set (Chroma Technology). Images were obtained with a SenSys-Cooled, charge-coupled high-resolution camera (Photometrics). IpLab Spectrum software (Signal Analytics) was used for image acquisition. For the ammonium chloride experiments, cells were treated with 5 mM NH4Cl for 16 h prior to indirect immunofluorescence microscopy using CFTR and mannose-6-phosphate receptor (M6PR) antibodies.
For confocal microscopy, CFBE41o-WT cells were grown on Transwell membrane support for 5 days and then processed for immunofluorescent staining as described above. Confocal imaging was performed using a Nikon 2000U inverted microscope (Melville, KY) equipped with a PerkinElmer UltraVIEW ERS 6FE-US spinning disk laser apparatus (Shelton, CT). Images were processed with Volocity 5 software (Improvision Inc., Waltham, MA).
Cell surface biotinylation
Cell surface CFTR was biotinylated as described previously [27].
Metabolic pulse-chase assay
CFBE41o-WT cells were pulse labeled with 100 μCi/ml of 35S-Methionine for 1 hour and chased for 0, 4, 8, 12, 16, or 24 h with growth medium as described previously [26]. The protein half-life (t1/2) was calculated as described previously [25].
Endocytosis assays
The endocytosis of cell surface CFTR was measured in internalization assays as described previously [18, 27].
siRNA-mediated depletion of Dab2, μ2, c-CBL and CHIP
siRNA duplexes corresponding to non-conserved regions of human Dab2, μ2 and c-CBL were purchased from Qiagen Inc. The specific sequence 5'-TAGAGCATGAACATCCAGTAA-3' was chosen as a target for the small interfering RNA (siRNA) depletion of Dab2. The targeting sequence for μ2 knockdown was 5'-TGCCATCGTGTGGAAGATCAA-3'. The targeting sequence for c-Cbl was 5'-CCCGCCGAACUCUCAGAUATT-3'. The double-stranded non-silencing control siRNA sequence 5'-AATTCTCCGAACGTGTCACGT-3', which has no significant homology to any other genes, was also purchased from Qiagen Inc. Depletion of human STUB1 (CHIP) was achieved by using siRNA oligos from Dharmacon as described previously [28]. Transfection of siRNA oligos was performed using siLentFect lipid reagent (Bio-Rad Laboratories) according to the manufacturer's instructions. Briefly, cells at ~70–80% confluency were transfected with the optimized transfection mixture. After 24 h incubation at 37°C, the transfection mixture was replaced with fresh cell culture medium. Experiments were conducted 3 to 6 days after transfection. The depletion efficiencies of individual genes were assessed by Western blotting.
Ussing chamber analyses
Short-circuit current (ISC) was measured under voltage clamp conditions using MC8 voltage clamps and P2300 Ussing chambers (Physiologic Instruments, San Diego, CA) as previously described [29]. Monolayers were initially bathed on both sides with identical Ringer's solutions containing (in mM) 115 NaCl, 25 NaHCO3, 2.4 KH2PO4, 1.24 K2HPO4, 1.2 CaCl2, 1.2 MgCl2, 10 D-glucose (pH 7.4). Bath solutions were vigorously stirred and gassed with 95%O2:5% CO2. Short-circuit current measurements were obtained using an epithelial voltage clamp (Physiologic Instruments). A one-second three-mV pulse was imposed every 10 s to monitor resistance calculated using Ohm's law. Where indicated, the mucosal bathing solution was changed to a low Cl− solution containing 1.2 mM NaCl and 115 mM Na+ gluconate, and all other components as above. Amiloride (100 μM) was added to block residual Na+ current, followed by the CFTR agonists forskolin (20 μM, Calbiochem) and genistein (50 μM, Sigma Aldridge) as indicated (minimum five-min observation at each concentration). CFTRInh-172 (10 μM) was added to the apical bathing solution at the end of experiments to block CFTR-dependent ISC. All chambers were maintained at 37 °C during experiments.
Transferrin recycling
Following siRNA depletion of Dab2, CFBE41o-WT cells were seeded onto coverslips and incubated at 37°C. 72 h after siRNA transfection, the cells were serum starved for 2 h and then pulsed with Alexa Fluor 594-conjugated transferrin (Alexa 594-Tfn, 20 μg/ml) for 5 min at 37°C. After multiple washing steps with ice-cold PBS to remove unbound Alexa 594-Tfn and inhibit transport, the coverslips were incubated with complete growth medium pre-warmed to 37°C for the time periods indicated. Then the coverslips were quickly washed with ice-cold PBS and fixed in 4% formaldehyde, mounted, and visualized using fluorescence microscopy.
Data analysis and statistics
All experiments were repeated at least 3 times, and the data were expressed as the mean ± standard deviation. Statistical analysis was performed using t test (2-tailed) in Microsoft Excel and significance was determined at the p<0.05 level.
Results
Depletion of AP-2 (μ2 subunit) and Dab2 in airway epithelial cells increases total CFTR levels
In order to determine the roles of AP-2 and Dab2 in CFTR trafficking at the cell surface, we first examined how siRNA KD of each of these adaptors affected CFTR expression in human airway epithelial cells (CFBE41o-WT). Using different concentrations of siRNA, we determined the maximum depletion conditions for each adaptor and their effects on total CFTR expression. Figure 1A illustrates that more than 90% of μ2 was depleted using siRNA KD and this results in a 2 to 3-fold increase in CFTR C band, the mature form of CFTR. Figure 1B shows that Dab2 levels were reduced more than 95% of the control and the CFTR C band was increased 5 to 6-fold. The core-glycosylated form (B band) of CFTR was also slightly increased upon Dab2 KD, whereas this was not seen in the μ2 KD. The results suggest that while depletion of μ2 increased total CFTR levels, the Dab2 depletion had a more pronounced effect on CFTR expression (Figure 1A and B). Interestingly, depletion of both adaptors did not have an additive effect, suggesting that the two adaptors were not acting independently (Figure 1C). Next, we performed coimmunoprecipitation experiments in order to confirm the interaction between CFTR and Dab2 by immunoprecipitating (IP) either CFTR and blotting for Dab2 or IP Dab2 and immunoblotting for CFTR (Figure 1D). The results confirm that CFTR and Dab2 are present in the same complex.
Figure 1. Increased CFTR levels in CFBE41o-WT cells following μ2 and Dab2 depletion and confirmation of CFTR and Dab2 interactions.

A. The consequences of μ2 depletion. B. The consequences of Dab2 depletion. CFBE41o-WT cells were transfected with the indicated amount of siRNA duplexes targeted specifically to either μ2 (A) or Dab2 (B). 72 h after transfection, 25 μg of cell lysates were separated by SDS-PAGE and immunoblotted with the antibodies indicated. CFTR levels increased when either μ2 or Dab2 were depleted. Mock control (M) is without any siRNA and indicates no significant difference in CFTR expression when compared to the siRNA control (C). C. The combination of Dab2 and μ2 depletion does not have an additive effect on CFTR expression levels. CFBE41o-WT cells were transfected with 20 nM control siRNA (C), μ2 or Dab2 siRNA alone, or the combination of μ2 and Dab2 siRNA oligos and cultured for 72 h. CFTR, Dab2 and μ2 were detected using anti-CFTR, anti-Dab2 and anti-μ2 antibodies. The combination of μ2 and Dab2 depletion did not increase the expression of CFTR when compared to Dab2 or μ2 depletion alone. The changes in μ2, Dab2 and CFTR levels following the siRNA depletion are indicated below the blots. D. CFTR and Dab2 interact with each other as demonstrated by co-immunoprecipitation. Cell lysates from CFBE41o- (without CFTR expression) and CFBE41o-WT cells were immunoprecipitated with anti-Dab2 (top panel) or anti- CFTR antibodies (bottom panel). Immunoprecipitated proteins were separated by SDS-PAGE and blotted with anti-CFTR or anti-Dab2 antibodies respectively. A fraction of CFTR was pulled down with anti-Dab2 antibody as indicated by the arrow (top panel) and Dab2 was pulled down by anti-CFTR antibodies (bottom panel). WCL: Western blots indicate CFTR and Dab2 expression in the cell lysates (5% of total whole cell lysates).
During its biogenesis, CFTR is first synthesized as a core glycosylated B band in the ER and then is further modified to the maturely glycoslylated C band as it passes through the Golgi complex. Because the total pool of CFTR was increased by the depletion of the adaptors, and the effects on the fully processed Band C CFTR were the most pronounced, we next examined how depletion of μ2 and Dab2 affected the surface pool of CFTR. To test this, we performed cell surface biotinylation and immunocytochemistry [25]. We first labeled the cell surface CFTR with biotin and measured the levels of biotinylated CFTR following either μ2 or Dab2 depletion. The results indicate that the cell surface CFTR was increased ~3 and ~5- fold when μ2 and Dab2 were depleted, respectively (Figure 2A and B). The increase of the cell surface CFTR is comparable to that of the total pool. To validate that the increased CFTR is on the cell surface, we performed confocal microscopy on polarized CFBE41o- cells grown on permeable supports (Figure 2C). Although μ2 KD increased the total (Figure 2C, top view) and the surface pool (Figure 2C, side view), the Dab2 depletion had a much more pronounced effect, suggesting that Dab2 might be affecting more than one step in the pathway. Importantly, these results established significant roles for AP-2 and Dab2 in the regulation of the cell surface and consequently the total CFTR pools in airway epithelial cells. These results are consistent with the idea that these adaptors regulate the endocytosis of CFTR. Furthermore, the more significant changes in CFTR surface levels following Dab2 depletion suggested divergent functions for these adaptors in the post endocytic compartments. To confirm this, we designed CFTR endocytosis, half-life, and functional and morphological studies in order to follow the fate of CFTR after depletion of the adaptors.
Figure 2. Increased cell surface CFTR levels following μ2 and Dab2 depletion.

A. The consequences of μ2 depletion. B. The consequences of Dab2 depletion. CFBE41o-WT cells were transfected with control (C), μ2 or Dab2 siRNA duplexes and cultured for 72 h. The efficiency of μ2 or Dab2 was measured by immunoblotting (top). Reduction in μ2 or Dab2 levels are indicated as % of control. β-actin was detected as loading control (middle). Cell surface CFTR levels were measured following cell surface biotinylation as described in the Experimental Procedures section. Changes in cell surface CFTR levels are indicated as fold change over control. C. Immunocytochemical detection of CFTR in CFBE41o-WT cells following μ2 or Dab2 depletion. Confocal images of control (left panel), μ2 (middle panel), or Dab2 depleted cells (right panel) grown on permeable supports are shown from the top and side views. CFTR was detected as described in the Experimental Procedures section and images were acquired under identical conditions for each sample.
AP-2 (μ2) and Dab2 are both required for CFTR endocytosis
Previous studies have shown that disruption of AP-2 function in non-polarized cells inhibits CFTR endocytosis [19, 23]. Here we examined this question in polarized human airway epithelial cells and directly compared the roles of AP-2 and Dab2 in CFTR endocytosis from the apical cell surface. First, we depleted μ2 or Dab2 and cultured the cells as polarized monolayers on filter supports. Next, we tested how depletion of μ2 or Dab2 affected CFTR internalization using a surface biotinylation assay to monitor endocytosis [25]. Immunoblotting experiments revealed that both adaptors were depleted >90% in all experiments. Importantly, the results indicated that both μ2 and Dab2 equally contribute to CFTR endocytosis (Figure 3). Specifically, in 2.5 min, 19.9 ± 2.1% (mean ± S. D.) of the cell surface CFTR was internalized in the control cells, while in μ2 depleted cells, CFTR internalization was reduced to 9.0 ± 1.8%, indicating a 55% decrease. Dab2 depletion had a similar effect (9.5 ± 1.3% endocytosis in 2.5 min), indicating a 53% inhibition of CFTR endocytosis. To determine if the effects of μ2 and Dab2 depletion were additive, we depleted both adaptors (Figure 3, bottom panels). In the double KD experiments, CFTR internalization was similar to the single μ2 or Dab2 depletion experiments (8.1 ± 2.5% in 2.5 min). These results suggested that the two adaptors do not work independently and both are necessary for efficient CFTR endocytosis.
Figure 3. Reduced CFTR endocytosis rates following μ2, Dab2 or μ2+Dab2 depletion.

CFBE41o-WT were transfected with control, μ2, Dab2 or μ2+Dab2 siRNA oligos as indicated. 24 h after transfection, the cells were transferred to Transwell filters and incubated for an additional 4 to 5 days under an air-liquid interface. The efficiency of μ2, Dab2 or μ2+Dab2 depletion was >95%. CFTR internalization assays were performed as described previously [18,27]. A. Representative gels of CFTR internalization assays. B. Quantitative analysis of CFTR internalization dynamics during a 7.5 min time period. The percentage of internalized CFTR was calculated from the loss of biotinylated CFTR during a 37°C incubation for time periods indicated under each condition, n=3. C. Quantitative analysis of CFTR internalization rates following μ2, Dab2 or μ2+Dab2 depletion in 2.5 minutes. Depletion of μ2, Dab2 or μ2+Dab2 significantly reduced CFTR internalization rates in a 2.5 min time period, n=3.
Dab2 depletion dramatically increases CFTR's protein half-life
Following endocytosis, CFTR either recycles to the cell surface or enters the degradative pathway [30]. To identify the roles of μ2 and Dab2 in the intracellular trafficking of CFTR following endocytosis, we tested whether depletion of these adaptors affected CFTR half-life. For these experiments, we monitored the half-life of CFTR using a metabolic pulse-chase protocol. We determined that in the control samples, the half-life of CFTR was 12 ± 0.6 h, which is consistent with previous reports [25]. In the μ2 depleted cells, the half-life of CFTR is 16 ± 2.8 h (Figure 4A), suggesting only a mild but significant effect. In contrast, in the Dab2-depleted cells, the half-life of CFTR was 37 ± 7.1 h, indicating that CFTR was 3-fold more stable in Dab2-depleted cells (Figure 4B). The results indicate that although μ2 and Dab2 are both required for the endocytosis of CFTR, Dab2 also plays a significant role in regulating CFTR's protein half-life.
Figure 4. Increased CFTR half-life in μ2 and Dab2 depleted cells.

A. The effects of μ2 depletion on CFTR half-life. B. The effects of Dab2 depletion on CFTR half-life. CFBE41o-WT cells were transfected with control, μ2 (A) or Dab2 (B) siRNA oligos. Depletion efficiencies were >95%. 72 h post-transfection, cells were metabolically labeled with 35S-methioline followed by chases in complete growth medium for the time periods indicated. CFTR was immunoprecipitated with anti-CFTR antibody (24–1) followed by SDS PAGE and autoradiography. CFTR half-lives were calculated as described in the Experimental Procedures. Representative gels are shown on the left, quantitative analysis of CFTR half-lives under each experimental condition is shown on the right. While μ2 depletion only slightly enhanced CFTR half-life, Dab2 depletion resulted in a very significant, ~3-fold increase, n=3.
The function of cell surface CFTR is increased in the absence of Dab2
CFTR is present in a functional complex at the cell surface [31]. Although numerous components of the complex have been identified, the role of Dab2 in the maintenance of the complex has not been examined. Since Dab2 depletion enhanced the cell surface levels of CFTR and might alter the composition and more importantly the functionality of the complex, we performed Ussing chamber studies to test whether higher CFTR levels resulted in enhanced CFTR function. Following Dab2 depletion or transfection with a control siRNA, monolayers were mounted into Ussing chambers and forskolin was used to indirectly elevate intracellular cAMP levels to activate CFTR (Figure 5). Genistein was added to maximally stimulate the channels and to determine the non-cAMP component activity. When the current reached a maximum, CF-inh172 was at added to block Isc. Under these conditions, the ΔIsc was 88.4 ± 7.4 μA/cm2 in control cells, and 175.4 ± 1.9 μA/cm2 in the Dab2-depleted cells. The results demonstrated that cell surface CFTR function was increased following Dab2 depletion and suggest that Dab2 is not required to keep CFTR in a functional complex since the ratio of the cAMP and genistein activated currents did not change compared to the control. Specifically, the results demonstrate that in Dab2 depleted cells, the CFTR currents were increased by ~2-fold, and this is consistent with the elevated cell surface levels of CFTR. Furthermore, the baseline currents were increased in the Dab2 depleted cells, and this is consistent with a previous study suggesting that the constitutive activity of CFTR increased as the expression levels increased [32].
Figure 5. Increased transepithelial chloride transport following Dab2 depletion in CFBE41o-WT monolayers.
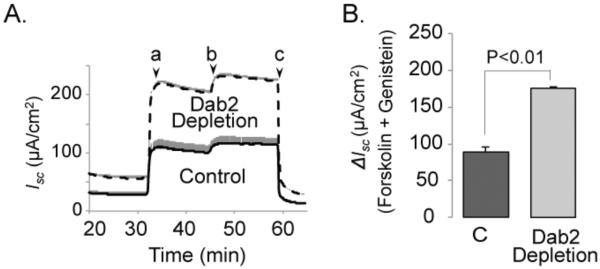
CFBE41o-WT cells were transfected with control or Dab2 siRNA oligos. 24 h after transfection, cells were lifted, seeded onto Transwell filters and cultured for additional 4 to 5 days. Dab2 depletion efficiency was >95%. The short-circuit currents (Isc) across the monolayers were measured in Ussing chambers as described in the Experimental Procedures section. A. Representative tracings from control and Dab2 depleted monolayers. Experiments were performed in the setting of a C1− gradient between the basolateral and apical compartments. Following addition of amiloride (100 μM), 20 μM forskolin (a), 50 μM genistein (b) and 10 μM CFTRInh-172 were added at times indicated. B. Forskiolin+genistein activated Isc. ΔIsc were calculated as an increase of Isc after forskolin and genistein addition over the base-line currents, n=4.
c-Cbl and CHIP depletion have no effect on CFTR endocytosis, but c-Cbl does affect CFTR protein half-life
Since CFTR half-life was dramatically increased by the Dab2 depletion, we tested if Dab2 depletion was somehow interfering with the function of two E3 ligases known to interact with CFTR, c-Cbl and CHIP [21, 33]. In the first experiment, we examined the role of c-Cbl that has been shown to regulate CFTR endocytosis and lysosomal targeting [21]. After siRNA KD of c-Cbl in polarized airway epithelia, we monitored CFTR endocytosis. Surprisingly, we saw no evidence that c-Cbl depletion affected CFTR internalization, even though c-Cbl depletion was greater than 90% (Figure 6A). In metabolic pulse-chase experiments, CFTR's half-life changed from 13.1 ± 2.4 h under control conditions to 18.5 ± 3.1 h upon c-Cbl KD, indicating a modest effect on CFTR stability (Figure 6B). These results indicate that while c-Cbl does not play a significant role in CFTR endocytosis, it may contribute to CFTR ubiquitination at a post-endocytic step. A similar analysis on CHIP KD revealed that CHIP depletion had no effect on either CFTR internalization or half-life (Figure 7A and B). In summary, the depletion of the two E3 ligases had no effect on CFTR internalization, and only c-Cbl depletion had a modest increase in CFTR stability, suggesting that the Dab2-associated effects on CFTR stability are independent of these two E3 ligases.
Figure 6. The effects of c-Cbl depletion on CFTR endocytosis rates (A) and protein half-life (B).

CFBE41o-WT cells were transfected with c-Cbl or control siRNA oligos. Top panels demonstrate the efficiency of c-Cbl depletion for each experiment. β-actin was detected as a loading control. The rate of CFTR endocytosis (A) and half-life (B) were measured as described in the Experimental Procedures section. Representative images from 3 experiments are shown in the middle panel. Depletion of c-Cbl did not affect the dynamics of CFTR internalization through a 7.5 minute time period (A, bottom). However, the half-life of CFTR was slightly longer following c-Cbl depletion (B, bottom).
Figure 7. CHIP depletion has no effect on CFTR endocytosis (A) or protein half-life (B).

CFBE41o-WT cells were transfected with CHIP or control siRNA oligos. The efficiency of CHIP depletion was assessed by Western blotting using anti-CHIP antibody. β-actin served as a loading control (A, top left panel). CFTR endocytosis (A) and protein half-life measurements (B) were performed as described in the Experimental Procedures. Representative gels from 3 experiments are shown on the left. Quantitation of CFTR endocytosis dynamics and half-life measurements are shown on the right. CHIP depletion did not affect the internalization rates or the half-life of cell surface CFTR.
Dab 2 depletion results in enlargement of the early endosomal compartments
Because Dab2 depletion dramatically increased CFTR half-life, next we investigated morphological changes in post-endocytic, intracellular compartments following Dab2 depletion by immunocytochemistry. CFTR staining was greatly elevated in Dab2 depleted cells (Figure 8A and B), but more interestingly, the vesicles of the EEA1 compartment, the early sorting endosome, were dramatically enlarged compared to control cells (compare Figure 8A and B, middle panels). Furthermore, the number of EEA1 positive vesicles in each cell was also increased as was the total amount of EEA1 protein as monitored by Western blot (Figure 8C). This suggests that Dab2 may be necessary for protein exit from the early endosome. Based on a recent study by Penheiter et al. [34] who demonstrated that Dab2 depletion in NIH-3T3 cells inhibited type II transforming growth factor-β receptor recycling, we tested Dab2 depletion on protein recycling to the cell surface. We used transferrin receptor (TFR) as our model, since it efficiently recycles to the cell surface [35]. We utilized a fluorescently labeled transferrin for these studies. The cells were pulsed for 5 min with fluorescent transferrin (Alexa 594-Tfn) and then, Tfn trafficking was monitored by immunofluorescence microscopy over a 30 min period. Tfn remains associated with the receptor until it is recycled to the cell surface and is released as apotransferrin, and therefore the loss of cell-associated fluorescence over time indicates recycling efficiency. The results indicate that in the control cells, the cell-associated Tfn was completely cleared after 30 min (Figure 9), whereas in the Dab2 depleted cells, much of the loaded fluorescent Tfn remained cell-associated for 30 min. This demonstrates that in the absence of Dab2, Tfn receptor recycling is dramatically compromised. The results indicate, therefore, that Dab2 is important for protein recycling.
Figure 8. Dab2 depletion leads to the enlargement of early endosomes.

CFBE41o-WT cells were transfected with control (A) or Dab2 (B) siRNA oligos. 72 h after transfection, the cells were fixed and processed for immunofluorescence microscopy using a rabbit polyclonal anti-CFTR antibody (green) and a mouse monoclonal anti-EEA1 antibody (red). The nuclei were stained with DAPI. In control cells (top), the intracellular CFTR partially co-localizes with the endosomal marker, EEA1 (top right panel, inlay). In Dab2 depleted cells, both the number and the size of the EEA1-containing endosomes increased and CFTR co-localization with EEA1 in the enlarged endosomes is evident, as indicated by the yellow color (bottom right panel, inlay). C. Increased levels of EEA1 in Dab2 depleted cells. CFBE41o-WT cells were transfected with control or Dab2 siRNAs and immunoblotting was performed using the antibodies indicated.
Figure 9. Dab2 depletion reduces transferrin recycling.

CFBE41o-WT cells were transfected with control (A) or Dab2 (B) siRNA oligos. The cells were then loaded with AlexaFluor-594-transferrin (Tfn) as described in the Experimental Procedures, followed by incubation at 37°C for the time periods indicated. During the incubations, the internalized AlexaFluor-594-Tfn is recycled back to the cell surface and the fluorescent Tfn is released to the media, resulting in a loss of fluorescence. Dab2 depletion resulted in the intracellular (vesicular) accumulation of Tfn (B), indicating a delay in Tfn recycling.
Delivery of CFTR to the late endosomal compartment is inhibited in Dab2 depleted cells
Although our results indicated that in Dab2-depleted cells, endocytosis and recycling were reduced, we reasoned that inhibition of delivery to the late endosomal/lysosomal compartments [30] must be inhibited if the CFTR half-life is extended during Dab2 depletion. To test this idea, we examined CFTR trafficking to the late endosomal compartment by monitoring CFTR co-localization with a marker of this compartment, the mannose-6-phosphate receptor (M6PR). To inhibit protease activity in this compartment, we treated the cells with a weak base, NH4Cl, to enhance visualization of CFTR in this compartment prior to degradation. We have successfully employed this method in the past to monitor transferrin receptor chimeras that were targeted to the lysosome [35]. The results indicate that under control conditions, CFTR and the M6PR do not co-localize (Figure 10A), whereas in the presence of protease inhibition, there is clear evidence that CFTR accumulated in these compartments (Figure 10B), presumably on its way to degradation. However, in Dab2 depleted cells, CFTR does not co-localize with the M6PR if proteases are inhibited, suggesting that CFTR trafficking to the late endosome is compromised in Dab2-depleted cells (Figure 10C). The results therefore suggest that CFTR delivery to the later stages of the endocytic pathways is compromised in Dab2-depleted cells, and this is consistent with the dramatically enhanced CFTR half-life that we monitored in the metabolic pulse-chase experiments.
Figure 10. CFTR delivery to the late endosomes is inhibited in Dab2 depleted cells.

CFBE41o-WT cells were treated with control (A, B) or Dab2-specific (C) siRNA oligos. 72 h after the transfection, one set of the control (B) and the Dab2 depleted cells (C) were treated with 5 mM NH4Cl for 16 h followed by immunofluorescent staining of CFTR and mannose-6-phosphate receptor (M6PR). A. CFTR and M6PR do not co-localize in control, untreated cells. B. NH4Cl treatment (inhibition of lysosomal degradation) resulted in CFTR and M6PR co-localization (right panel, yellow). C. Dab2 depletion and NH4Cl treatment together enhanced CFTR staining, however no co-localization of CFTR and M6PR is apparent.
Discussion
A major focus of this study was to determine the role of two adaptor molecules, AP-2 (μ2) and Dab2, in CFTR endocytosis, recycling, and degradation from the cell surface. Understanding the processes that regulate wild type CFTR cell surface levels will provide a background for understanding the cell surface instability of rescued ΔF508 CFTR [25]. We examined two endocytic adaptor molecules, AP-2 and Dab2, and two E3 ligases, CHIP and c-Cbl. c-Cbl has also been reported to serve as an endocytic adaptor molecule [21]. Since AP-2 and Dab2 recognize different target recognition sequences, e.g., YDSI versus FXNPXY, it was unclear how two different adaptors function in CFTR endocytosis. If both adaptors were used interchangeably, then inhibition of one might only have limited effects on CFTR endocytosis. In contrast, we see similar and very significant effects of both AP-2 and Dab2 KDs on CFTR internalization. Furthermore, since depletion of both μ2 and Dab2 resulted in a similar effect as the depletion of one, these results are consistent with the hypothesis that they work in concert and disruption of one of them eliminates the function of the other during endocytosis.
Our results are consistent with a recent report indicating that in intestinal epithelial cells, Dab2 directly interacts with the α-2 subunit of AP-2, but not CFTR [23]. In the model by Collaco et al. [23], the α2 subunit of AP-2 interacts directly with both CFTR and Dab2, and Dab2 interacts with myosin VI, which interacts with the actin cytoskeleton. Thus Dab2 forms a bridge between AP-2 and myosin VI and does not bind directly to CFTR. Therefore, Dab2 does not serve as a CFTR adaptor in the classic sense. This model is consistent with the earlier observations by Morris et al. [36,37] that Dab2 interacts with the α2 subunit of AP-2 and myosin VI. Our CFTR internalization data in μ2 and Dab2 depleted cells are consistent with the model proposed by Collaco et al. [23] for CFTR endocytosis. Since both the μ2 and the α2 subunits interact with CFTR [19, 23], this suggests that both subunits of AP-2 have critical contact sites for CFTR. In the Collaco et al. [23] studies, depletion of the α2 subunit interfered with CFTR endocytosis, while we found similar results following KD of the μ2 subunit of AP-2. These two findings are also consistent with the idea that depletion of either AP-2 subunit interferes with the assembly of the AP-2 molecule and therefore inhibits AP-2 function. It will be interesting to see which region of CFTR is recognized by the α2 chain of AP-2.
Because our studies indicated that Dab2 depletion dramatically increased the half-life of CFTR, we tested the idea that c-Cbl might be mediating this effect. We based our hypothesis on a recent report indicating that in human airway epithelial cells depletion of c-Cbl enhanced CFTR's half-life and also inhibited endocytosis [21]. These results suggested that c-Cbl, a known E3 ligase also functioned as an adaptor molecule. We tested whether c-Cbl and μ2 were interchangeable during endocytosis. Surprisingly, c-Cbl depletion (~90%) did not affect CFTR endocytosis. The only effect of c-Cbl depletion was a modest increase in CFTR half-life. This supports the view that c-Cbl functions at a post-endocytic step [21]. Therefore, our results in combination with those of Collaco et al. [23] confirm that the only adaptor for CFTR endocytosis is AP-2, as was originally proposed by Weixel et al. [38]. However, the most important finding of these studies is that we determined the differences in the function of AP-2 and Dab2.
Because we observed a dramatic increase in the half-life of CFTR following Dab2 depletion, we wanted to understand the mechanism of this effect. Our first analysis indicated that two E3 ligases known to associate with CFTR, c-Cbl and CHIP [21, 33] were not necessary for CFTR endocytosis and more importantly depletion of these E3 ligases did not dramatically increase CFTR half-life. However, by examining the morphology of the endocytic compartments following Dab2 depletion, we observed that the sorting endosome or EEA1 compartment was enlarged in these cells. This result is consistent with those of Penheiter et al. [34] who were examining type II transforming growth factor-beta (TGF-β) receptor recycling in NIH-3T3 cells. They studied the role of Dab2 in TGF-β receptor endocytosis and found that siRNA KD of Dab2 had no effect on TGF-β receptor internalization. Dab2 depletion did, however, result in enlarged EEA1-positive endosomes, and interfered with TGF-β receptor trafficking to the Rab11-positive recycling endosome and TGF-β receptor recycling [34]. How this affected TGF-β receptor half-life, however, was not examined in these studies. This sorting block is also consistent with the results of Chibalina et al. [39] who demonstrated that siRNA depletion of myosin VI or its interacting protein, lemur tyrosine kinase 2 (LMTK2), blocked protein transport out of the sorting endosome, resulting in enlarged endosomes and a delay in transferrin receptor recycling. Our studies in Dab2 depleted cells indicate a similar phenotype of enlarged endosomes, with a corresponding inhibition of transferrin receptor recycling. Therefore, our results presented herein in combination with those discussed above indicate significant roles for myosin VI, Dab2, and LMTK2 in sorting of proteins from the early endosome to the recycling endosome. Our results also establish that Dab2 is necessary for both endocytosis and recycling.
In previous studies we examined mutations in CFTR that both enhanced [27] and inhibited endocytosis [18], and neither one of these mutations affected CFTR protein half-life. Furthermore, in the present studies, μ2 depletion only had a modest effect on CFTR half-life. Therefore it is unclear why the block at the sorting endosome stage would have a dramatic effect on CFTR's half-life in Dab2 depleted cells. This was even more surprising since previous studies reported that blocking the exit from the recycling endosome has no effect on CFTR expression levels [40], suggesting that the half-life was not affected. Therefore in order to gain a deeper understanding of this mechanism, we examined how CFTR transport to the late endosome was affected in Dab2 depleted cells. The results indicated that Dab2 is necessary for CFTR delivery into the degradation compartment. Interestingly, it has been demonstrated that depletion of the C-terminal Eps15 homology domain 4 (EDH4) by siRNA leads to the generation of enlarged early endosomal structures that contain Rab5 and EEA1 and accumulation of LDL particles destined for degradation [41]. Because the LDL particles accumulated rather than being delivered to the late endosome and lysosome, the authors suggested that EDH4 KD indicated that EDH4 was not only involved in the recycling of molecules through the sorting endosome, but also in delivery of cargo destined for degradation. Based on the similar phenotype of the enlargement of the sorting endosome where recycling receptors and cargo are trapped, it is tempting to speculate that the myosin VI and Dab2 are required for efficient exit from the sorting endosome for delivery to both the recycling compartment and the late endosome, and therefore Dab2 is required at multiple steps in the endocytic pathway.
In summary, the results of our study indicate the both μ2 and Dab2 are necessary for CFTR endocytosis, however, only Dab2 is essential for the post-endocytic trafficking of CFTR either for recycling or for delivery to the late endosome. Based on this, we propose a model (Figure 11) that illustrates 3 sites in the endocytic pathway are affected by Dab2 depletion: 1) internalization from the cell surface; 2) delivery from the sorting endosome to the recycling endosome; and 3) delivery to the late endosome. Given the complexity of the trafficking of the wild-type CFTR protein in the endocytic pathway, it will be intriguing to see how these types of manipulations with siRNA KDs will affect the trafficking of a rescued mutant protein such as ΔF508 CFTR.
Figure 11. A model for Dab2 function in CFTR trafficking.
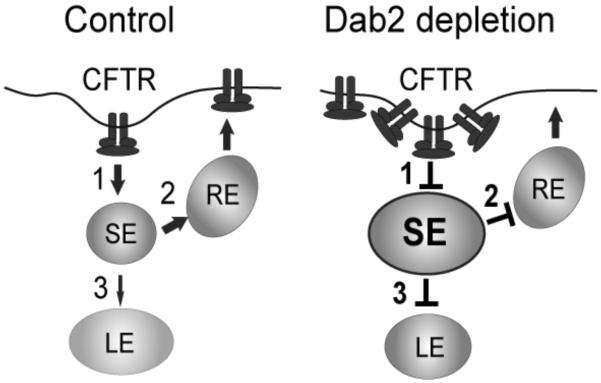
Dab2 regulates three steps of CFTR trafficking within the endocytic pathway: 1) internalization from the cell surface; 2) exit from the sorting endosome to the recycling compartment; and 3) delivery to the late endosomal compartment. This block to the later stages of the endocytic pathway reduces the degradation rate of CFTR.
Acknowledgements
This work was supported by NIH grants R01 DK60065 (JFC), R01 HL076587 (ZB) and K23 DK075788 (SMR) and RG82840N (LF) from the American Lung Association. This work was also supported by Dr. Eric Sorscher and grants to the Gregory Fleming James Cystic Fibrosis Research Center (P30 DK 072482 and R474-CR11).
Abbreviations
- c-Cbl
casitas B-lineage lymphoma
- CHIP
carboxy terminus of Hsp70-interacting protein
- cAMP
cyclic AMP
- co-IP
co-immunoprecipitation
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- Dab2
Disabled-2
- HRP
horseradish peroxidase
- IGEPAL
octylphenoxypolyethoxyethanol
- IP
immunoprecipitation
- Isc
short-circuit current
- KD
knockdown
- M6PR
mannose-6-phosphate receptor
- PVDF
Polyvinylidene fluoride
- TGF-β
Transforming growth factor beta
- Tfn
transferrin
- TR
transferrin receptor
References
- 1.Trowbridge IS, Collawn JF, Hopkins CR. Signal-dependent membrane protein trafficking in the endocytic pathway. Annu. Rev. Cell Biol. 1993;9:129–161. doi: 10.1146/annurev.cb.09.110193.001021. [DOI] [PubMed] [Google Scholar]
- 2.Pearse BM. Receptors compete for adaptors found in plasma membrane coated pits. EMBO J. 1988;7:3331–3336. doi: 10.1002/j.1460-2075.1988.tb03204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kirchhausen T. Adaptors for clathrin-mediated traffic. Annu. Rev. Cell Dev. Biol. 1999;15:705–732. doi: 10.1146/annurev.cellbio.15.1.705. [DOI] [PubMed] [Google Scholar]
- 4.Owen DJ, Vallis Y, Pearse BM, McMahon HT, Evans PR. The structure and function of the beta 2-adaptin appendage domain. EMBO J. 2000;19:4216–4227. doi: 10.1093/emboj/19.16.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Traub LM, Downs MA, Westrich JL, Fremont DH. Crystal structure of the alpha appendage of AP-2 reveals a recruitment platform for clathrin-coat assembly. Proc. Natl. Acad. Sci. U.S.A. 1999;96:8907–8912. doi: 10.1073/pnas.96.16.8907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maurer ME, Cooper JA. The adaptor protein Dab2 sorts LDL receptors into coated pits independently of AP-2 and ARH. J. Cell. Sci. 2006;119:4235–4246. doi: 10.1242/jcs.03217. [DOI] [PubMed] [Google Scholar]
- 7.Collawn JF, Stangel M, Kuhn LA, Esekogwu V, Jing SQ, Trowbridge IS, Tainer JA. Transferrin receptor internalization sequence YXRF implicates a tight turn as the structural recognition motif for endocytosis. Cell. 1990;63:1061–1072. doi: 10.1016/0092-8674(90)90509-d. [DOI] [PubMed] [Google Scholar]
- 8.Chen WJ, Goldstein JL, Brown MS. NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low density lipoprotein receptor. J. Biol. Chem. 1990;265:3116–3123. [PubMed] [Google Scholar]
- 9.Traub LM. Sorting it out: AP-2 and alternate clathrin adaptors in endocytic cargo selection. J. Cell Biol. 2003;163:203–208. doi: 10.1083/jcb.200309175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohn JA, Strong TV, Picciotto MR, Nairn AC, Collins FS, Fitz JG. Localization of the cystic fibrosis transmembrane conductance regulator in human bile duct epithelial cells. Gastroenterology. 1993;105:1857–1864. doi: 10.1016/0016-5085(93)91085-v. [DOI] [PubMed] [Google Scholar]
- 11.Strong TV, Boehm K, Collins FS. Localization of cystic fibrosis transmembrane conductance regulator mRNA in the human gastrointestinal tract by in situ hybridization. J. Clin. Invest. 1994;93:347–354. doi: 10.1172/JCI116966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kreda SM, Mall M, Mengos A, Rochelle L, Yankaskas J, Riordan JR, Boucher RC. Characterization of wild-type and deltaF508 cystic fibrosis transmembrane regulator in human respiratory epithelia. Mol. Biol. Cell. 2005;16:2154–2167. doi: 10.1091/mbc.E04-11-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertrand CA, Frizzell RA. The role of regulated CFTR trafficking in epithelial. Am. J. Physiol. Cell Physiol. 2003;285:C1–18. doi: 10.1152/ajpcell.00554.2002. [DOI] [PubMed] [Google Scholar]
- 14.Ameen N, Silvis M, Bradbury NA. Endocytic trafficking of CFTR in health and disease. J. Cyst. Fibros. 2007;6:1–14. doi: 10.1016/j.jcf.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okiyoneda T, Lukacs GL. Cell surface dynamics of CFTR: the ins and outs. Biochim. Biophys. Acta. 2007;1773:476–479. doi: 10.1016/j.bbamcr.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 16.Prince LS, Peter K, Hatton SR, Zaliauskiene L, Cotlin LF, Clancy JP, Marchase RB, Collawn JF. Efficient endocytosis of the cystic fibrosis transmembrane conductance regulator requires a tyrosine-based signal. J Biol Chem. 1999;274:3602–3609. doi: 10.1074/jbc.274.6.3602. [DOI] [PubMed] [Google Scholar]
- 17.Hu W, Howard M, Lukacs GL. Multiple endocytic signals in the C-terminal tail of the cystic fibrosis transmembrane conductance regulator. Biochem. J. 2001;354:561–572. doi: 10.1042/0264-6021:3540561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peter K, Varga K, Bebok Z, McNicholas-Bevensee CM, Schwiebert L, Sorscher EJ, Schwiebert EM, Collawn JF. Ablation of internalization signals in the carboxyl-terminal tail of the cystic fibrosis transmembrane conductance regulator enhances cell surface expression. J. Biol. Chem. 2002;277:49952–49957. doi: 10.1074/jbc.M209275200. [DOI] [PubMed] [Google Scholar]
- 19.Weixel KM, Bradbury NA. Mu 2 binding directs the cystic fibrosis transmembrane conductance regulator to the clathrin-mediated endocytic pathway. J. Biol. Chem. 2001;276:46251–46259. doi: 10.1074/jbc.M104545200. [DOI] [PubMed] [Google Scholar]
- 20.Swiatecka-Urban A, Boyd C, Coutermarsh B, Karlson KH, Barnaby R, Aschenbrenner L, Langford GM, Hasson T, Stanton BA. Myosin VI regulates endocytosis of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 2004;279:38025–38031. doi: 10.1074/jbc.M403141200. [DOI] [PubMed] [Google Scholar]
- 21.Ye S, Cihil K, Stolz DB, Pilewski JM, Stanton BA, Swiatecka-Urban A. c-Cbl facilitates endocytosis and lysosomal degradation of cystic fibrosis transmembrane conductance regulator in human airway epithelial cells. J. Biol. Chem. 2010;285:27008–27018. doi: 10.1074/jbc.M110.139881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spudich G, Chibalina MV, Au JS, Arden SD, Buss F, Kendrick-Jones J. Myosin VI targeting to clathrin-coated structures and dimerization is mediated by binding to Disabled-2 and PtdIns(4,5)P2. Nat. Cell Biol. 2007;9:176–183. doi: 10.1038/ncb1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collaco A, Jakab R, Hegan P, Mooseker M, Ameen N. Alpha-AP-2 directs myosin VI-dependent endocytosis of cystic fibrosis transmembrane conductance regulator chloride channels in the intestine. J. Biol. Chem. 2010;285:17177–17187. doi: 10.1074/jbc.M110.127613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bebok Z, Collawn JF, Wakefield J, Parker W, Li Y, Varga K, Sorscher EJ, Clancy JP. Failure of cAMP agonists to activate rescued deltaF508 CFTR in CFBE41o- airway epithelial monolayers. J. Physiol. 2005;569:601–615. doi: 10.1113/jphysiol.2005.096669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varga K, Goldstein RF, Jurkuvenaite A, Chen L, Matalon S, Sorscher EJ, Bebok Z, Collawn JF. Enhanced cell-surface stability of rescued DeltaF508 cystic fibrosis transmembrane conductance regulator (CFTR) by pharmacological chaperones. Biochem. J. 2008;410:555–564. doi: 10.1042/BJ20071420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fu L, Gao YS, Sztul E. Transcriptional repression and cell death induced by nuclear aggregates of non-polyglutamine protein. Neurobiol. Dis. 2005;20:656–665. doi: 10.1016/j.nbd.2005.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jurkuvenaite A, Varga K, Nowotarski K, Kirk KL, Sorscher EJ, Li Y, Clancy JP, Bebok Z, Collawn JF. Mutations in the amino terminus of the cystic fibrosis transmembrane conductance regulator enhance endocytosis. J Biol Chem. 2006;281:3329–3334. doi: 10.1074/jbc.M508131200. [DOI] [PubMed] [Google Scholar]
- 28.Okiyoneda T, Barriere H, Bagdany M, Rabeh WM, Du K, Hohfeld J, Young JC, Lukacs GL. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science. 2010;329:805–810. doi: 10.1126/science.1191542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rowe SM, Pyle LC, Jurkevante A, Varga K, Collawn J, Sloane PA, Woodworth B, Mazur M, Fulton J, Fan L, Li Y, Fortenberry J, Sorscher EJ, Clancy JP. DeltaF508 CFTR processing correction and activity in polarized airway and non-airway cell monolayers. Pulm. Pharmacol. Ther. 2010;23:268–278. doi: 10.1016/j.pupt.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gentzsch M, Chang XB, Cui L, Wu Y, Ozols VV, Choudhury A, Pagano RE, Riordan JR. Endocytic trafficking routes of wild type and DeltaF508 cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell. 2004;15:2684–2696. doi: 10.1091/mbc.E04-03-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naren AP, Cobb B, Li C, Roy K, Nelson D, Heda GD, Liao J, Kirk KL, Sorscher EJ, Hanrahan J, Clancy JP. A macromolecular complex of beta 2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc. Natl. Acad. Sci. U.S.A. 2003;100:342–346. doi: 10.1073/pnas.0135434100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stutts MJ, Gabriel SE, Olsen JC, Gatzy JT, O'Connell TL, Price EM, Boucher RC. Functional consequences of heterologous expression of the cystic fibrosis transmembrane conductance regulator in fibroblasts. J. Biol. Chem. 1993;268:20653–20658. [PubMed] [Google Scholar]
- 33.Meacham GC, Patterson C, Zhang W, Younger JM, Cyr DM. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 2001;3:100–105. doi: 10.1038/35050509. [DOI] [PubMed] [Google Scholar]
- 34.Penheiter SG, Singh RD, Repellin CE, Wilkes MC, Edens M, Howe PH, Pagano RE, Leof EB. Type II transforming growth factor-beta receptor recycling is dependent upon the clathrin adaptor protein Dab2. Mol. Biol. Cell. 2010;21:4009–4019. doi: 10.1091/mbc.E09-12-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.White S, Hatton SR, Siddiqui MA, Parker CD, Trowbridge IS, Collawn JF. Analysis of the structural requirements for lysosomal membrane targeting using transferrin receptor chimeras. J. Biol. Chem. 1998;273:14355–14362. doi: 10.1074/jbc.273.23.14355. [DOI] [PubMed] [Google Scholar]
- 36.Morris SM, Cooper JA. Disabled-2 colocalizes with the LDLR in clathrin-coated pits and interacts with AP-2. Traffic. 2001;2:111–123. doi: 10.1034/j.1600-0854.2001.020206.x. [DOI] [PubMed] [Google Scholar]
- 37.Morris SM, Arden SD, Roberts RC, Kendrick-Jones J, Cooper JA, Luzio JP, Buss F. Myosin VI binds to and localises with Dab2, potentially linking receptor-mediated endocytosis and the actin cytoskeleton. Traffic. 2002;3:331–341. doi: 10.1034/j.1600-0854.2002.30503.x. [DOI] [PubMed] [Google Scholar]
- 38.Weixel KM, Bradbury NA. The carboxyl terminus of the cystic fibrosis transmembrane conductance regulator binds to AP-2 clathrin adaptors. J. Biol. Chem. 2000;275:3655–3660. doi: 10.1074/jbc.275.5.3655. [DOI] [PubMed] [Google Scholar]
- 39.Chibalina MV, Seaman MN, Miller CC, Kendrick-Jones J, Buss F. Myosin VI and its interacting protein LMTK2 regulate tubule formation and transport to the endocytic recycling compartment. J. Cell Science. 2007;120:4278–4288. doi: 10.1242/jcs.014217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Picciano JA, Ameen N, Grant BD, Bradbury NA. Rme-1 regulates the recycling of the cystic fibrosis transmembrane conductance regulator. Am. J. Physiol. Cell Physiol. 2003;285:C1009–1018. doi: 10.1152/ajpcell.00140.2003. [DOI] [PubMed] [Google Scholar]
- 41.Sharma M, Naslavsky N, Caplan S. A role for EHD4 in the regulation of early endosomal transport. Traffic. 2008;9:995–1018. doi: 10.1111/j.1600-0854.2008.00732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]